
Genetic Structure and Geographical Differentiation of Larix sibirica Ledeb. in the Urals

 , , , , and
, , , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Microsatellite Amplification
3. Results
3.1. Polymorphic SSR Primers
3.2. Polymorphism of Microsatellite Markers in 15 Populations of L. sibirica
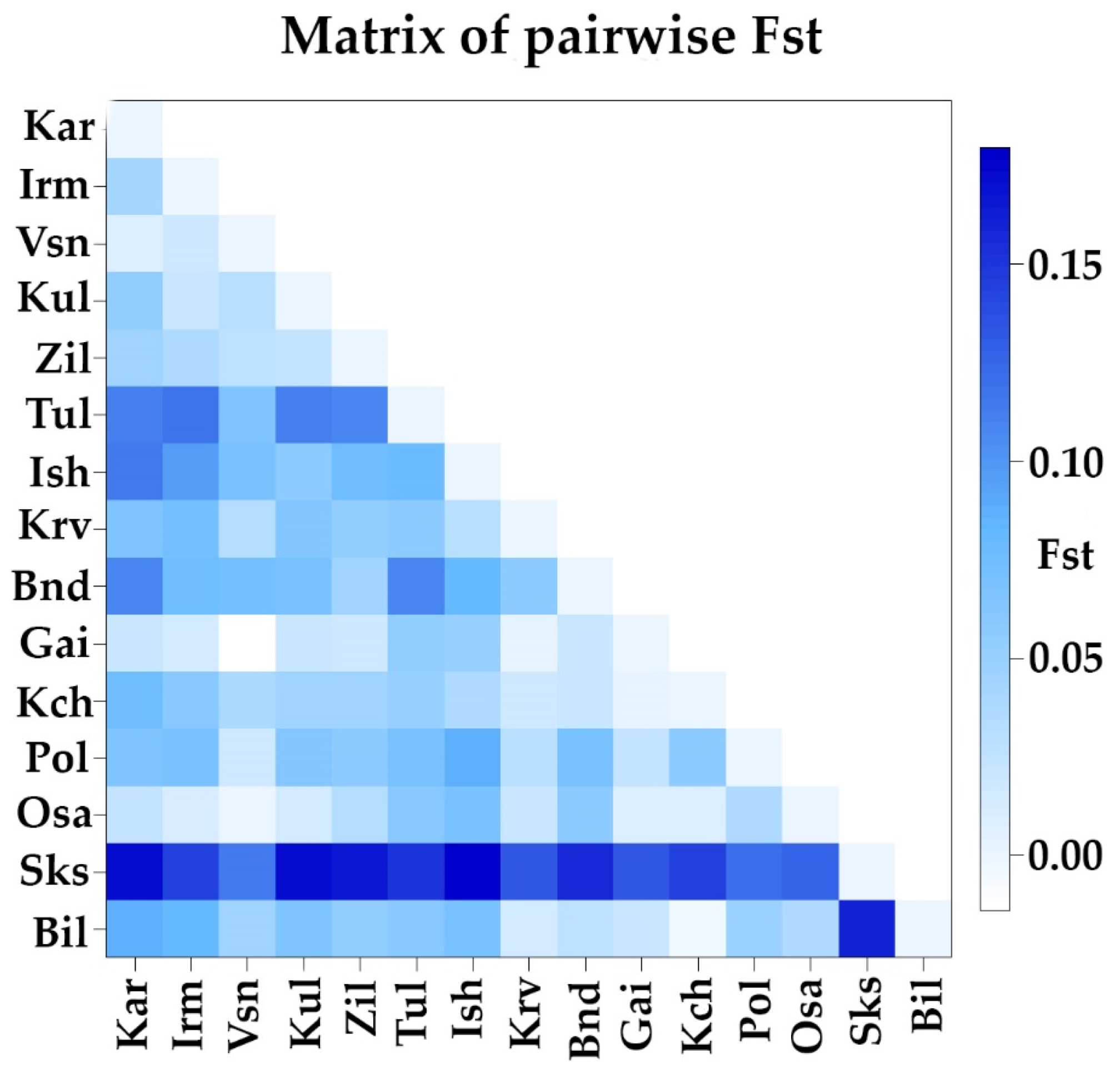
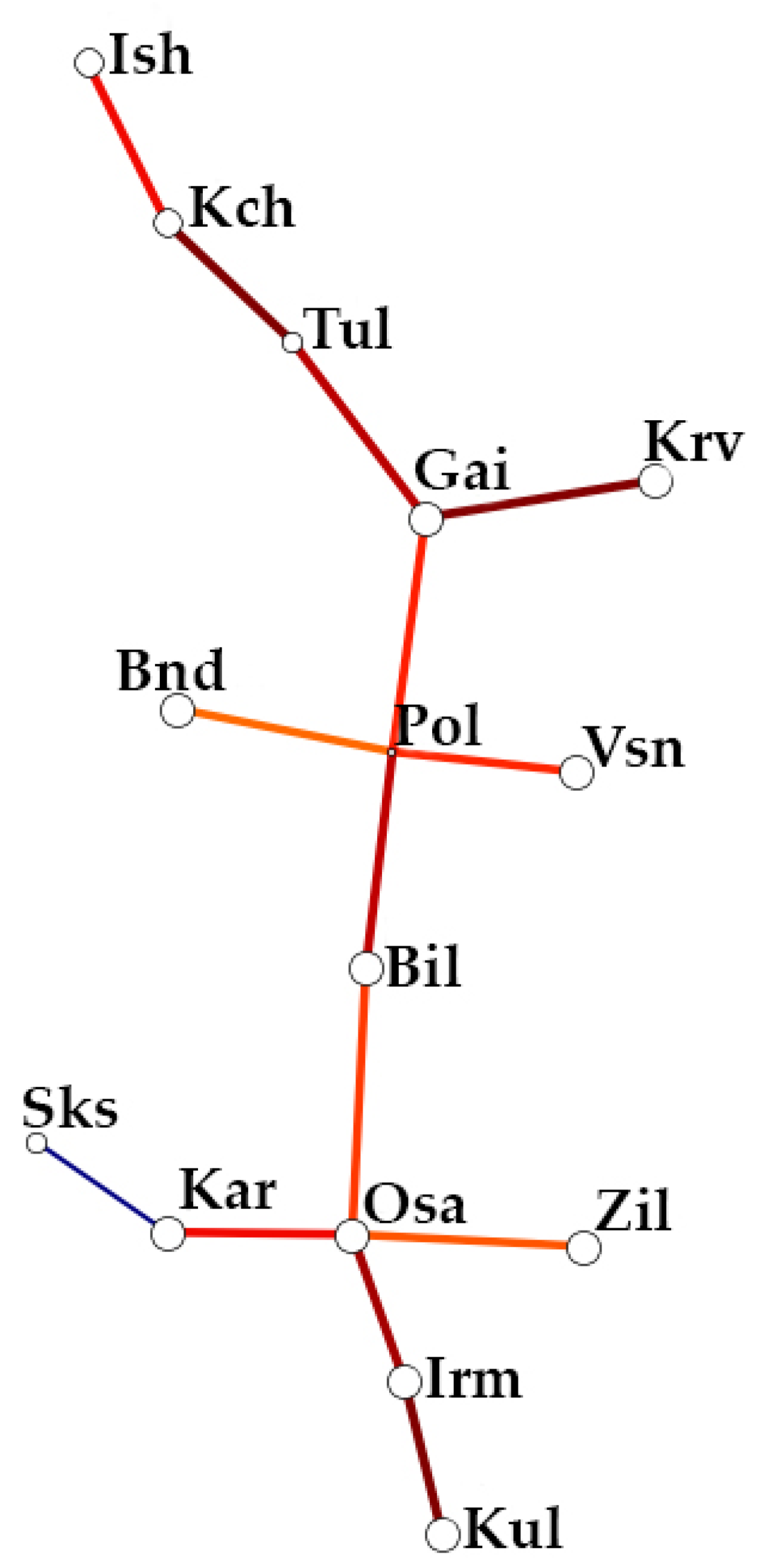
3.3. Analysis of the Genetic Structure and Differentiation of the Studied Populations of Siberian Larch
4. Discussion
4.1. Polymorphism of Microsatellite Markers in 15 Populations of L. sibirica
4.2. Analysis of the Genetic Structure and Differentiation of the Populations of Siberian Larch Studied
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hamrick, J.L.; Godt, M.J.W.; Gonzales, E. Conservation of genetic diversity in old-growth forest communities of the southeastern United States. Appl. Veg. Sci. 2006, 9, 51–58. [Google Scholar] [CrossRef]
- FAO. Global Forest Resources Assessment: Main Report; FAOSTAT: Rome, Italy, 2020; p. 184. [Google Scholar]
- Balloux, F.; Lugon-Moulin, N. The estimation of population differentiation with microsatellite markers. Mol. Ecol. 2002, 11, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Porth, I.; El-Kassaby, Y. Assessment of the Genetic Diversity in Forest Tree Populations Using Molecular Markers. Diversity 2014, 6, 283–295. [Google Scholar] [CrossRef]
- Yanbaev, Y.; Sultanova, R.; Blonskaya, L.; Bakhtina, S.; Tagirova, A.; Tagirov, V.; Kulagin, A. Gene pool of Scots pine (Pinus sylvestris L.) under reforestation in extreme environment. Wood Res. 2020, 65, 459–470. [Google Scholar] [CrossRef]
- Schmidt, W.C. Around the World with Larix: An Introduction. Ecol. Manag. Larix For. Look Ahead 1995, 6–18. [Google Scholar]
- Brandt, J.P.; Flannigan, M.D.; Maynard, D.G.; Thompson, I.D.; Volney, W.J.A. An introduction to Canada’s boreal zone: Ecosystem processes, health, sustainability, and environmental issues. Environ. Rev. 2013, 21, 207–226. [Google Scholar] [CrossRef]
- Semerikov, V.L. Population structure and molecular systematics of Larix Mill. species. Ext. Abstr. Dr. (Biol.) Diss. 2006. [Google Scholar]
- Fedorov, V.S.; Ryazanova, T.V. Bark of Siberian Conifers: Composition, Use, and Processing to Extract Tannin. Forests 2021, 12, 1043. [Google Scholar] [CrossRef]
- Liu, X.; Chen, W.; Liu, Q.; Dai, J. Abietic acid suppresses non-small-cell lung cancer cell growth via blocking IKKbeta/NF-kappaB signaling. Onco. Targets Ther. 2019, 12, 4825–4837. [Google Scholar] [CrossRef]
- Talevi, A.; Cravero, M.S.; Castro, E.A.; Bruno-Blanch, L.E. Discovery of anticonvulsant activity of abietic acid through application of linear discriminant analysis. Bioorg. Med. Chem. Lett. 2007, 17, 1684–1690. [Google Scholar] [CrossRef]
- Ito, Y.; Ito, T.; Yamashiro, K.; Mineshiba, F.; Hirai, K.; Omori, K.; Yamamoto, T.; Takashiba, S. Antimicrobial and antibiofilm effects of abietic acid on cariogenic Streptococcus mutans. Odontology 2020, 108, 57–65. [Google Scholar] [CrossRef]
- Cheliak, W.M.; Pitel, J.A. Inheritance and Linkage of Allozymes in Larix laricina. Silvae Genet. 1985, 34, 142–147. [Google Scholar]
- Fins, L.; Seeb, L.W. Genetic Variation in Allozymes of Western Larch. Can. J. For. Res. 1986, 16, 1013–1018. [Google Scholar] [CrossRef]
- Lewandowski, A.; Berczyk, J.; Mejnartowicz, L. Genetic Structure and the Mating System in an Old Stand of Polish Larch. Silvae Genet. 1991, 40, 75–79. [Google Scholar]
- Semerikov, V.L.; Matveev, A.V. Investigation of Genetic Variation of Allozyme Loci in Siberian Larch Larix sibirica Ledb. Russ. J. Genet. 1995, 34, 944–949. [Google Scholar]
- Shigapov, Z.K.; Putenikhin, V.P.; Shigapova, A.I.; Urazbakhtina, K.A. Genetic Structure of the Ural Populations of Larix sukaczewii. Russ. J. Genet. 1998, 34, 54–63. [Google Scholar]
- Larionova, A.Y.; Yakhneva (Oreshkova), N.V.; Kuz’mina, N.A. Genetic Variation of Siberian Larch in the Lower Angara River Basin. Lesovedenie 2003, 4, 17–22. [Google Scholar]
- Oreshkova, N.V.; Barchenkov, A.P. Genetic Peculiarities and Morphological Variability of Siberian Larch in Altai–Sayany Mountain Region. Vestn. Krasn. Gos. Agrar. Univ. 2004, 40, 1127–1134. [Google Scholar]
- Larionova, A.; Iakhneva, N.V.; Abaimov, A.P. Genetic diversity and differentiation of Gmelin larch Larix gmelinii populations from Evenkia (Central Siberia). Genetika 2004, 40, 1370–1377. [Google Scholar] [CrossRef] [PubMed]
- Babushkina, E.A.; Vaganov, E.A.; Grachev, A.M.; Oreshkova, N.V.; Belokopytova, L.V.; Kostyakova, T.V.; Krutovsky, K.V. The effect of individual genetic heterozygosity on general homeostasis, heterosis and resilience in Siberian larch (Larix sibirica Ledeb.) using dendrochronology and microsatellite loci genotyping. Dendrochronologia 2016, 38, 26–37. [Google Scholar] [CrossRef]
- Oreshkova, N.V.; Putintseva, Y.A.; Sharov, V.V.; Kuzmin, D.A.; Krutovsky, K.V. Development of microsatellite genetic markers in Siberian larch (Larix sibirica Ledeb.) based on the de novo whole genome sequencing. Russ. J. Genet. 2017, 53, 1194–1199. [Google Scholar] [CrossRef][Green Version]
- Li, S.; Ramakrishnan, M.; Vinod, K.K.; Kalendar, R.; Yrjälä, K.; Zhou, M. Development and Deployment of High-Throughput Retrotransposon-Based Markers Reveal Genetic Diversity and Population Structure of Asian Bamboo. Forests 2019, 11, 31. [Google Scholar] [CrossRef]
- Kalendar, R.; Muterko, A.; Boronnikova, S. Retrotransposable Elements: DNA Fingerprinting and the Assessment of Genetic Diversity. Methods Mol. Biol. 2021, 2222, 263–286. [Google Scholar] [CrossRef]
- Voronova, A.; Rendon-Anaya, M.; Ingvarsson, P.; Kalendar, R.; Rungis, D. Comparative Study of Pine Reference Genomes Reveals Transposable Element Interconnected Gene Networks. Genes 2020, 11, 1216. [Google Scholar] [CrossRef]
- Kalendar, R.; Schulman, A.H. Transposon-based tagging: IRAP, REMAP, and iPBS. Methods Mol. Biol. 2014, 1115, 233–255. [Google Scholar] [CrossRef]
- Oreshkova, N.V.; Belokon, M.M.; Jamiyansuren, S. Genetic diversity, population structure, and differentiation of Siberian larch, Gmelin larch, and Cajander larch on SSR-marker data. Russ. J. Genet. 2013, 49, 178–186. [Google Scholar] [CrossRef]
- Wagner, S.; Gerber, S.; Petit, R.J. Two highly informative dinucleotide SSR multiplexes for the conifer Larix decidua (European larch). Mol. Ecol. Resour. 2012, 12, 717–725. [Google Scholar] [CrossRef]
- Chen, X.B.; Xie, Y.H.; Sun, X.M. Development and characterization of polymorphic genic-SSR markers in Larix kaempferi. Molecules 2015, 20, 6060–6067. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Wang, Z.; He, Q.; Zhao, J.; Fan, Z.; Zhang, J. Development of EST-SSR markers in Larix principis-rupprechtii Mayr and evaluation of their polymorphism and cross-species amplification. Trees 2018, 32, 1559–1571. [Google Scholar] [CrossRef]
- Gramazio, P.; Plesa, I.M.; Truta, A.M.; Sestras, A.F.; Vilanova, S.; Plazas, M.; Vicente, O.; Boscaiu, M.; Prohens, J.; Sestras, R.E. Highly informative SSR genotyping reveals large genetic diversity and limited differentiation in European larch (Larix decidua) populations from Romania. Turk. J. Agric. For. 2018, 42, 165–175. [Google Scholar] [CrossRef]
- Liewlaksaneeyanawin, C.; Ritland, C.E.; El-Kassaby, Y.A.; Ritland, K. Single-copy, species-transferable microsatellite markers developed from loblolly pine ESTs. Theor. Appl. Genet. 2004, 109, 361–369. [Google Scholar] [CrossRef]
- Dylis, N.V. Siberian larch. In Materials for Taxonomy, Geography and History; Izdatelstvo MOIP: Moscow, Russia, 1947; p. 137. [Google Scholar]
- Igoshina, K.N. Larch in the Urals: Materials on the History of Flora and Vegetation of the USSR; Nauka: Leningrad, Russia, 1963. [Google Scholar]
- Putenikhin, V.P.; Martinsson, O. Present distribution of Larix sukaczewii Dyl. In Russia; The Swedish University of Agricultural Sciences, SLU: Umea, Russia, 1995. [Google Scholar]
- Putenikhin, V.P.; Farukshina, G.G.; Shigapov, Z.K. Sukachev Larch in the Urals. In Variability and Population Genetic Structure; Nauka: Moscow, Russia, 2004. [Google Scholar]
- Polezhayeva, M.A.; Semerikov, V.L. Genetic Diversity of cpSSR Loci in Larix Genus over the Far East Areas; Bulletin of the North-East Scientific Center, Russian Academy of Sciences, Far East Branch: Moscow, Russia, 2009; Volume 2, pp. 75–83. [Google Scholar]
- Zhang, L.; Zhang, H.G.; Li, X.F. Analysis of genetic diversity in Larix gmelinii (Pinaceae) with RAPD and ISSR markers. Genet. Mol. Res. 2013, 12, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Adrianova, I.Y. Genetic variability and differentiation of Kamchatka, Sakhalin and Kuril larch trees. Sib. For. J. 2014, 4, 110–116. [Google Scholar]
- Vasileva, Y.; Prishnivskaya, Y.; Chertov, N.; Zhulanov, A. Analysis of genetic diversity and structure of Urals populations of Western Race of Siberian larch (Larix sibirica Ledeb.) based on intermicrosatellite markers polymorphism. Bull. Sci. Pract. 2018, 4, 113–124. [Google Scholar] [CrossRef]
- Vasilyeva, Y.S.; Sboeva, Y.V.; Boronnikova, S.V.; Chertov, N.V.; Beltyukova, N.N. Genetic diversity, genetic structure and differentiation of Siberian larch populations in the Urals. Turczaninowia 2020, 23, 67–82. [Google Scholar] [CrossRef]
- Kalendar, R.; Boronnikova, S.; Seppanen, M. Isolation and Purification of DNA from Complicated Biological Samples. Methods Mol. Biol. 2021, 2222, 57–67. [Google Scholar] [CrossRef]
- Isoda, K.; Watanabe, A. Isolation and characterization of microsatellite loci from Larix kaempferi. Mol. Ecol. Notes 2006, 6, 664–666. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Peakall, R.O.D.; Smouse, P.E. genalex 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef]
- Goldstein, D.B.; Ruiz Linares, A.; Cavalli-Sforza, L.L.; Feldman, M.W. An evaluation of genetic distances for use with microsatellite loci. Genetics 1995, 139, 463–471. [Google Scholar] [CrossRef]
- Kivela, M.; Arnaud-Haond, S.; Saramaki, J. EDENetworks: A user-friendly software to build and analyse networks in biogeography, ecology and population genetics. Mol. Ecol. Resour. 2015, 15, 117–122. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Hijmans, R.J. Raster: Geographic Data Analysis and Modeling. In 2021. Available online: https://cran.r-project.org/web/packages/raster/index.html (accessed on 22 August 2021).
- Verity, R.; Nichols, R.A. Estimating the Number of Subpopulations (K) in Structured Populations. Genetics 2016, 203, 1827–1839. [Google Scholar] [CrossRef] [PubMed]
- Amyaga, E.; Nifontov, S. Selection of nuclear microsatellite loci for specific identification of Larix gmélinii Rupr. and comparison of genetic profiles of Larix to solve agricultural problems. IOP Conf. Ser. Earth Environ. Sci. 2019, 316, 012016. [Google Scholar] [CrossRef]
- Decision, On measures for the development of non-ferrous metallurgy region for 1997–2005. In Decision of the Government of the Chelyabinsk Oblast; Government of the Chelyabinsk Oblast: Chelyabinsk, Russia, 1997; p. 16.
- Kremer, A.; Hipp, A.L. Oaks: An evolutionary success story. New Phytol. 2020, 226, 987–1011. [Google Scholar] [CrossRef] [PubMed]
- Cortes, A.J.; Restrepo-Montoya, M.; Bedoya-Canas, L.E. Modern Strategies to Assess and Breed Forest Tree Adaptation to Changing Climate. Front. Plant. Sci. 2020, 11, 583323. [Google Scholar] [CrossRef] [PubMed]
- Vasilyeva, Y.; Chertov, N.; Nechaeva, Y.; Sboeva, Y.; Pystogova, N.; Boronnikova, S.; Kalendar, R. Genetic Structure, Differentiation and Originality of Pinus sylvestris L. Populations in the East of the East European Plain. Forests 2021, 12, 999. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| SSR Locus | Primer Sequence (5′-3′) | Ta (°C) | A | Repeat Motif | Lfr (bp) |
|---|---|---|---|---|---|
| bcLK189 | F: M13-ACCATACGCATACCCAATAGA | 58 | 17 | (AG)17AT(AG)6 | 155–170 |
| R: AGTTTTCCTTTCCCACACAAT | |||||
| Ld101 | F: M13-ACACCAAGGACTCTCTGACTAC | 58 | 16 | (AC)12 | 189–225 |
| R: GGTGATTCCAGAAGCAGGTG | |||||
| bcLK228 | F: M13-CCCTAACCCTAGAATCCAATAA | 61 | 12 | (AG)18 | 179–215 |
| R: GAGGAAGGCGACAAGTCATT | |||||
| Ld56 | F: M13-AGCCATCGTGGTTCTTCTTTG | 58 | 9 | (AC)16 | 227–243 |
| R: CTTGTAACTGTGCACCCACC | |||||
| Ld50 | F: M13-GAAGGCGACTTTACATGCCC | 58 | 6 | (CA)18 | 161–189 |
| R: TCCATCTTTATGTCTCTTCCATGC | |||||
| bcLK253 | F: M13-AACACCATAGTGCAATGTGC | 58 | 4 | (AG)17 | 199–204 |
| R: TCCTCTTGTTGATGCCACTT | |||||
| Ld42 | F: M13-TCGTATGCATTGTCCAAATTTCC | 58 | 8 | (TG)14 | 167–186 |
| R: TCCAAGTGAGGTCACACGAG | |||||
| bcLK263 | F: M13-CGATTGGTATAGTGGTCATTGT | 58 | 37 | (TC)20 | 191–254 |
| R: CCATCATACCTTCTTGAAGAG |
| Populations | Number of Alleles (Na) | Number of Effective Alleles (Ne) | Observed Heterozygosity (Ho) | Expected Heterozygosity (He) |
|---|---|---|---|---|
| Kar | 7.000 | 3.263 | 0.522 | 0.587 |
| Irm | 8.250 | 4.998 | 0.567 | 0.676 |
| Vsn | 8.500 | 5.276 | 0.484 | 0.643 |
| Kul | 7.375 | 4.034 | 0.500 | 0.629 |
| Zil | 6.750 | 3.929 | 0.460 | 0.632 |
| Tul | 6.375 | 3.825 | 0.387 | 0.634 |
| Ish | 4.625 | 2.762 | 0.366 | 0.563 |
| Krv | 7.750 | 4.266 | 0.433 | 0.612 |
| Bnd | 6.875 | 4.357 | 0.463 | 0.635 |
| Gai | 8.125 | 4.740 | 0.428 | 0.682 |
| Kch | 8.375 | 4.584 | 0.479 | 0.666 |
| Pol | 5.875 | 3.662 | 0.562 | 0.622 |
| Osa | 7.000 | 4.008 | 0.418 | 0.626 |
| Sks | 4.250 | 2.283 | 0.396 | 0.520 |
| Bil | 7.000 | 4.273 | 0.458 | 0.624 |
| For a total sample of 15 populations | 6.942 | 4.017 | 0.461 | 0.623 |
| Locus | Fis | Fit | Fst |
|---|---|---|---|
| bcLK189 | 0.138 | 0.198 | 0.070 |
| Ld101 | 0.577 | 0.609 | 0.077 |
| bcLK228 | 0.595 | 0.628 | 0.080 |
| Ld56 | 0.351 | 0.427 | 0.117 |
| bcLK253 | 0.083 | 0.150 | 0.073 |
| Ld50 | 0.357 | 0.428 | 0.111 |
| bcLK263 | 0.014 | 0.083 | 0.070 |
| Ld42 | −0.045 | 0.075 | 0.115 |
| Total | 0.259 | 0.325 | 0.089 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chertov, N.; Vasilyeva, Y.; Zhulanov, A.; Nechaeva, Y.; Boronnikova, S.; Kalendar, R. Genetic Structure and Geographical Differentiation of Larix sibirica Ledeb. in the Urals. Forests 2021, 12, 1401. https://doi.org/10.3390/f12101401
Chertov N, Vasilyeva Y, Zhulanov A, Nechaeva Y, Boronnikova S, Kalendar R. Genetic Structure and Geographical Differentiation of Larix sibirica Ledeb. in the Urals. Forests. 2021; 12(10):1401. https://doi.org/10.3390/f12101401
Chicago/Turabian StyleChertov, Nikita, Yulia Vasilyeva, Andrei Zhulanov, Yulia Nechaeva, Svetlana Boronnikova, and Ruslan Kalendar. 2021. "Genetic Structure and Geographical Differentiation of Larix sibirica Ledeb. in the Urals" Forests 12, no. 10: 1401. https://doi.org/10.3390/f12101401
APA StyleChertov, N., Vasilyeva, Y., Zhulanov, A., Nechaeva, Y., Boronnikova, S., & Kalendar, R. (2021). Genetic Structure and Geographical Differentiation of Larix sibirica Ledeb. in the Urals. Forests, 12(10), 1401. https://doi.org/10.3390/f12101401