
Transcriptome Analysis of Ginkgo biloba L. Leaves across Late Developmental Stages Based on RNA-Seq and Co-Expression Network


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials, RNA Extraction, and Library Construction
2.2. Sequencing, Assembly, and Annotation
2.3. SNP Calling and Differential Analysis
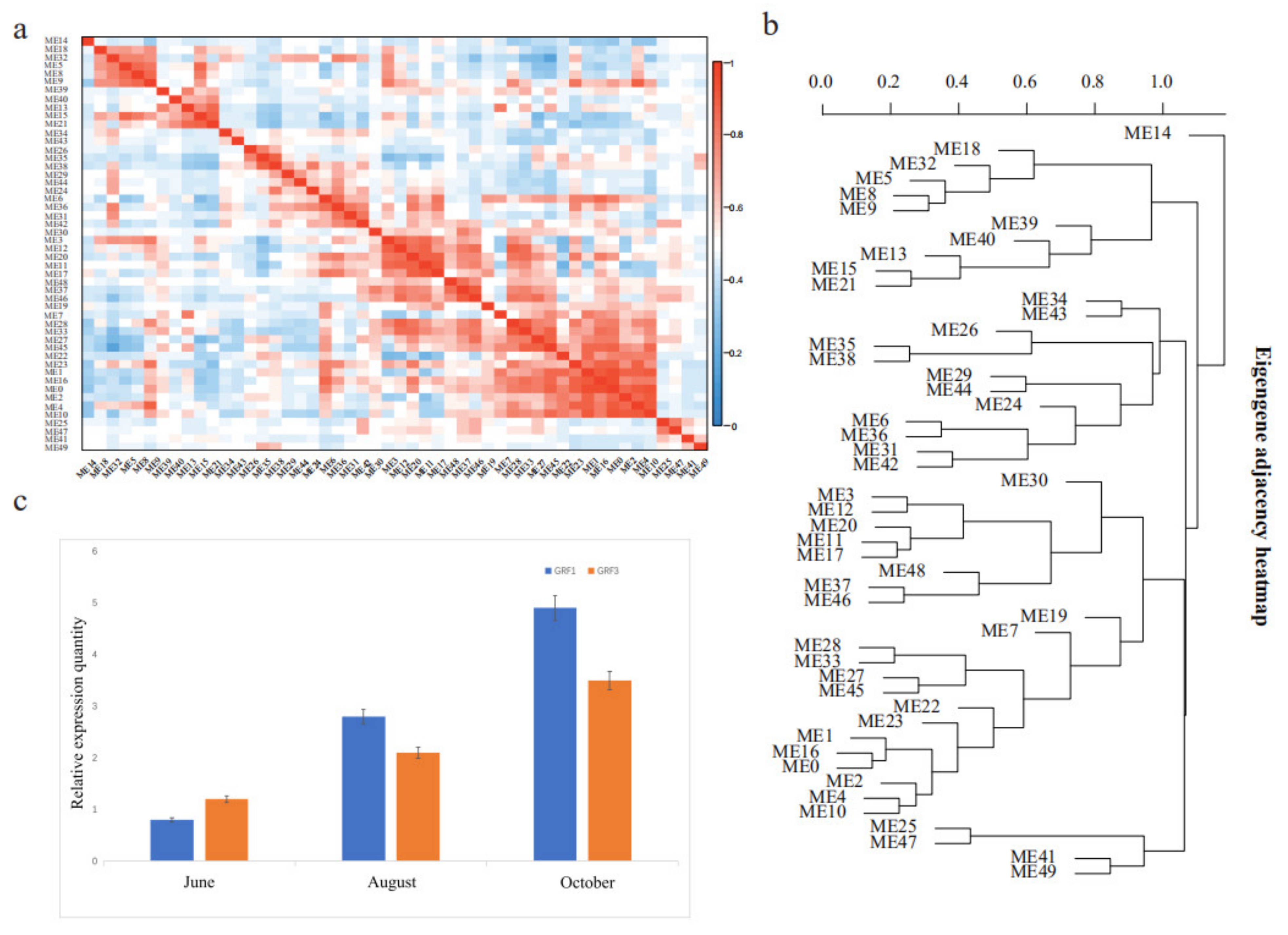
2.4. WGCNA Analysis and Quantitative Real-Time PCR
3. Results and Discussion
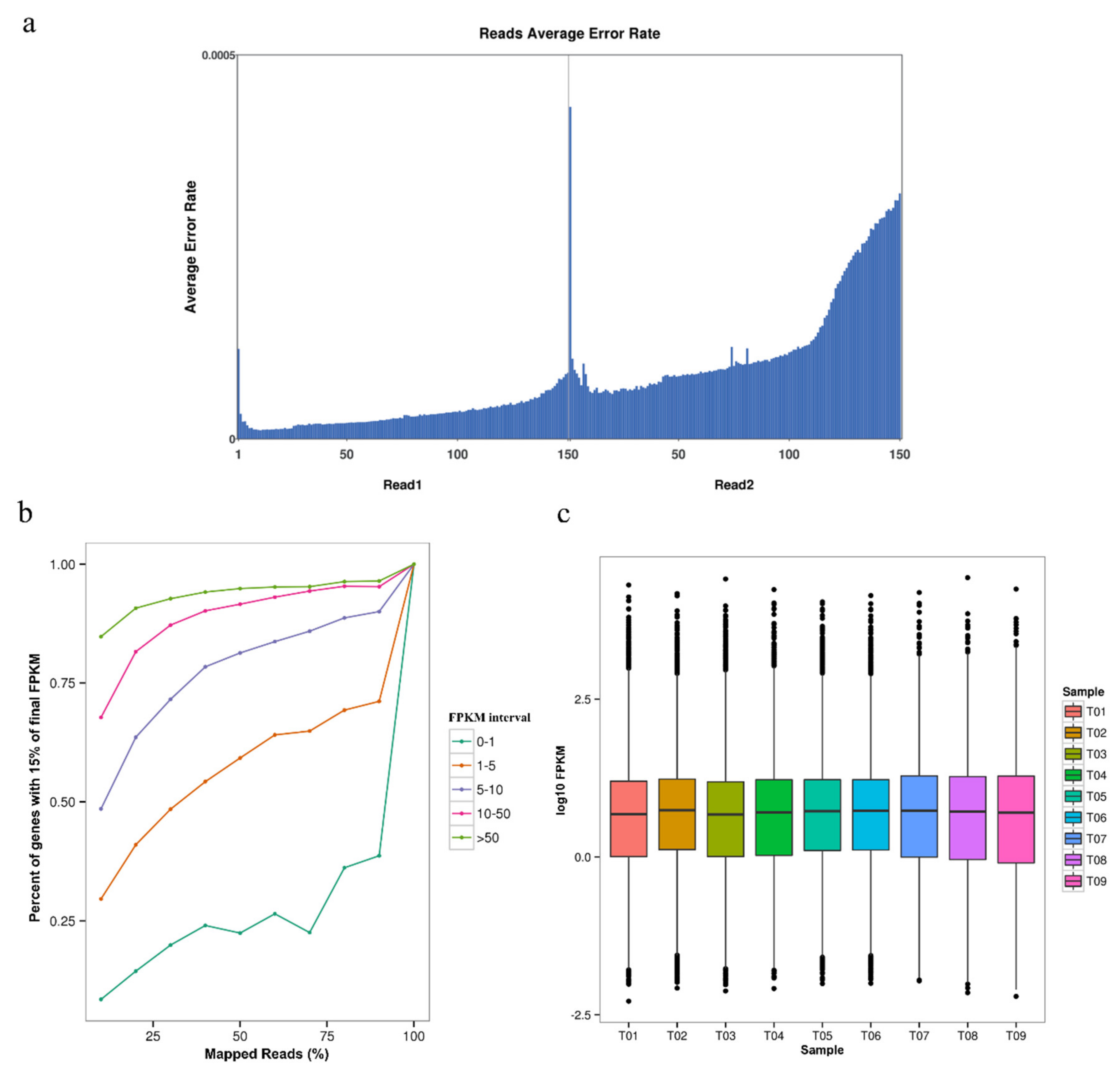
3.1. Overall Characteristics and Quality Evaluation in G. biloba Transcriptome
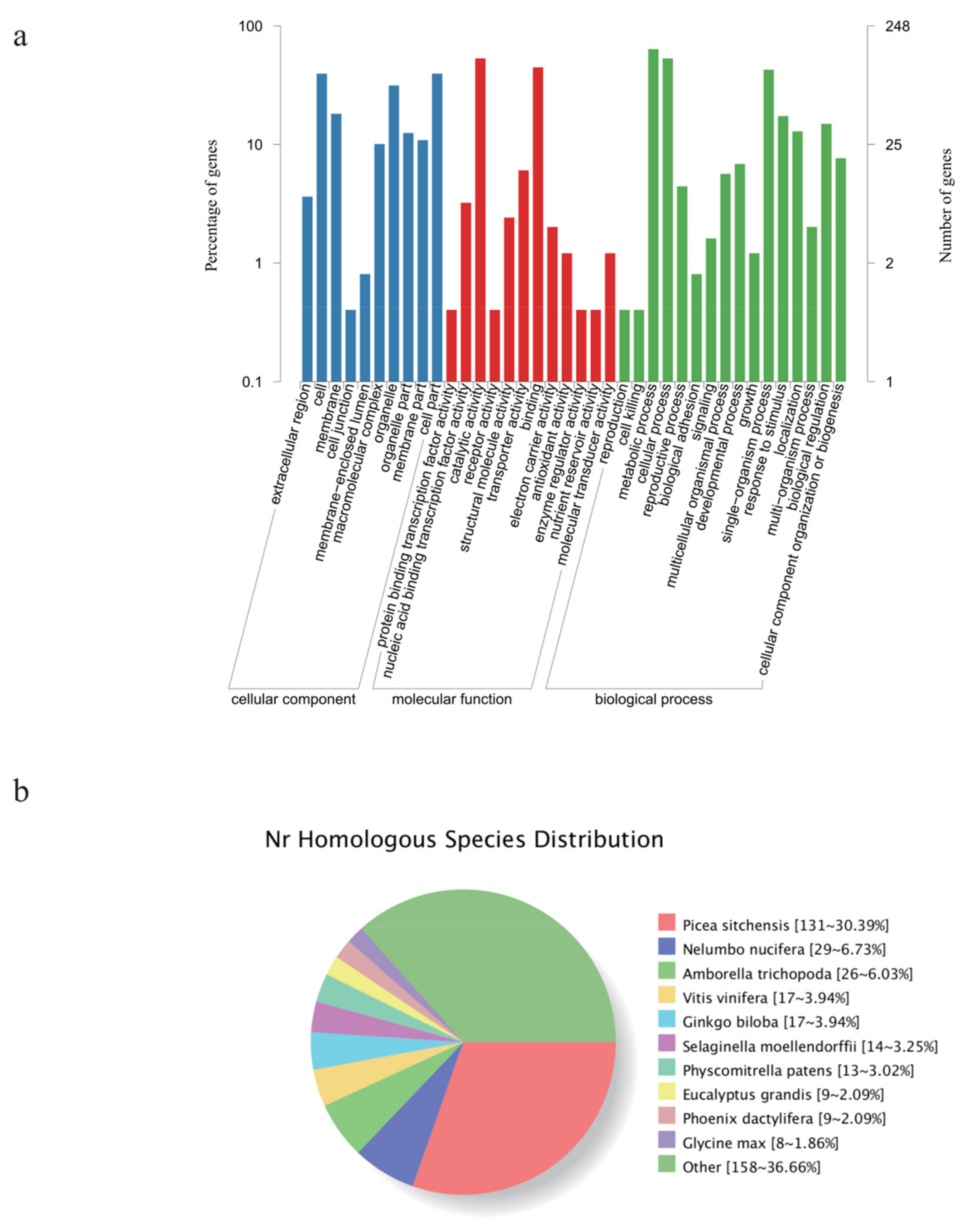
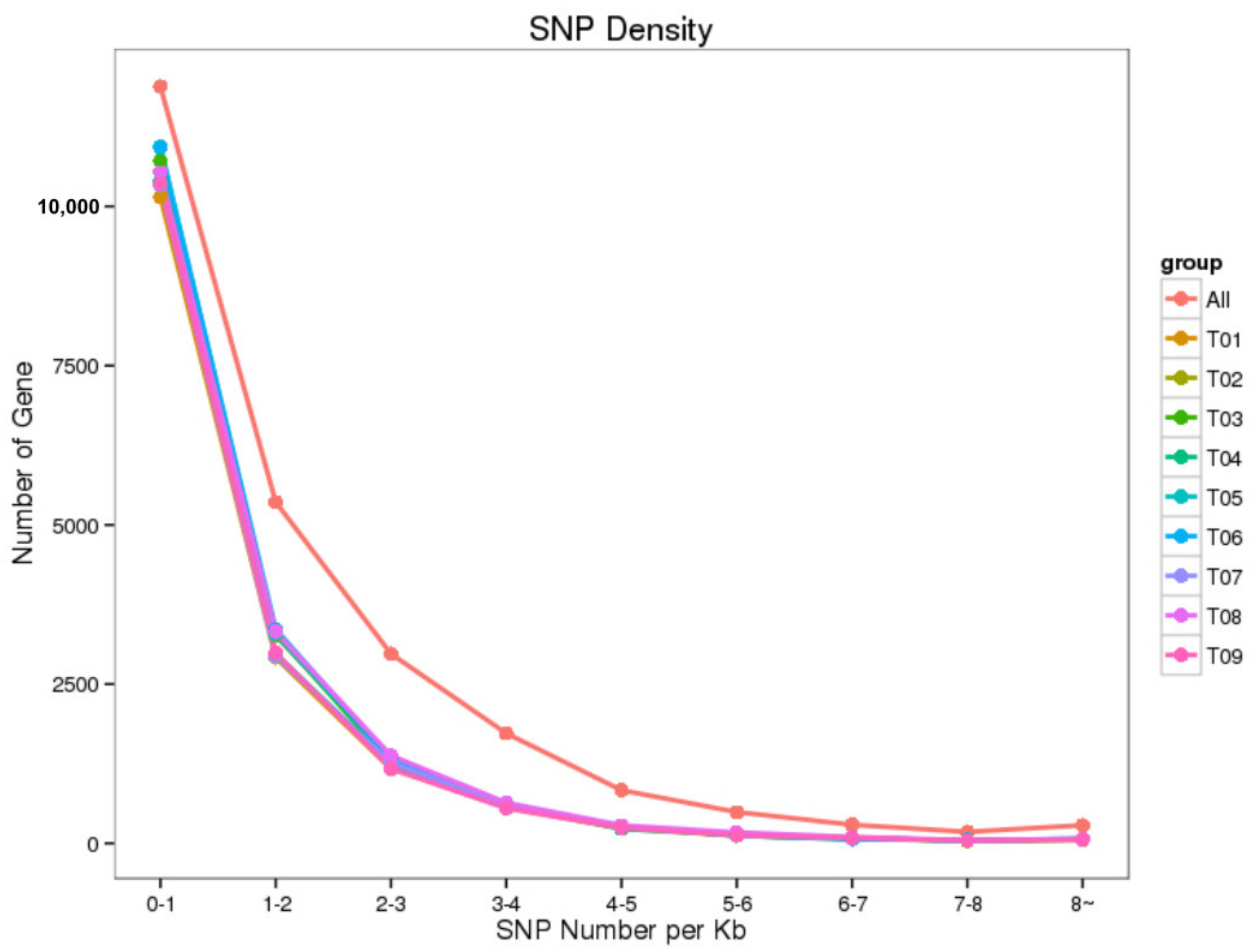
3.2. Identification of New Genes and Potential SNPs
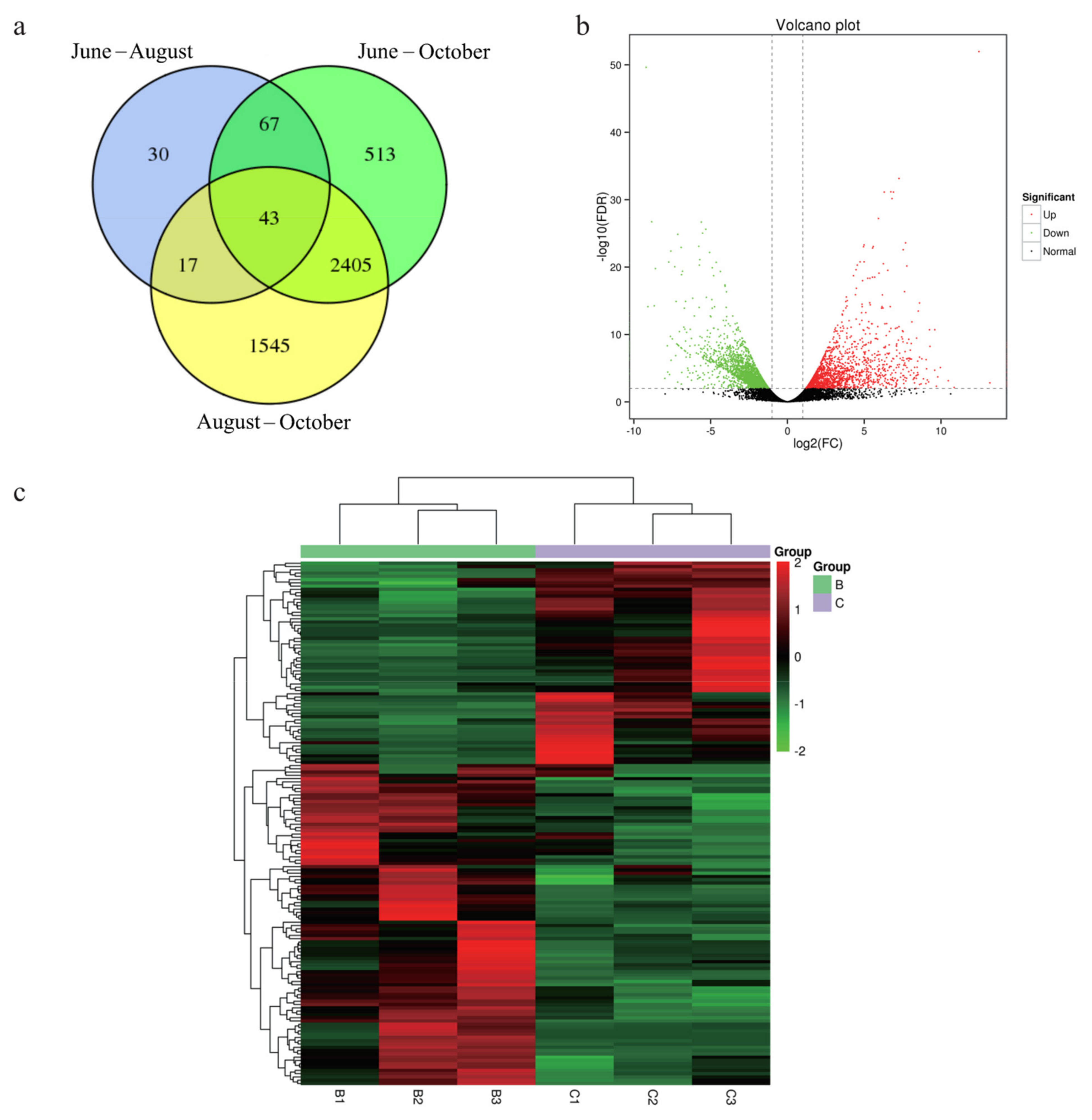
3.3. Differential Expression Analysis
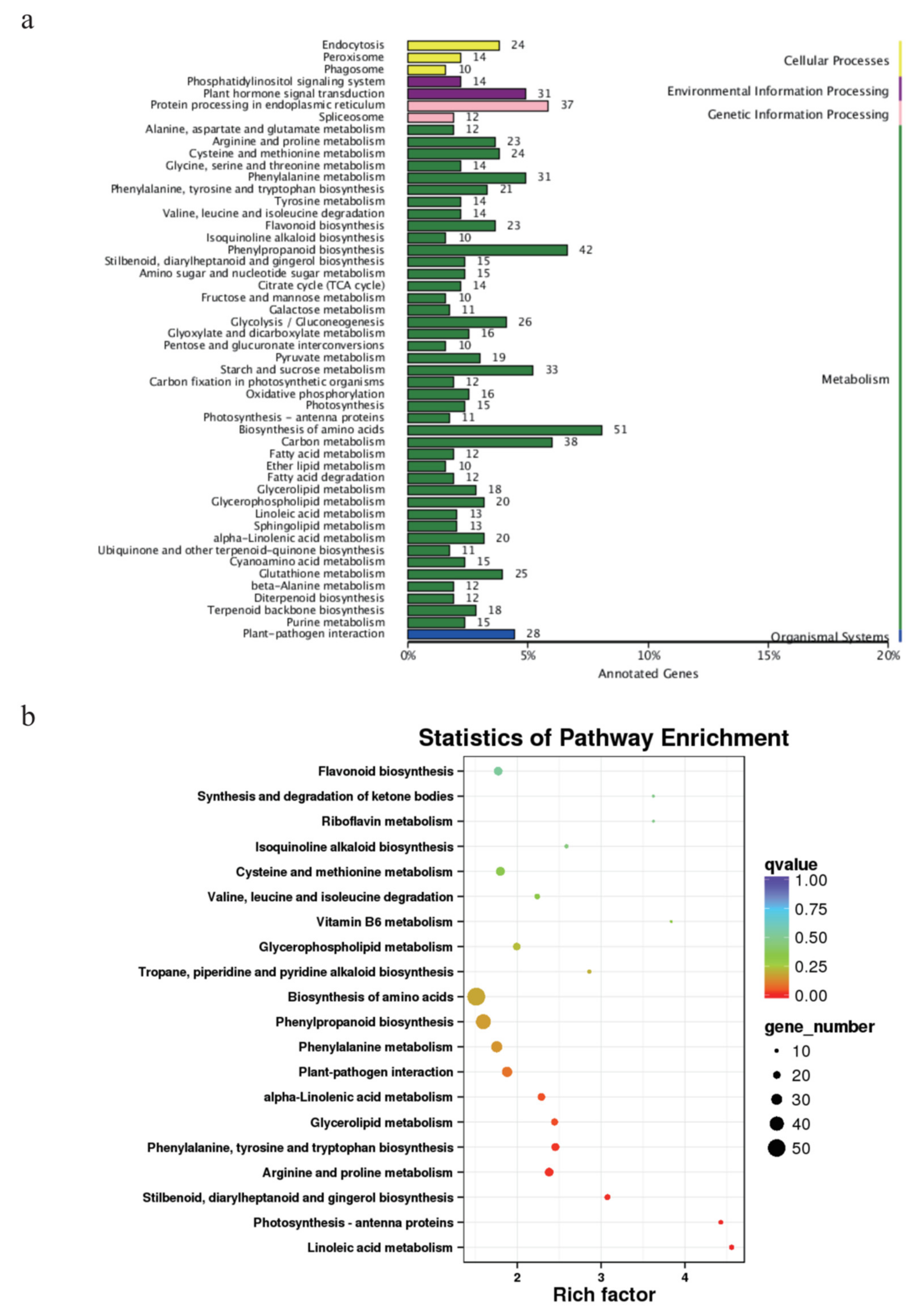
3.4. Growth-Related DEGs in G. biloba Leaves Based on Enrichment and WGCNA Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhou, Z.; Zheng, S. The missing link in Ginkgo evolution. Nature 2003, 423, 821–822. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, T.A.; Montoro, P. Chemical analysis and quality control of Ginkgo biloba leaves, extracts, and phytopharmaceuticals. J. Chromatogr. A 2009, 1216, 2002–2032. [Google Scholar] [CrossRef]
- Bastianetto, S.; Zheng, W.H.; Quirion, R. The Ginkgo biloba extract (EGb 761) protects and rescues hippocampal cells against nitric oxide-induced toxicity: Involvement of its flavonoid constituents and protein kinase C. J. Neurochem. 2000, 74, 2268–2277. [Google Scholar] [CrossRef]
- Ihl, R.; Bachinskaya, N.; Korczyn, A.D.; Vakhapova, V.; Tribanek, M.; Hoerr, R.; Napryeyenko, O.; Group, G.S. Efficacy and safety of a once-daily formulation of Ginkgo biloba extract EGb 761 in dementia with neuropsychiatric features: A randomized controlled trial. Int. J. Geriatr. Psychiatry 2011, 26, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hao, W.; Qin, Y.; Decker, Y.; Wang, X.; Burkart, M.; Schotz, K.; Menger, M.D.; Fassbender, K.; Liu, Y. Long-term treatment with Ginkgo biloba extract EGb 761 improves symptoms and pathology in a transgenic mouse model of Alzheimer’s disease. Brain Behav. Immun. 2015, 46, 121–131. [Google Scholar] [CrossRef]
- Nelissen, H.; Gonzalez, N.; Inze, D. Leaf growth in dicots and monocots: So different yet so alike. Curr. Opin. Plant Biol. 2016, 33, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Hepworth, J.; Lenhard, M. Regulation of plant lateral-organ growth by modulating cell number and size. Curr. Opin. Plant Biol. 2014, 17, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.; Dengler, N. Leaf Vascular Pattern Formation. Plant Cell 1997, 9, 1121–1135. [Google Scholar] [CrossRef]
- Baute, J.; Herman, D.; Coppens, F.; De Block, J.; Slabbinck, B.; Dell’Acqua, M.; Pe, M.E.; Maere, S.; Nelissen, H.; Inze, D. Combined Large-Scale Phenotyping and Transcriptomics in Maize Reveals a Robust Growth Regulatory Network. Plant Physiol. 2016, 170, 1848–1867. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.H.; Sun, J.; Oh, D.H.; Zielinski, R.E.; Clouse, S.D.; Huber, S.C. Enhancing Arabidopsis leaf growth by engineering the BRASSINOSTEROID INSENSITIVE1 receptor kinase. Plant Physiol. 2011, 157, 120–131. [Google Scholar] [CrossRef]
- Qin, X.; Liu, J.H.; Zhao, W.S.; Chen, X.J.; Guo, Z.J.; Peng, Y.L. Gibberellin 20-oxidase gene OsGA20ox3 regulates plant stature and disease development in rice. Mol. Plant Microbe Interact. 2013, 26, 227–239. [Google Scholar] [CrossRef]
- He, B.; Gu, Y.; Xu, M.; Wang, J.; Cao, F.; Xu, L.A. Transcriptome analysis of Ginkgo biloba kernels. Front. Plant Sci. 2015, 6, 819. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, L.; Wu, Q.; Men, X.; Yao, L.; Xing, S. Transcriptome profile analysis reveals the ontogenesis of rooted chichi in Ginkgo biloba L. Gene 2018, 669, 8–14. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2, accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.; Zhao, Y.; Zhang, H.; Fan, G.; Liu, X.; Zhou, W.; Shi, C.; Wang, J.; Liu, W.; Liang, X.; et al. Draft genome of the living fossil Ginkgo biloba. Gigascience 2016, 5, 49. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernandez-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Liu, C.G.; Zhang, G.Q. Single nucleotide polymorphism (SNP) and its application in rice. Yi Chuan 2006, 28, 737–744. [Google Scholar]
- Schmid, K.J.; Sorensen, T.R.; Stracke, R.; Torjek, O.; Altmann, T.; Mitchell-Olds, T.; Weisshaar, B. Large-scale identification and analysis of genome-wide single-nucleotide polymorphisms for mapping in Arabidopsis thaliana. Genome Res. 2003, 13, 1250–1257. [Google Scholar] [CrossRef]
- He, P.; Huang, S.; Xiao, G.; Zhang, Y.; Yu, J. Abundant RNA editing sites of chloroplast protein-coding genes in Ginkgo biloba and an evolutionary pattern analysis. BMC Plant Biol. 2016, 16, 257. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, D.; Kende, H. The AtGRF family of putative transcription factors is involved in leaf and cotyledon growth in Arabidopsis. Plant J. 2003, 36, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Kuijt, S.J.; Greco, R.; Agalou, A.; Shao, J.; t Hoen, C.C.; Overnas, E.; Osnato, M.; Curiale, S.; Meynard, D.; van Gulik, R.; et al. Interaction between the growth-regulating factor and knotted1-like homeobox families of transcription factors. Plant Physiol. 2014, 164, 1952–1966. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, H.; Chan, J.; Sanchez-Pulido, L.; Andrade-Navarro, M.A.; Doonan, J.H.; Lloyd, C.W. Microtubule-associated AIR9 recognizes the cortical division site at preprophase and cell-plate insertion. Curr. Biol. 2006, 16, 1938–1943. [Google Scholar] [CrossRef]
- Stracke, R.; Werber, M.; Weisshaar, B. The R2R3-MYB gene family in Arabidopsis thaliana. Curr. Opin. Plant Biol. 2001, 4, 447–456. [Google Scholar] [CrossRef]
- Arlotta, C.; Puglia, G.D.; Genovese, C.; Toscano, V.; Karlova, R.; Beekwilder, J.; De Vos, R.C.H.; Raccuia, S.A. MYB5-like and bHLH influence flavonoid composition in pomegranate. Plant Sci. 2020, 298, 110563. [Google Scholar] [CrossRef] [PubMed]
- Clouse, S.D.; Sasse, J.M. Brassinosteroids: Essential Regulators of Plant Growth and Development. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1998, 49, 427–451. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.H.; Li, J. BRI1/BAK1, a receptor kinase pair mediating brassinosteroid signaling. Cell 2002, 110, 203–212. [Google Scholar] [CrossRef]
- Geilfus, C.M.; Ober, D.; Eichacker, L.A.; Muhling, K.H.; Zorb, C. Down-regulation of ZmEXPB6 (Zea mays beta-expansin 6) protein is correlated with salt-mediated growth reduction in the leaves of Z. mays L. J. Biol. Chem. 2015, 290, 11235–11245. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.T.; Cosgrove, D.J. Altered expression of expansin modulates leaf growth and pedicel abscission in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2000, 97, 9783–9788. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cao, H.; Sun, Y.; Li, X.; Chen, F.; Carles, A.; Li, Y.; Ding, M.; Zhang, C.; Deng, X.; et al. Arabidopsis paired amphipathic helix proteins SNL1 and SNL2 redundantly regulate primary seed dormancy via abscisic acid-ethylene antagonism mediated by histone deacetylation. Plant Cell 2013, 25, 149–166. [Google Scholar] [CrossRef]
- Huarte, H.R.; Puglia, G.D.; Prjibelski, A.D.; Raccuia, S.A. Seed Transcriptome Annotation Reveals Enhanced Expression of Genes Related to ROS Homeostasis and Ethylene Metabolism at Alternating Temperatures in Wild Cardoon. Plants 2020, 9, 1225. [Google Scholar] [CrossRef]
- Puglia, G.D.; Prjibelski, A.D.; Vitale, D.; Bushmanova, E.; Schmid, K.J.; Raccuia, S.A. Hybrid transcriptome sequencing approach improved assembly and gene annotation in Cynara cardunculus (L.). BMC Genom. 2020, 21, 317. [Google Scholar] [CrossRef]
- Smita, S.; Katiyar, A.; Chinnusamy, V.; Pandey, D.M.; Bansal, K.C. Transcriptional Regulatory Network Analysis of MYB Transcription Factor Family Genes in Rice. Front. Plant Sci. 2015, 6, 1157. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Clean Reads | Mapped Reads | GC (%) | N (%) | Q20 (%) | Q30 (%) |
|---|---|---|---|---|---|---|
| T01 | 40,843,502 | 32,241,189 (78.94%) | 45.83 | 0.01 | 95.68 | 90.09 |
| T02 | 45,536,702 | 36,635,846 (80.45%) | 45.36 | 0.01 | 95.27 | 89.37 |
| T03 | 43,261,360 | 34,735,785 (80.29%) | 46.00 | 0.01 | 95.61 | 89.94 |
| T04 | 41,269,614 | 33,568,286 (81.34%) | 45.33 | 0.01 | 95.52 | 89.77 |
| T05 | 48,103,982 | 39,385,780 (81.88%) | 45.92 | 0.01 | 95.63 | 89.96 |
| T06 | 46,977,092 | 38,531,630 (82.02%) | 44.70 | 0.01 | 95.21 | 89.27 |
| T07 | 54,312,460 | 44,998,707 (82.85%) | 44.46 | 0.01 | 95.34 | 89.46 |
| T08 | 55,105,380 | 45,855,776 (83.21%) | 44.57 | 0.01 | 95.58 | 89.92 |
| T09 | 58,075,760 | 48,387,657 (83.32%) | 44.57 | 0.01 | 95.68 | 90.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Han, X.; Ruan, J.; Xu, L.; He, B. Transcriptome Analysis of Ginkgo biloba L. Leaves across Late Developmental Stages Based on RNA-Seq and Co-Expression Network. Forests 2021, 12, 315. https://doi.org/10.3390/f12030315
Liu H, Han X, Ruan J, Xu L, He B. Transcriptome Analysis of Ginkgo biloba L. Leaves across Late Developmental Stages Based on RNA-Seq and Co-Expression Network. Forests. 2021; 12(3):315. https://doi.org/10.3390/f12030315
Chicago/Turabian StyleLiu, Hailin, Xin Han, Jue Ruan, Lian Xu, and Bing He. 2021. "Transcriptome Analysis of Ginkgo biloba L. Leaves across Late Developmental Stages Based on RNA-Seq and Co-Expression Network" Forests 12, no. 3: 315. https://doi.org/10.3390/f12030315
APA StyleLiu, H., Han, X., Ruan, J., Xu, L., & He, B. (2021). Transcriptome Analysis of Ginkgo biloba L. Leaves across Late Developmental Stages Based on RNA-Seq and Co-Expression Network. Forests, 12(3), 315. https://doi.org/10.3390/f12030315