
Expression Patterns and Regulation of Non-Coding RNAs during Synthesis of Cellulose in Eucalyptus grandis Hill


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Growth Conditions
2.2. Measurement of Cellulose, Lignin, and Hemicellulose Content
2.2.1. Determination of Neutral Detergent Fiber (NDF) with Neutral Detergent
2.2.2. Determination of Cellulose with Acid Detergent
2.3. Total RNA Extraction and Detection
2.4. LncRNA Extract
2.5. Identification and Classification of lncRNAs
2.6. Screening of Differentially Expressed lncRNAs
2.7. MiRNA Extraction and Identification Analysis
2.8. MiRNA Expression Analysis
2.9. Identification of circRNAs
2.10. Expression Analysis and Target Relationship Prediction of circRNAs
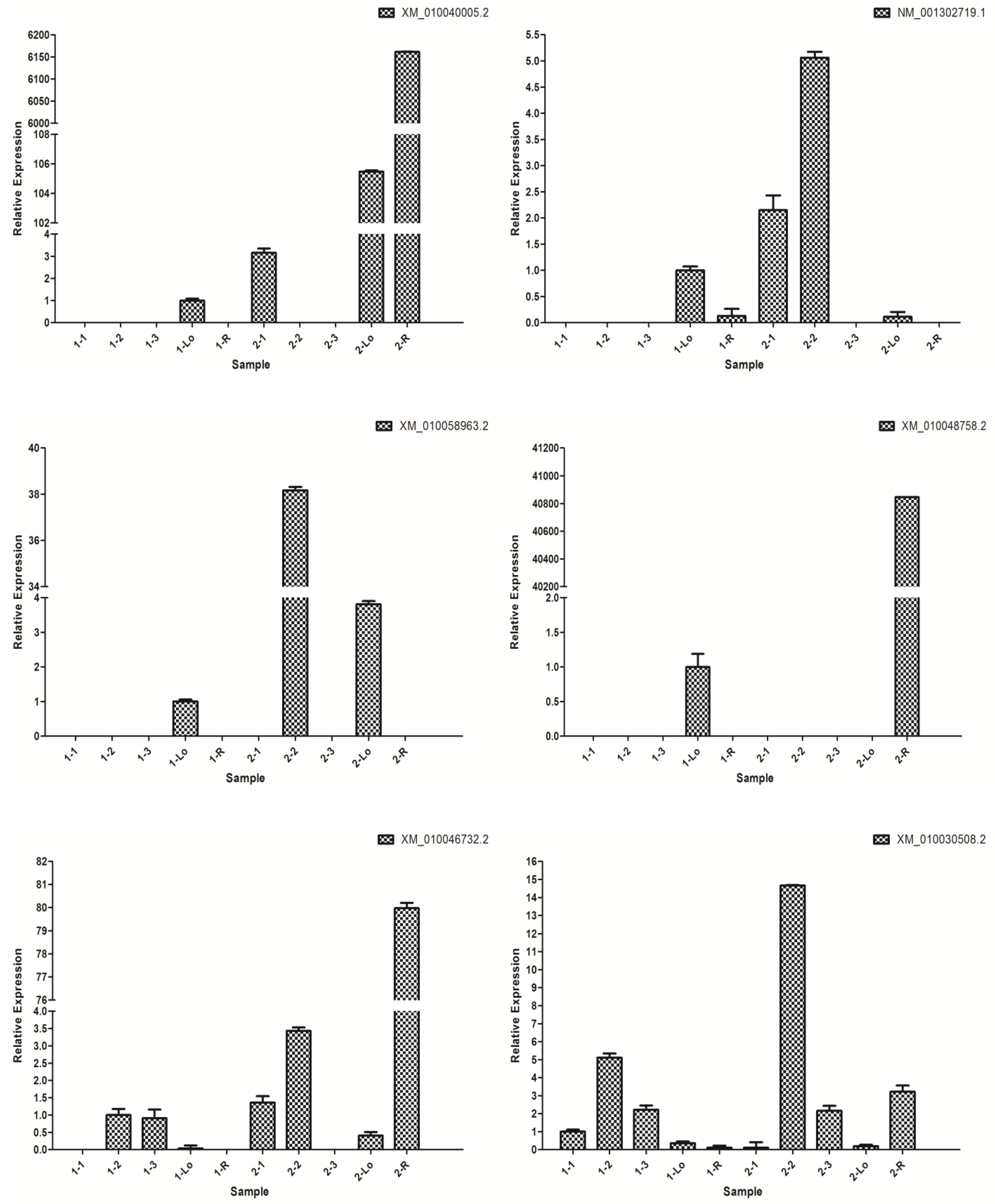
2.11. RNA Reverse Transcription and q RT-PCR
2.12. Co-Expression of ceRNA Regulatory Network
2.13. Extraction of Total RNA from Immature Xylem of E. grandis and Reverse Transcription of cDNA
2.14. Gene Cloning and Vector Construction
2.15. Assembly of Plasmid Constructs for Plant Genetic Transformation
2.16. Positive Transgenic Plant Screening and Phenotype Analysis
3. Results
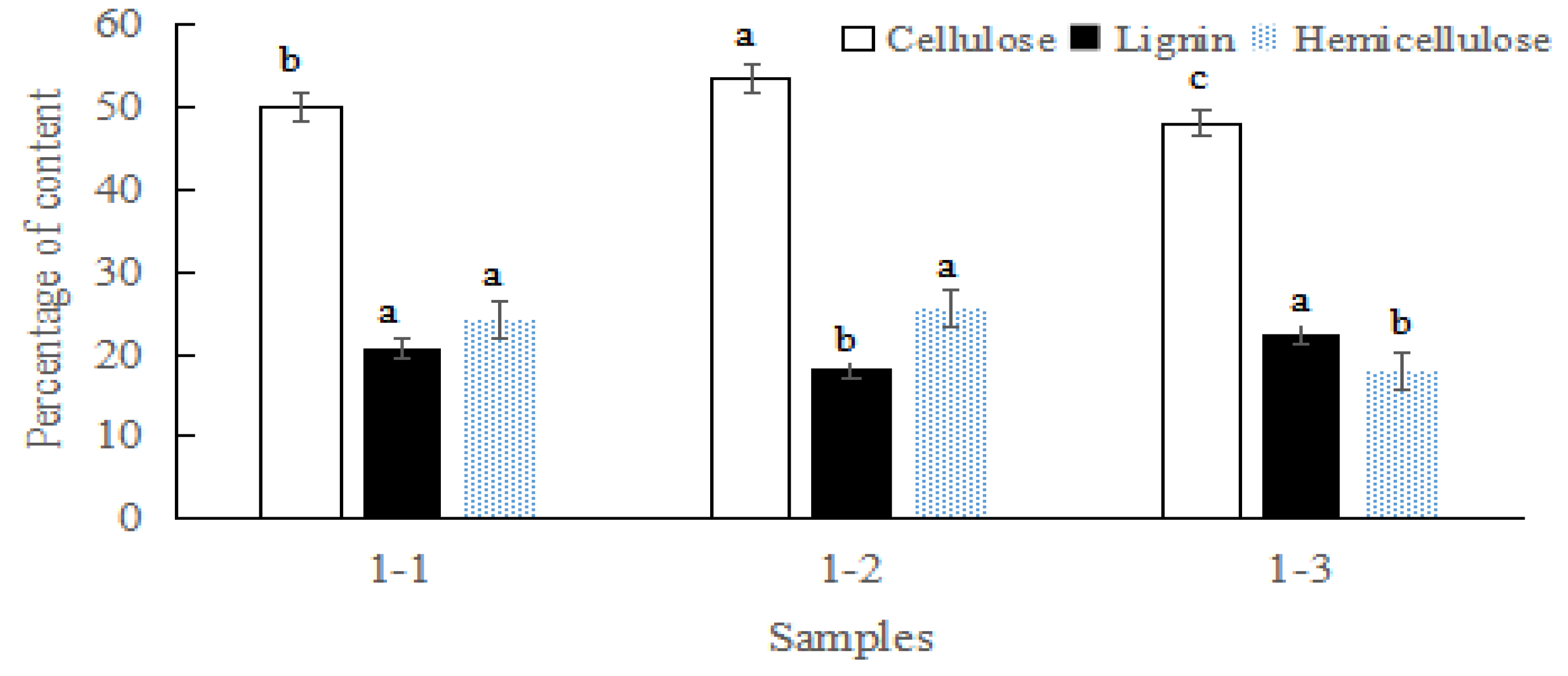
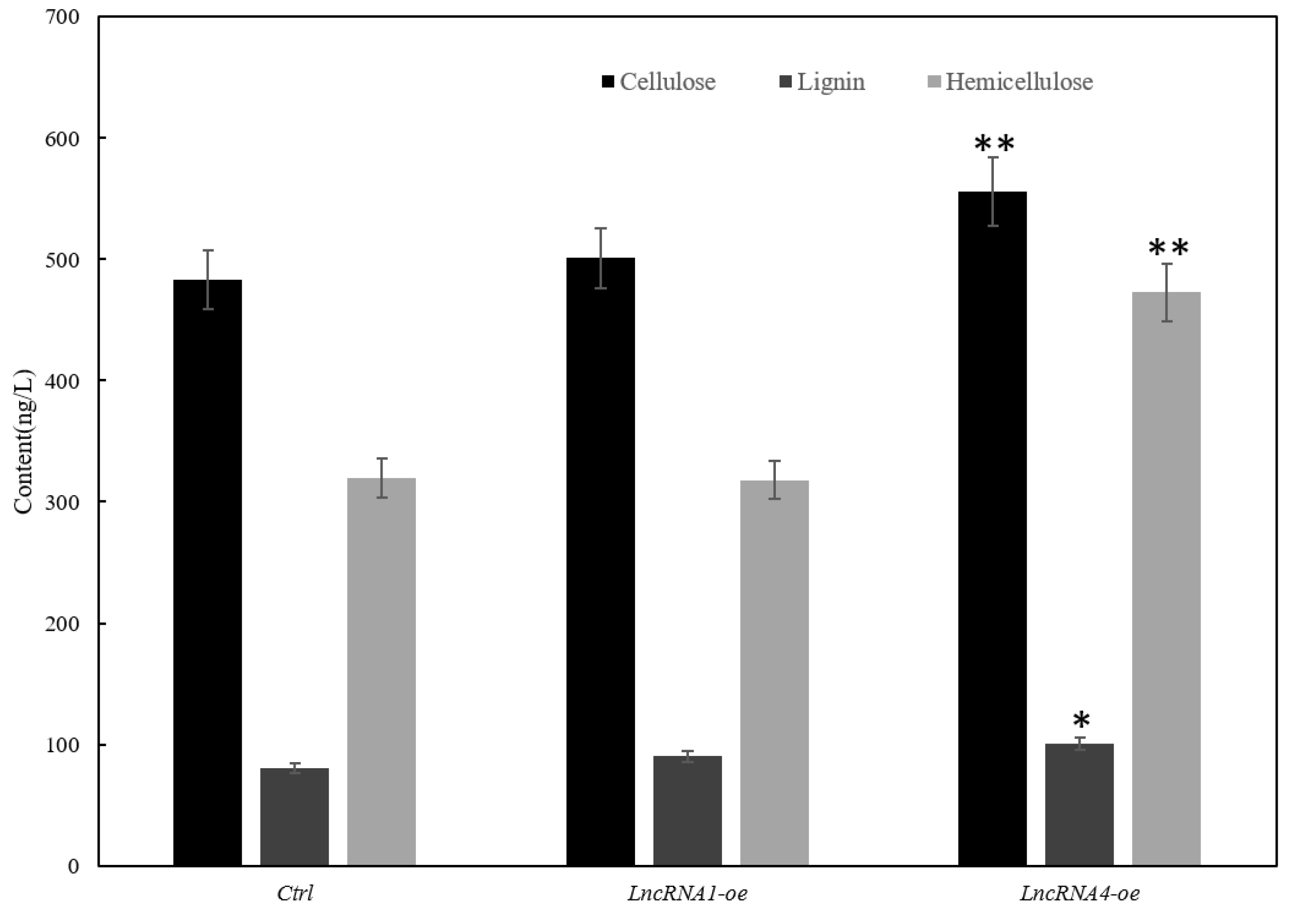
3.1. The Content of Cellulose, Lignin, and Hemicellulose
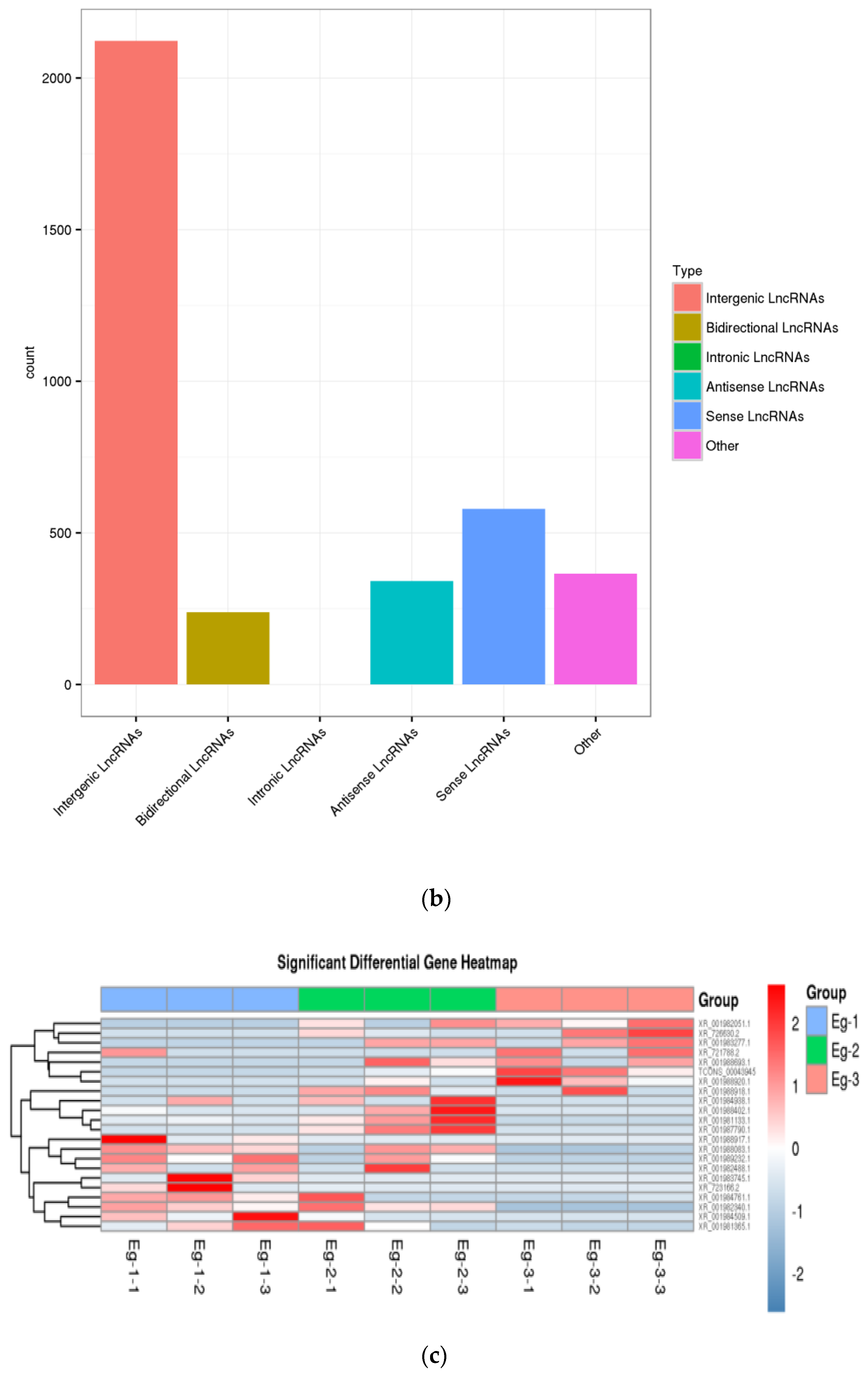
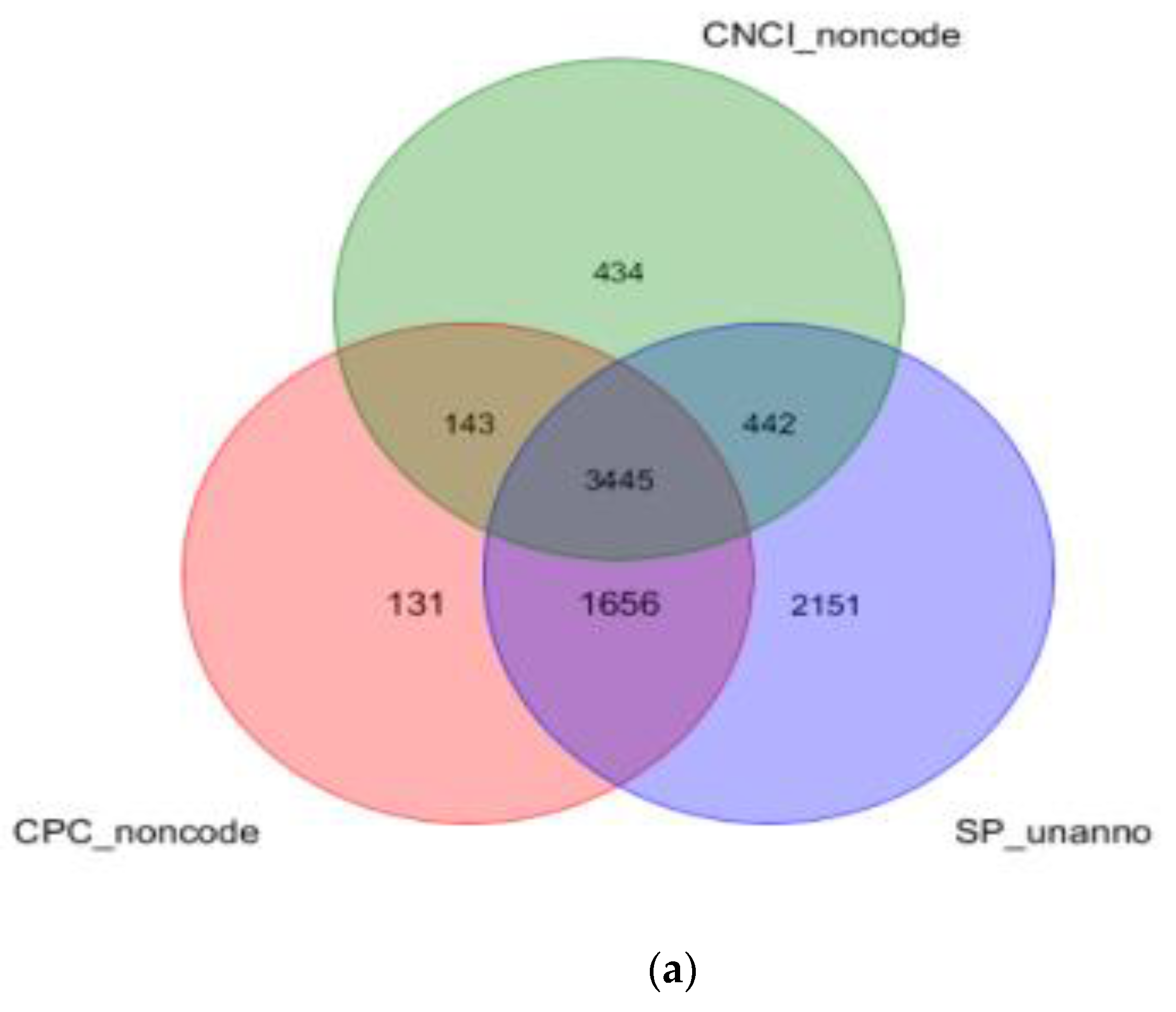
3.2. Identification and Analysis of lncRNAs in E. grandis
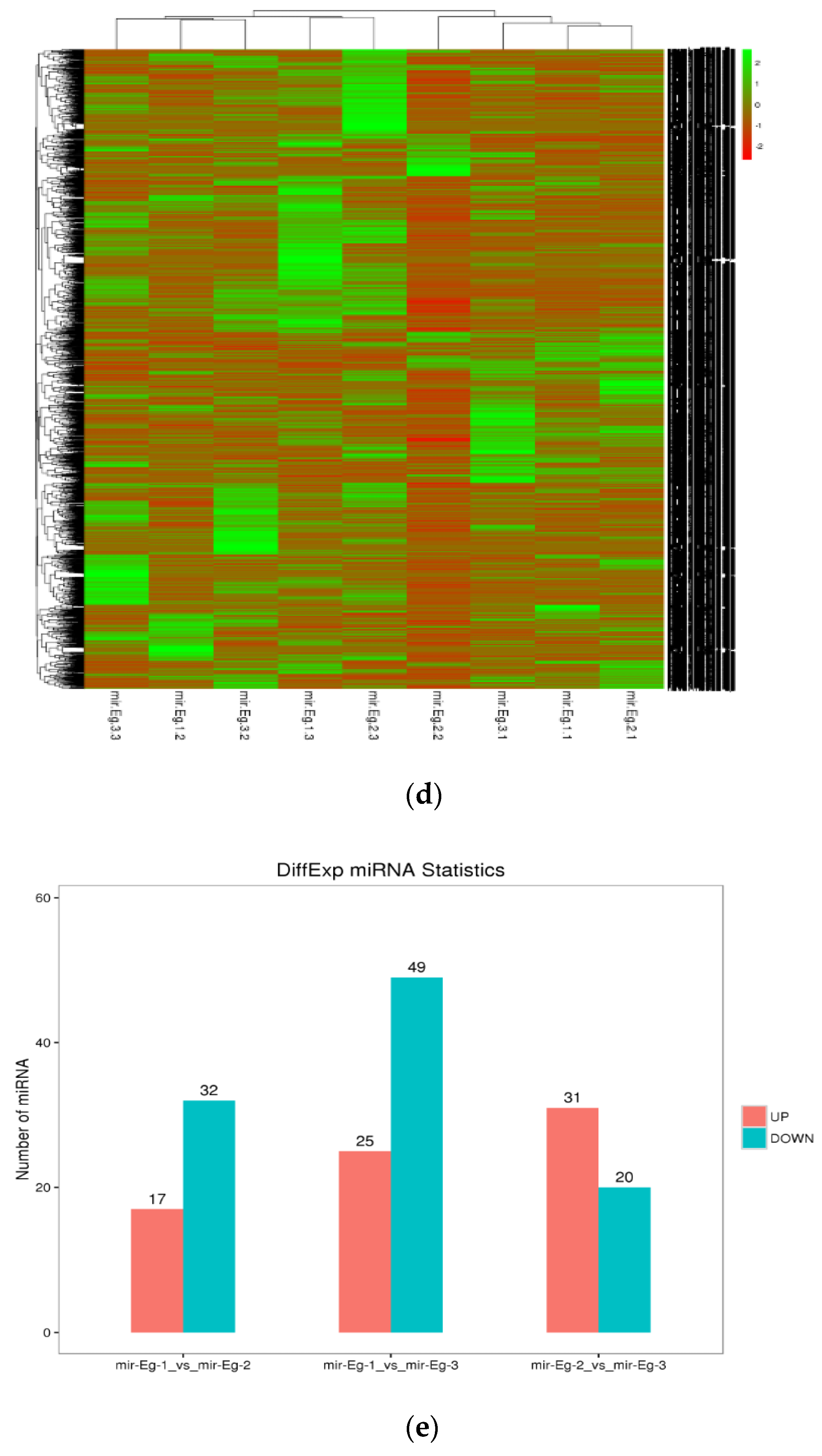
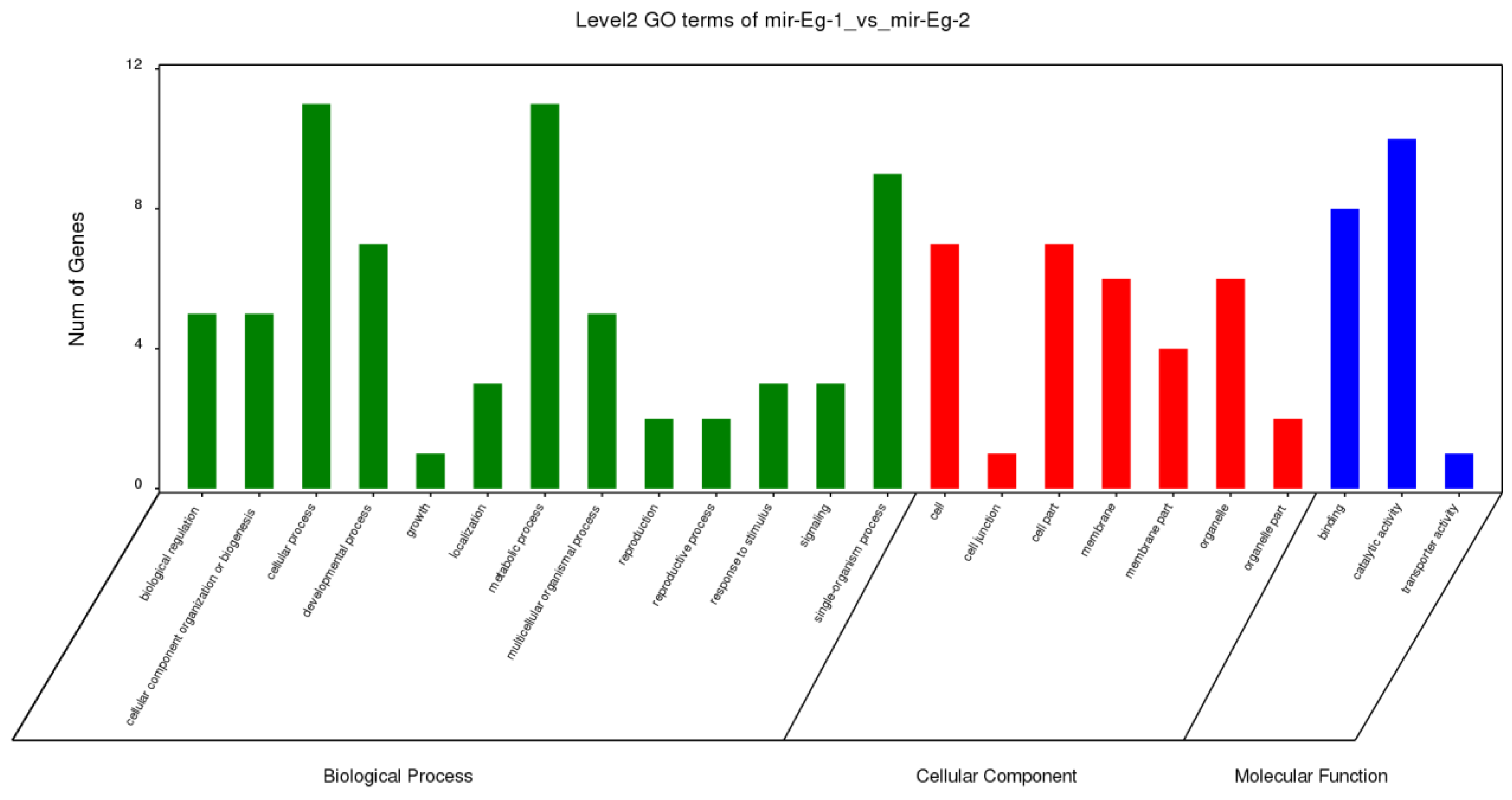
3.3. Identification and Analysis of miRNA in E. grandis
3.4. Circular RNA Statistical Analysis
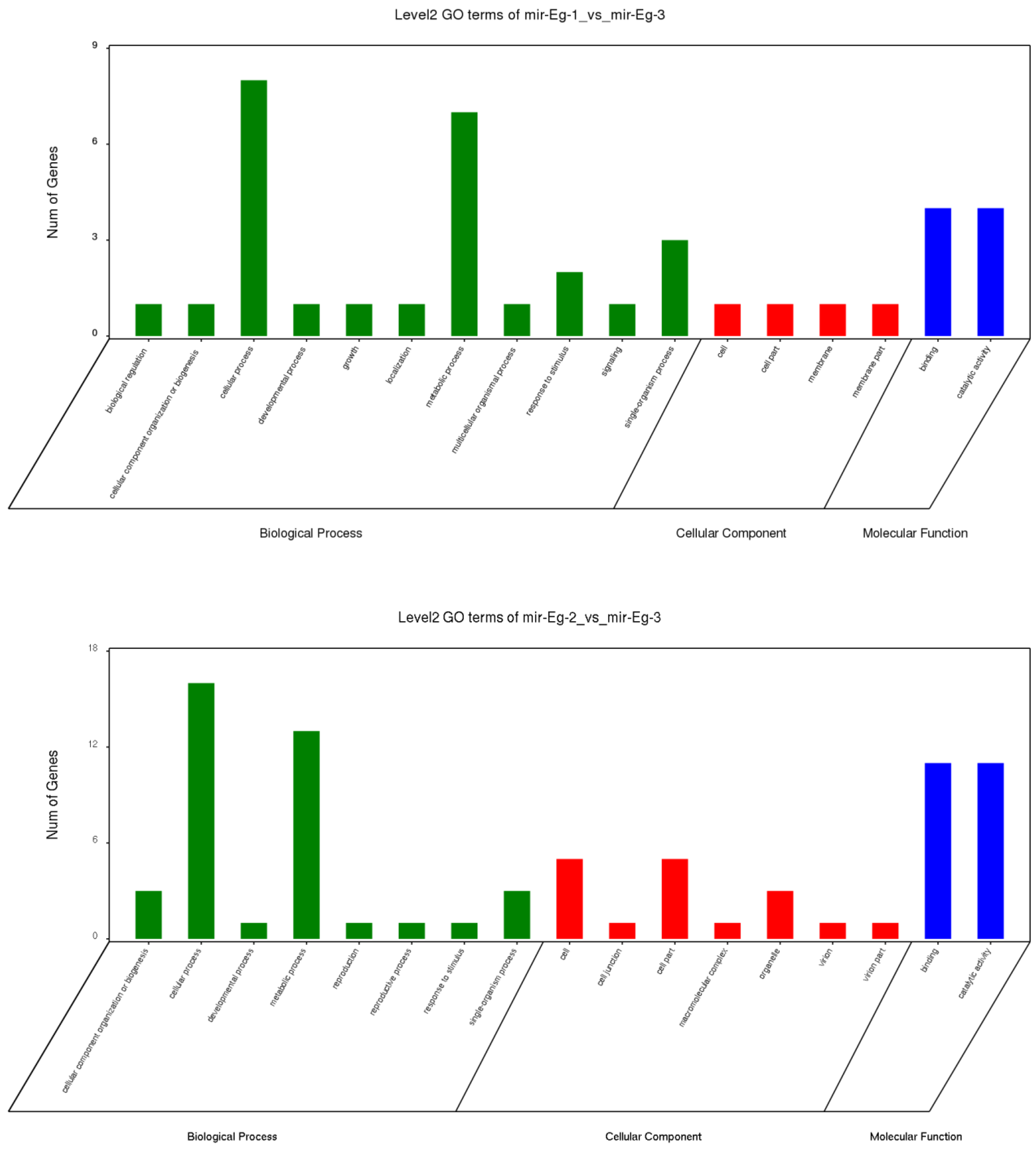
3.5. Screening of Genes Related to Cellulose Synthesis in E. grandis
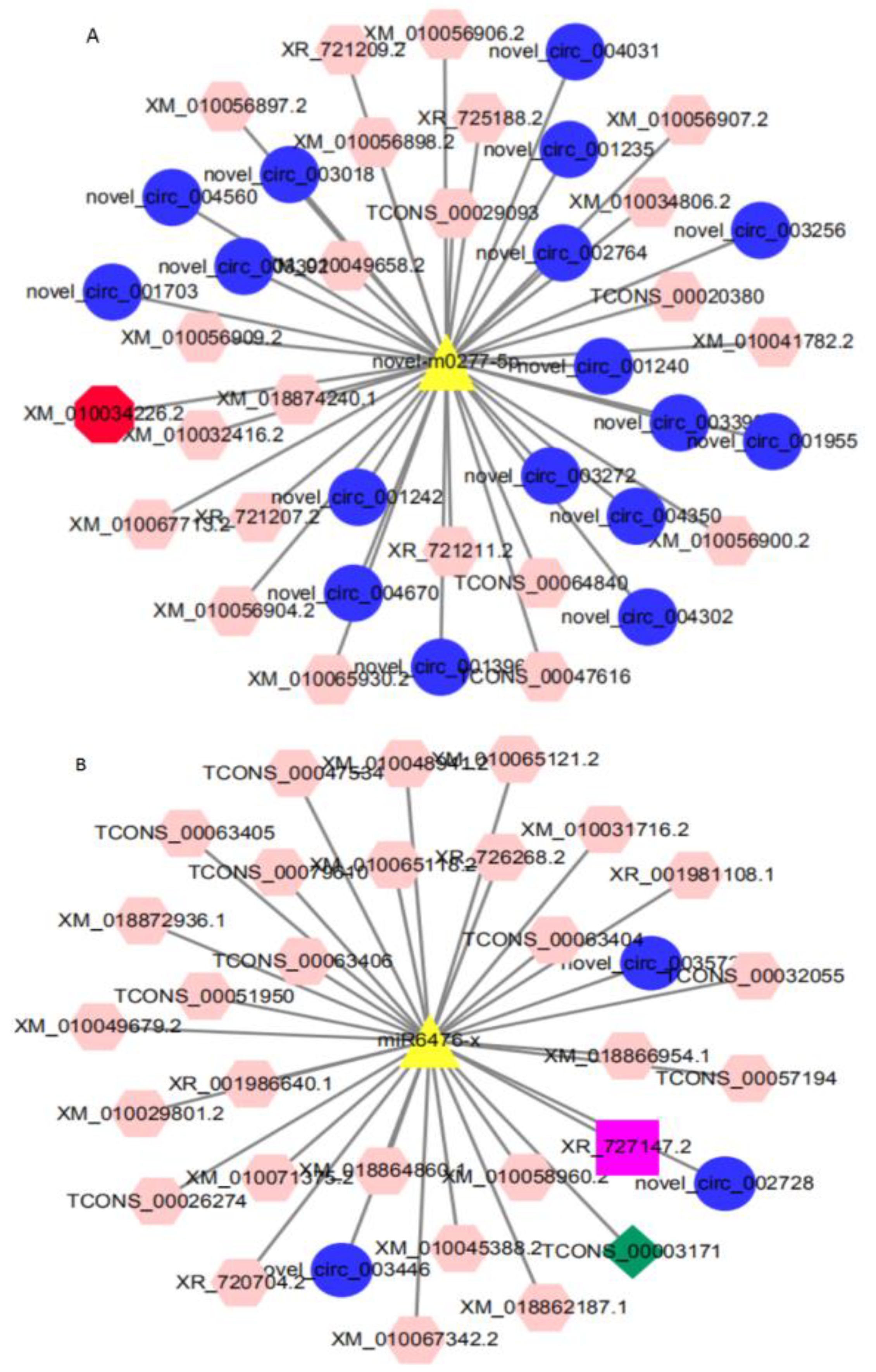
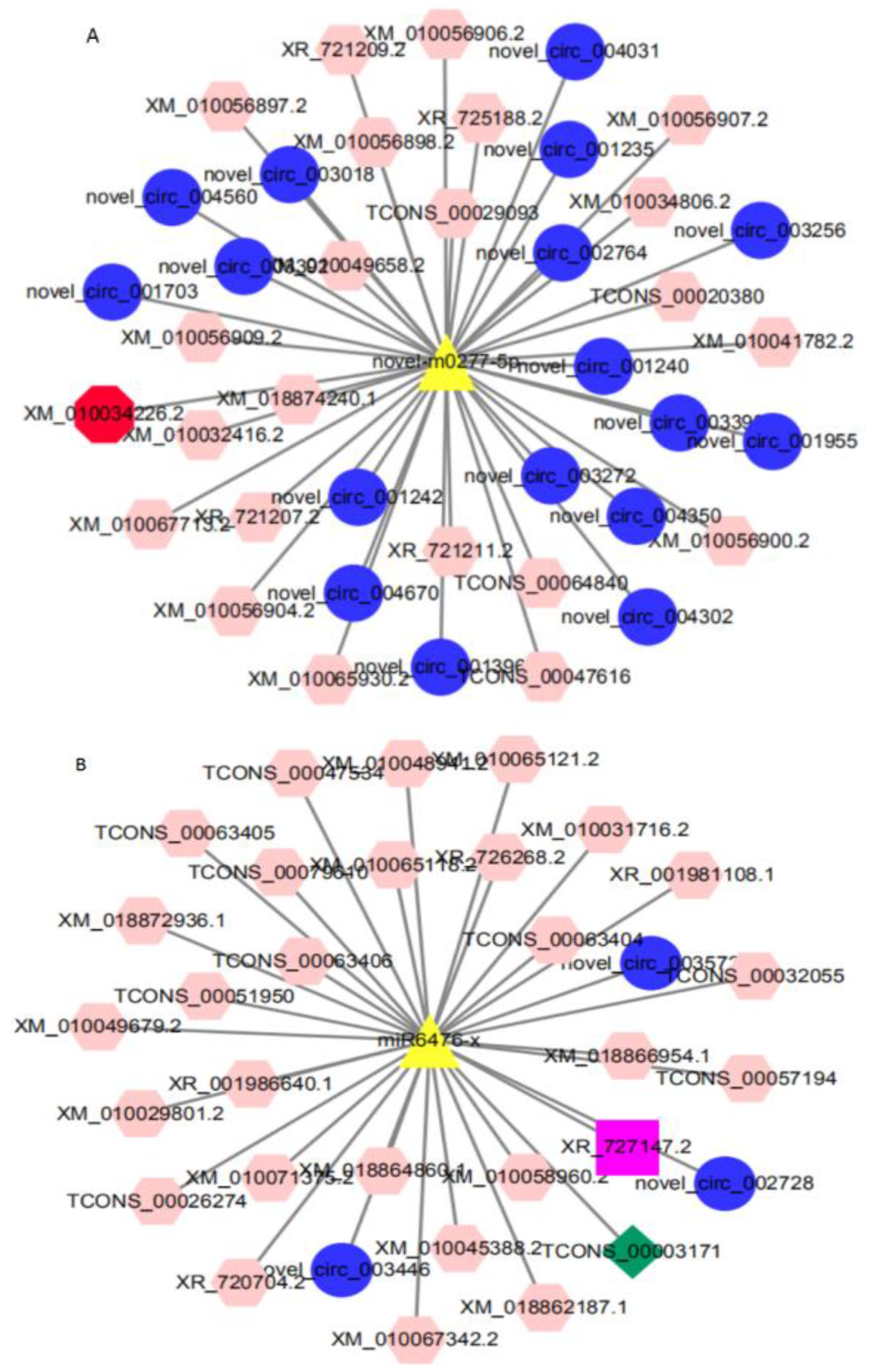
3.6. Construction of ceRNA Co-Expression Regulatory Network
3.7. Gene Cloning and Vector Construction
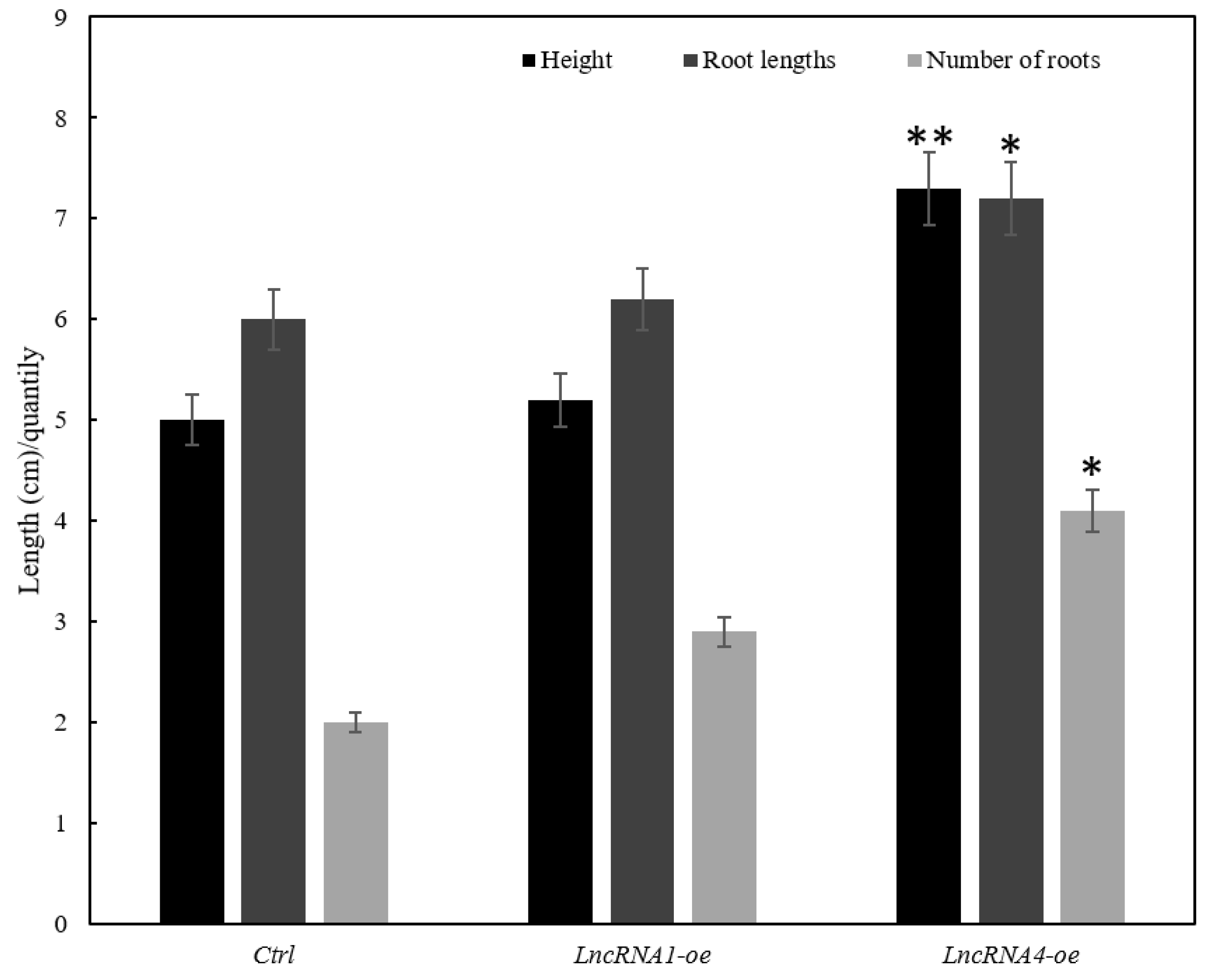
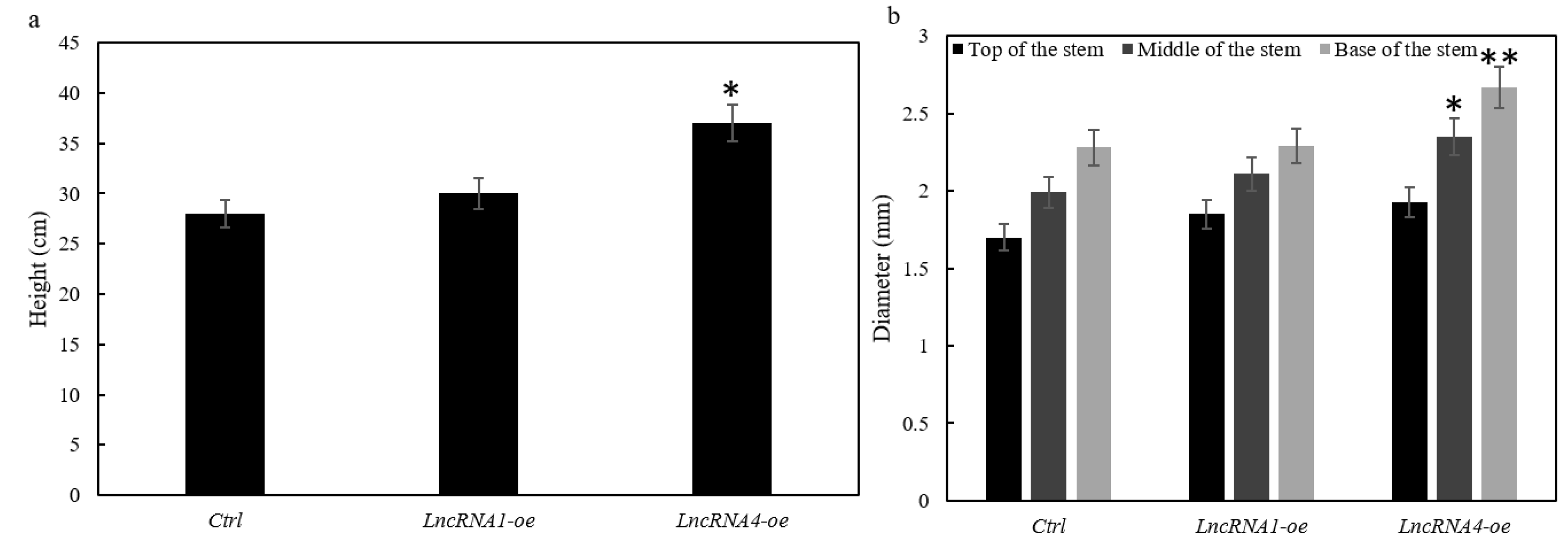
3.8. Effects of Overexpression of lncRNA1 and lncRNA4 on Plant Growth
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tredenick, E.C.; Farquhar, G.D. Dynamics of Moisture Transport in Plant Cuticles: The Role of Cellulose. arXiv 2021, arXiv:2102.08666v1. [Google Scholar]
- Plomion, C.; Leprovost, G.; Stokes, A. Wood formation in trees. Plant Physiol. 2015, 127, 1513–1523. [Google Scholar] [CrossRef]
- Steck, E.; Boeuf, S.; Gabler, J.; Werth, N.; Schnatzer, P.; Diederichs, S.; Richter, W. Regulation of H19 and its encoded microRNA-675 in osteoarthritis and under anabolic and catabolic in vitro conditions. J. Mol. Med. 2012, 90, 1185–1195. [Google Scholar] [CrossRef]
- Batista, P.J.; Chang, H.Y. Long non-coding RNAs: Cellular address codes in development and disease. Cell 2013, 152, 1298–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Zhang, H.; Guo, H.; Hu, W.; Ji, W. The emerging role of long non-coding RNAs in plant defense against fungal stress. Int. J. Mol. Sci. 2020, 21, 2659. [Google Scholar] [CrossRef]
- Wilusz, J.E. Long noncoding RNAs: Re-writing dogmas of RNA processing and stability. Biochim. Biophys. Acta BBA-Gene Regul. Mech. 2016, 1859, 128–138. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Tao, Y.; Liao, Q. Long noncoding RNA: A crosslink in biological regulatory network. Brief. Bioinform. 2017, 19, 930–945. [Google Scholar] [CrossRef]
- Wang, H.; Chung, P.J.; Liu, J.; Jang, I.-C.; Kean, M.J.; Xu, J.; Chua, N.-H. Genome-wide identification of long noncoding natural antisense transcripts and their responses to light in Arabidopsis. Genome Res. 2014, 24, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.-W.; Xu, L.; Wu, Y.-R.; Chen, X.-A.; Liu, Y.; Zhu, S.-H.; Ding, W.-N.; Wu, P.; Yi, K.-K. OsGLU3, a putative membrane-bound endo-1, 4-beta-glucanase, is required for root cell elongation and division in rice (Oryza sativa L.). Mol. Plant 2012, 5, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Cagirici, H.B.; Alptekin, B.; Budak, H. RNA sequencing and co-expressed long non-coding RNA in modern and wild wheats. Sci. Rep. 2017, 7, 10670. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256–264. [Google Scholar] [CrossRef]
- Lv, Y.; Liang, Z.; Ge, M.; Qi, W.; Zhang, T.; Lin, F.; Peng, Z.; Zhao, H. Genome-wide identification and functional prediction of nitrogen-responsive intergenic and intronic long non-coding RNAs in maize (Zea mays L.). BMC Genom. 2016, 17, 350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Chen, X.; Mu, M.; Wang, J.; Wang, X.; Wang, D.; Yin, Z.; Fan, W.; Wang, S.; Guo, L. Genome-wide analysis of long noncoding RNAs and their responses to drought stress in cotton (Gossypium hirsutum L.). PLoS ONE 2016, 11, e0156723. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Chen, L.; Zhang, C.; Hao, P.; Jing, X.; Li, X. Global transcriptome analysis reveals extensive gene remodeling, alternative splicing and differential transcription profiles in non-seed vascular plant Selaginella moellendorffii. BMC Genom. 2017, 18, 1042. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Du, Q.; Chen, J.; Wang, Q.; Zhang, D. Identification and allelic dissection uncover roles of lncRNAs in secondary growth of Populus tomentosa. DNA Res. 2017, 24, 473–486. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Quan, M.; Du, Q.; Zhang, D. The interactions between the long non-coding RNA NERDL and its target gene affect wood formation in Populus tomentosa. Front. Plant Sci. 2017, 8, 1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.; Sun, J.; Dong, Y.; Wang, C.; Xiao, S.; Mo, L.; Jiao, Z. Comparative transcriptome analysis uncovers regulatory roles of long non-coding RNAs involved in resistance to powdery mildew in melon. BMC Genom. 2020, 21, 125. [Google Scholar] [CrossRef]
- Lin, Z.; Long, J.; Yin, Q.; Wang, B.; Li, H.; Luo, J.; Wang, H.C.; Wu, A.-M. Identification of novel lncRNAs in Eucalyptus grandis. Ind. Crop. Prod. 2019, 129, 309–317. [Google Scholar] [CrossRef]
- Axtell, M.J. Classification and comparison of small RNAs from plants. Annu. Rev. Plant Biol. 2013, 64, 137–159. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-M.; Peter, M.E. microRNAs and death receptors. Cytokine Growth Factor Rev. 2008, 19, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Li, A.; Yu, B.; Li, S. Interplay between miRNAs and lncRNAs: Mode of action and biological roles in plant development and stress adaptation. Comput. Struct. Biotechnol. J. 2021, 19, 2567–2574. [Google Scholar] [CrossRef]
- Sun, X.; Wang, C.; Xiang, N.; Li, X.; Yang, S.; Du, J.; Yang, Y.; Yang, Y. Activation of secondary cell wall biosynthesis by miR319-targeted TCP 4 transcription factor. Plant Biotechnol. J. 2017, 15, 1284–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinhart, B.J.; Weinstein, E.G.; Rhoades, M.W.; Bartel, B.; Bartel, D.P. MicroRNAs in plants. Genes Dev. 2002, 16, 1616–1626. [Google Scholar] [CrossRef] [Green Version]
- Mica, E.; Gianfranceschi, L.; Pe, M.E. Characterization of five microRNA families in maize. J. Exp. Bot. 2006, 57, 2601–2612. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Du, H.; Wuyun, T.-N. Genome-wide identification of microRNAs and their targets in the leaves and fruits of Eucommia ulmoides using high-throughput sequencing. Front. Plant Sci. 2016, 7, 1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappas, M.; Reis, A.; Farinell, L.; Pasquali, G.; Pappas, G.; Grattapaglia, D. Interspecific discovery and expression profiling of Eucalyptus micro RNAs by deep sequencing. BMC Proc. 2011, 5, P173. [Google Scholar] [CrossRef] [Green Version]
- Nigro, J.M.; Cho, K.R.; Fearon, E.R.; Kern, S.E.; Ruppert, J.M.; Oliner, J.D.; Kinzler, K.W.; Vogelstein, B. Scrambled exons. Cell 2016, 64, 607–613. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, X.; Hu, X.; Dai, L.; Fu, X.; Zhang, J.; Ao, Y. Circular RNA related to the chondrocyte ECM regulates MMP13 expression by functioning as a MiR-136 ‘Sponge’in human cartilage degradation. Sci. Rep. 2016, 6, 22572. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.-L.; Yang, L. Regulation of circRNA biogenesis. RNA Biol. 2015, 12, 381–388. [Google Scholar] [CrossRef]
- Andreeva, K.; Cooper, N.G. Circular RNAs: New players in gene regulation. Adv. Biosci. Biotechnol. 2015, 6, 433. [Google Scholar] [CrossRef] [Green Version]
- Ye, C.-Y.; Zhang, X.; Chu, Q.; Liu, C.; Yu, Y.; Jiang, W.; Zhu, Q.-H.; Fan, L.; Guo, L. Full-length sequence assembly reveals circular RNAs with diverse non-GT/AG splicing signals in rice. RNA Biol. 2016, 14, 1055–1063. [Google Scholar] [CrossRef] [Green Version]
- Darbani, B.; Noeparvar, S.; Borg, S. Identification of circular RNAs from the parental genes involved in multiple aspects of cellular metabolism in barley. Front. Plant Sci. 2016, 7, 776. [Google Scholar] [CrossRef] [Green Version]
- Zuo, J.; Wang, Q.; Zhu, B.; Luo, Y.; Gao, L. Deciphering the roles of circRNAs on chilling injury in tomato. Biochem. Biophys. Res. Commun. 2016, 479, 132–138. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.C.; Fang, S.S.; Wu, Y.; Zhang, J.H.; Chen, Y.; Liu, J.; Wu, B.; Wu, J.R.; Li, E.M.; Xu, L.Y.; et al. CNIT: A fast and accurate web tool for identifying protein-coding and long non-coding transcripts based on intrinsic sequence composition. Nucleic Acids Res. 2019, 47, W516–W522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.N.; Liu, Y.; Wang, Y.F. MiRdetector: A Computational Tool to Predict and Detect miRNA Genes. J. Shanghai Univ. 2006, 12, 376–382. [Google Scholar]
- Antheia, K.; Jon, J.; Tomas, L.L.; Ab Dimajid, O. Biological coefficient of variation of samples S1, S2 and S3 as estimated by TopHat/HTSeq/edgeR software. PLoS ONE 2013, 8, e81809. [Google Scholar]
- Chen, L.; Yu, Y.; Zhang, X.; Liu, C.; Ye, C.; Fan, L. PcircRNA_finder: A software for circRNA prediction in plants. Bioinformatics 2016, 32, 3528–3529. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆Ct method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Zhu, B.Q.; Zhang, Y.B.; Wang, H.Y.; Ba, C.F. The research of applying primer premier 5.0 to design PCR primer. J. Jinzhou Med Coll. 2004, 25, 43–46. [Google Scholar]
- Liu, B.; Zhang, J.; Wang, L.; Li, J.; Zheng, H.; Chen, J.; Lu, M. A survey of Populus PIN-FORMED family genes reveals their diversified expression patterns. J. Exp. Bot. 2014, 65, 2437–2448. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Shao, F.; Macmillan, C.; Wilson, I.W.; Van der Merwe, K.; Hussey, S.G.; Myburg, A.A.; Dong, X.; Qiu, D. Genomewide analysis of the lateral organ boundaries domain gene family in Eucalyptus grandis reveals members that differentially impact secondary growth. Plant Biotechnol. J. 2018, 16, 124–136. [Google Scholar] [CrossRef] [Green Version]
- Etchells, J.P.; Mishra, L.S.; Kumar, M.; Campbell, L.; Turner, S.R. Wood formation in trees is increased by manipulating PXY-regulated cell division. Curr. Biol. 2015, 25, 1050–1055. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Sun, Z.; Xu, M. Identification and characterization of long non-coding RNAs involved in the formation and development of poplar adventitious roots. Ind. Crop. Prod. 2018, 118, 334–346. [Google Scholar] [CrossRef]
- Ding, Z.; Tie, W.; Fu, L.; Yan, Y.; Liu, G.; Yan, W.; Li, Y.; Wu, C.; Zhang, J.; Hu, W. Strand-specific RNA-seq based identification and functional prediction of drought-responsive lncRNAs in cassava. BMC Genom. 2019, 20, 214. [Google Scholar] [CrossRef] [Green Version]
- Long, Y.; Wang, X.; Youmans, D.T.; Cech, T.R. How do lncRNAs regulate transcription? Sci. Adv. 2017, 3, eaao2110. [Google Scholar] [CrossRef] [Green Version]
- McNair, G.R. Whole-Tree and Tension Wood-Associated Expression Profiles of microRNAs in Eucalyptus Trees. Ph.D. Thesis, University of Pretoria, Pretoria, South Africa, 2009. [Google Scholar]
- Lin, Z.; Li, Q.; Yin, Q.; Wang, J.; Zhang, B.; Gan, S.; Wu, A.-M. Identification of novel miRNAs and their target genes in Eucalyptus grandis. Tree Genet. Genomes 2018, 14, 60. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.-O.; Chen, T.; Xiang, J.-F.; Yin, Q.-F.; Xing, Y.-H.; Zhu, S.; Yang, L.; Chen, L.-L. Circular intronic long noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Tsukahara, T. A view of pre-mRNA splicing from RNase R resistant RNAs. Int. J. Mol. Sci. 2014, 15, 9331–9342. [Google Scholar] [CrossRef] [Green Version]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talhouarne, G.J.; Gall, J.G. Lariat intronic RNAs in the cytoplasm of Xenopus tropicalis oocytes. RNA 2014, 20, 1476–1487. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Zheng, S.; Deng, X.; Yang, A.; Xie, X.; Tang, H.; Xie, X. The role of circular RNA CDR1as/ciRS-7 in regulating tumor microenvironment: A pan-cancer analysis. Biomolecules 2019, 9, 429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Filonov, G.S.; Noto, J.J.; Schmidt, C.A.; Hatkevich, T.L.; Wen, Y.; Jaffrey, S.R.; Matera, A.G. Metazoan tRNA introns generate stable circular RNAs in vivo. RNA 2015, 21, 1554–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | lncRNA | Target Gene | Symbol | Description |
|---|---|---|---|---|
| LncRNA1 | XR_001980078.1 | XM_010061899.2 | RhGT1 | PREDICTED: UDP-glycosyltransferase 88B1 [Eucalyptus grandis] |
| LncRNA2 | XR_720796.2 | XM_010030945.2 | GATL9 | PREDICTED: probable galacturonosyltransferase-like 9 [Eucalyptus grandis] |
| LncRNA3 | XR_727233.2 | XM_018877681.1 | UXS6 | PREDICTED: UDP-glucuronic acid decarboxylase 5 [Eucalyptus grandis] |
| LncRNA4 | XR_001985124.1 | XM_010048758.2 | CESA7 | PREDICTED: cellulose synthase A catalytic subunit 7 [UDP-forming]-like [Eucalyptus grandis] |
| miRNA | Target Gene | Symbol | Description |
|---|---|---|---|
| miR5298-y | XM_018877681.1 | UXS6 | PREDICTED: UDP-glucuronic acid decarboxylase 5 [Eucalyptus grandis] |
| miR3951-x | XM_010030945.2 | GATL9 | PREDICTED: probable galacturonosyltransferase-like 9 [Eucalyptus grandis] |
| miR5198-y | NM_001302719.1 | CESA5 | probable cellulose synthase A catalytic subunit 5 [Eucalyptus grandis] |
| miR5156-x | XM_010055035.2 | UTR4 | PREDICTED: UDP-galactose/UDP-glucose transporter 2 [Eucalyptus grandis] |
| miR5298-y | XM_018877682.1 | UXS6 | PREDICTED: UDP-glucuronic acid decarboxylase 5 [Eucalyptus grandis] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhan, N.; Wang, Z.; Xie, Y.; Shang, X.; Liu, G.; Wu, Z. Expression Patterns and Regulation of Non-Coding RNAs during Synthesis of Cellulose in Eucalyptus grandis Hill. Forests 2021, 12, 1565. https://doi.org/10.3390/f12111565
Zhan N, Wang Z, Xie Y, Shang X, Liu G, Wu Z. Expression Patterns and Regulation of Non-Coding RNAs during Synthesis of Cellulose in Eucalyptus grandis Hill. Forests. 2021; 12(11):1565. https://doi.org/10.3390/f12111565
Chicago/Turabian StyleZhan, Ni, Zhen Wang, Yaojian Xie, Xiuhua Shang, Guo Liu, and Zhihua Wu. 2021. "Expression Patterns and Regulation of Non-Coding RNAs during Synthesis of Cellulose in Eucalyptus grandis Hill" Forests 12, no. 11: 1565. https://doi.org/10.3390/f12111565
APA StyleZhan, N., Wang, Z., Xie, Y., Shang, X., Liu, G., & Wu, Z. (2021). Expression Patterns and Regulation of Non-Coding RNAs during Synthesis of Cellulose in Eucalyptus grandis Hill. Forests, 12(11), 1565. https://doi.org/10.3390/f12111565