
Comparative Transcriptome Analysis of Stage-Specific Changes in Gene Expression during Larval Development in Monochamus alternatus Hope

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Insects
2.2. RNA Extraction
2.3. cDNA Library Construction and Illumina Sequencing
2.4. Sequence Assembly
2.5. Functional Annotation
2.6. Differentially Expressed Gene (DEG) Analysis Based on FPKM Values
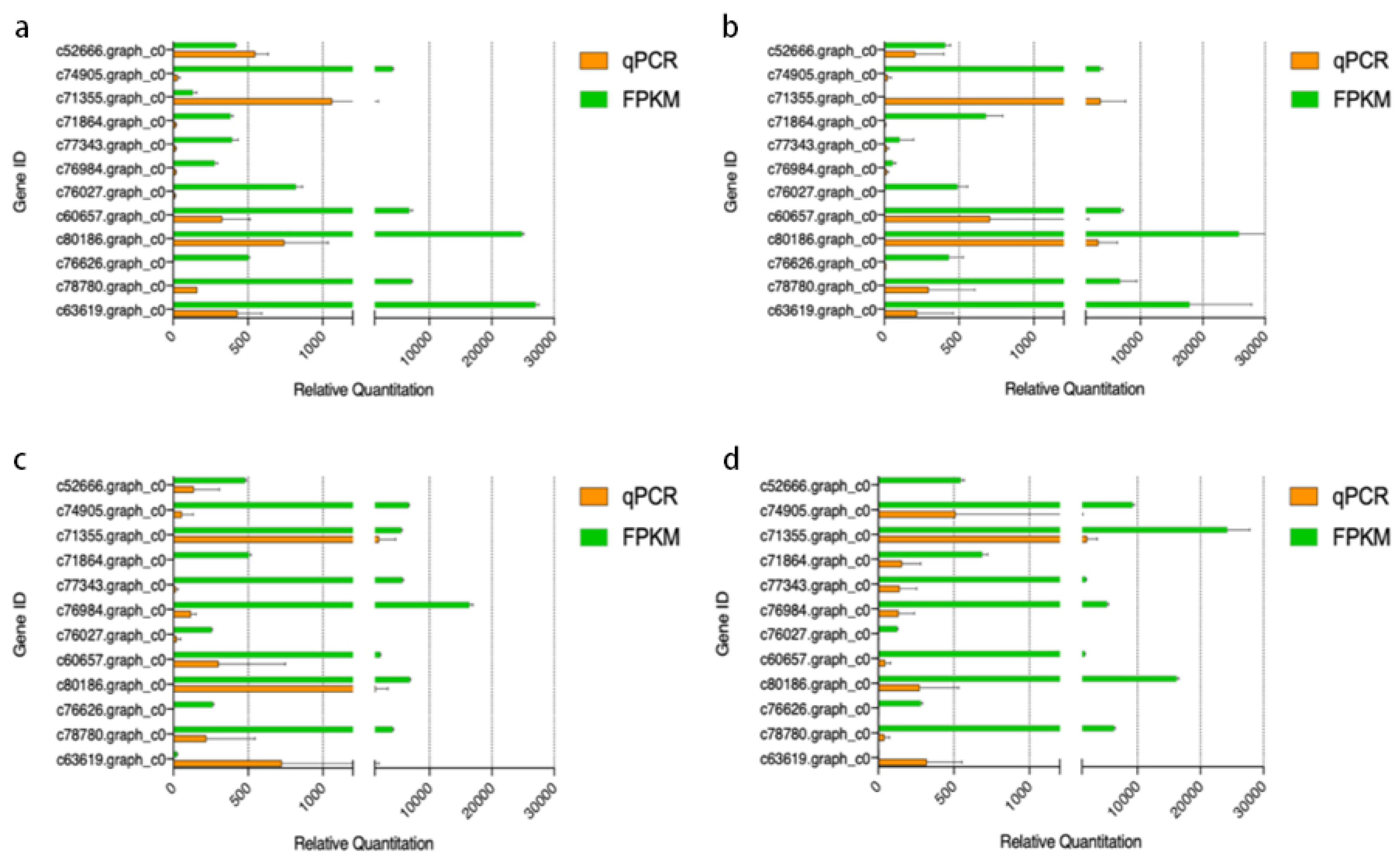
2.7. Validation of Gene Expression by qPCR
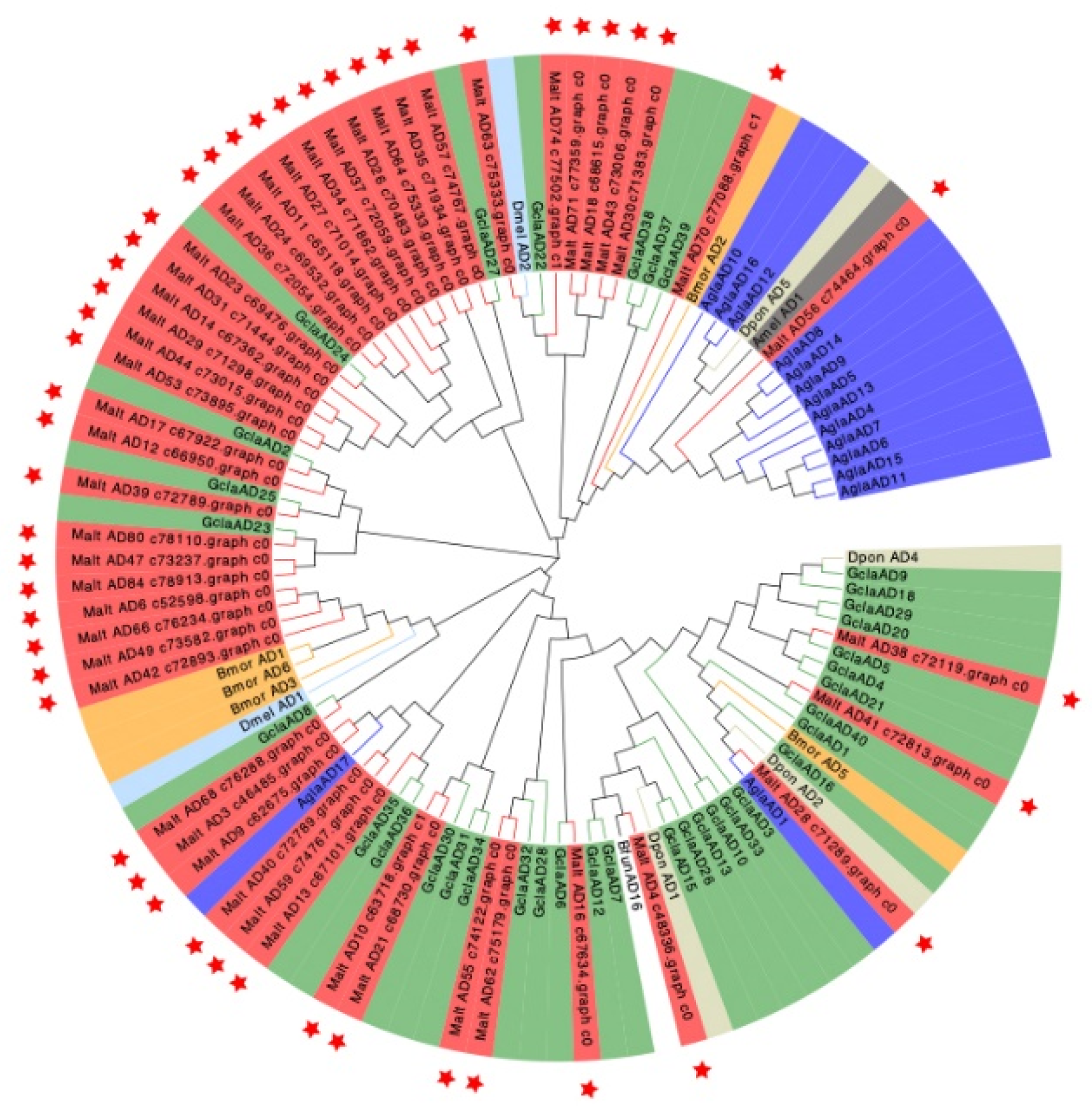
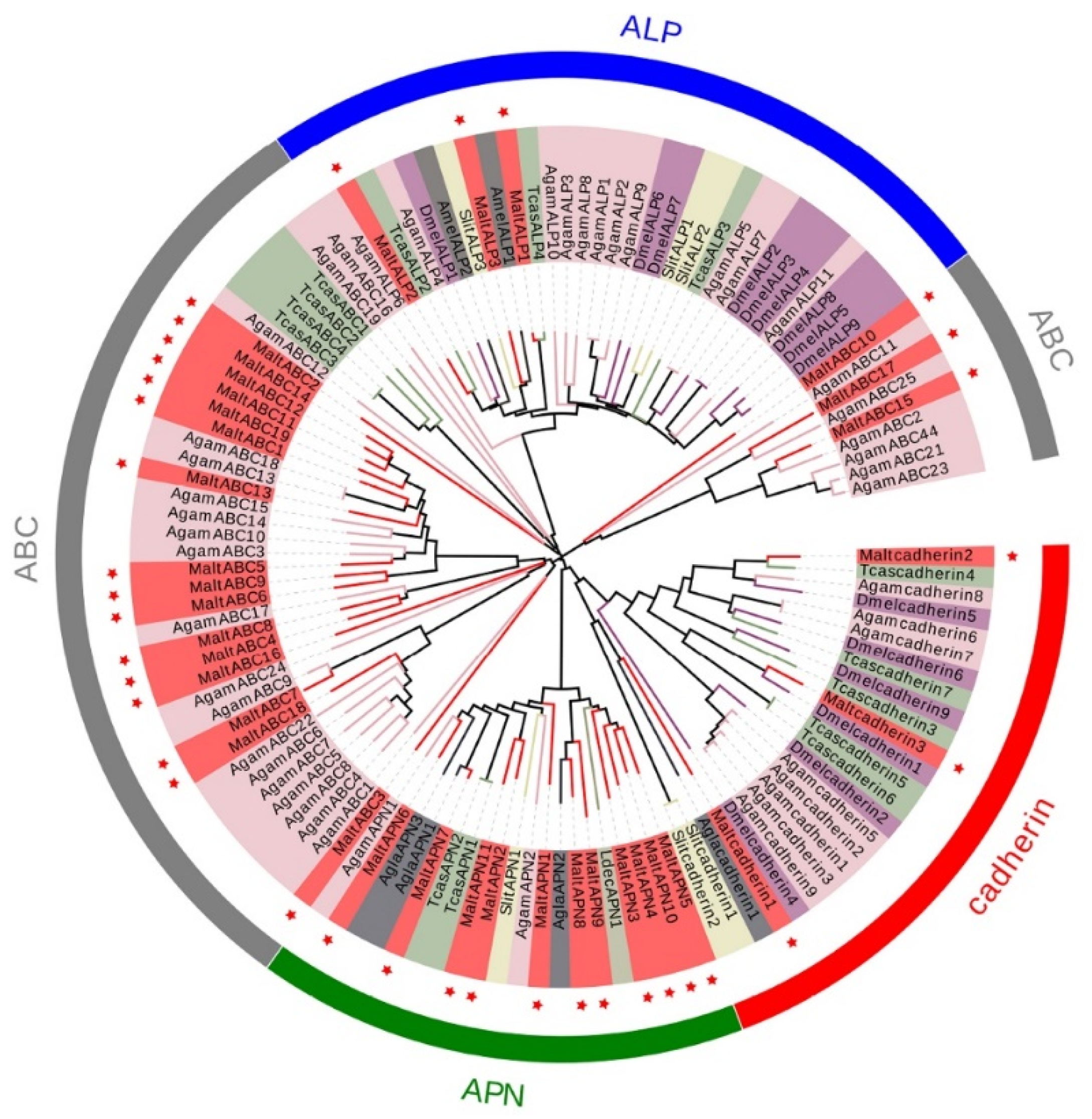
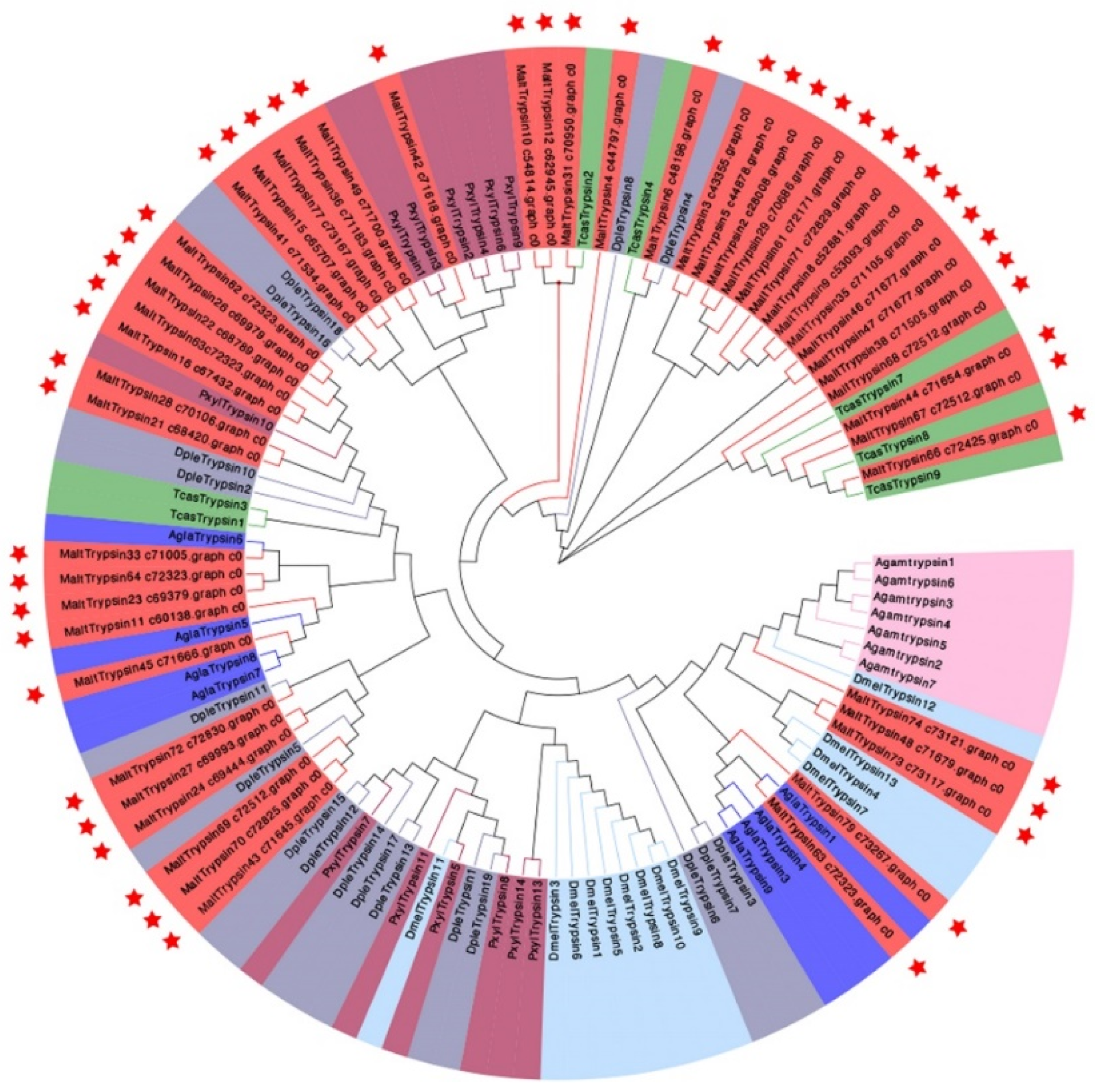
2.8. Construction of a Phylogenetic Tree and Heat Map
2.9. Statistical Analysis
3. Results
3.1. Illumina Sequencing and de Novo Assembly
3.2. Functional Annotation and Differential Expression of Unigenes in the Larvae of M. alternatus
3.3. Analysis of Differentially Expressed Genes Based on FPKM Values
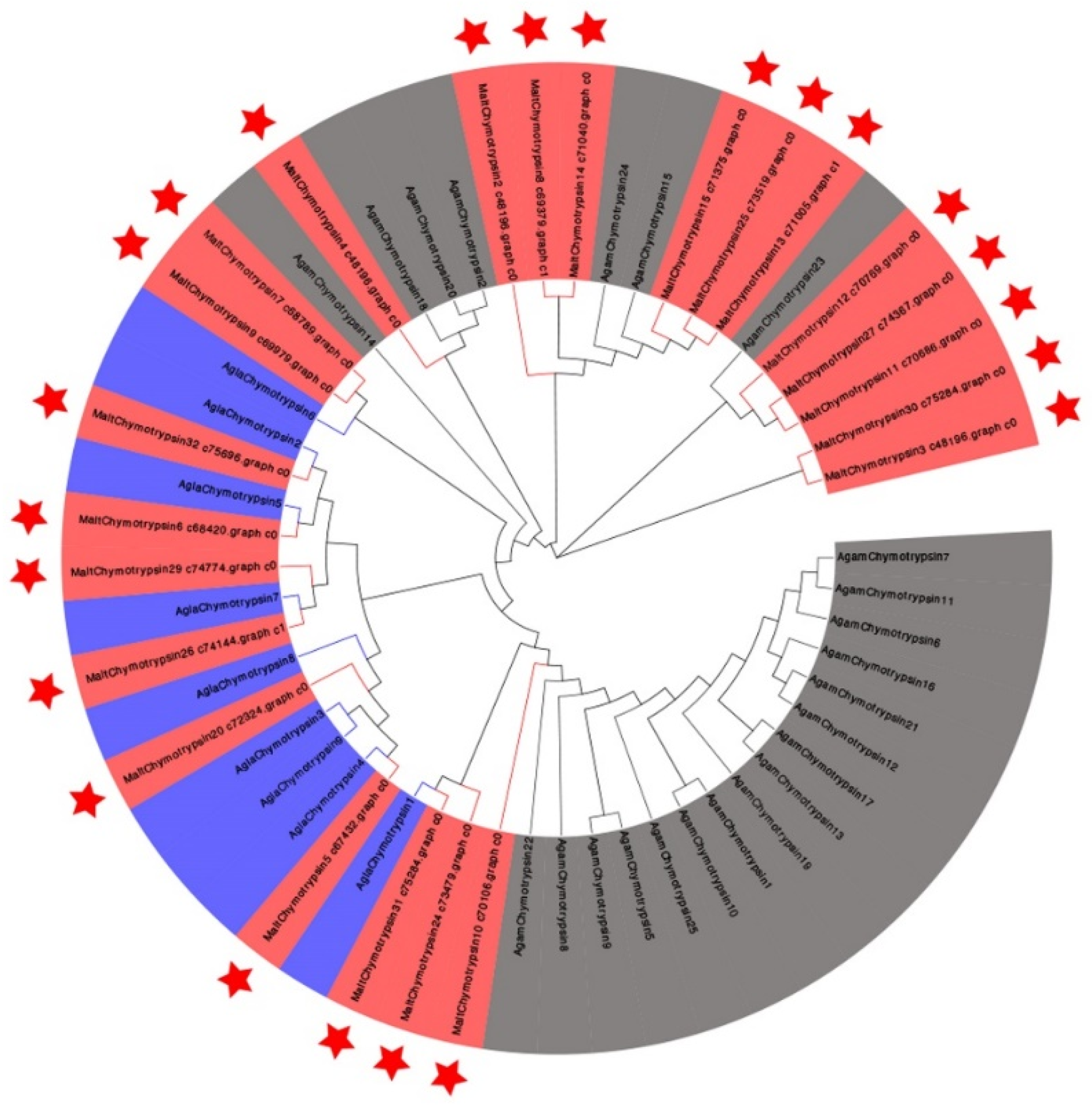
3.4. Candidate Genes Related to Lignocellulose
3.5. Candidate Genes Related to Bt Receptors
3.6. Candidate Genes Related to the Digestion Process
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suh, D.Y.; Jung, J.K.; Lee, S.K.; Seo, S.T. Effect of aerial spraying of thiacloprid on pine sawyer beetles (Monochamus alternatus) and honey bees (Apis mellifera) in pine forests. Entomol. Res. 2020, 51, 83–89. [Google Scholar] [CrossRef]
- Zhao, B.G. Pine Wilt Disease. Springer 2008, 17, 459. [Google Scholar]
- Suzuki, K. Pine wilt disease-a threat to pine forest in Europe. Dendrobiology 2004, 48, 59–67. [Google Scholar]
- Wang, Y.; Chen, F.; Wang, L.; Li, M. Investigation of beetle species that carry the pine wood nematode, Bursaphelenchus xylophilus (Steiner and Buhrer) Nickle, in China. J. For. Res. 2020, 32, 1745–1751. [Google Scholar] [CrossRef]
- Bulletin of the National Forestry and Grassland Administration (No. 5 of 2021) (Pine Wood Nematode Disease Epidemic Areas in 2021). Available online: http://www.forestry.gov.cn/main/3457/20210329/151957233445926.html (accessed on 17 July 2021).
- Xun, L.I.; Dai, L.; Shen, S. Technology, Study on the biological characteristics of Monochamus alternatus in Chenzhou City. Hunan For. Sci. Technol. 2010, 37, 24–26. [Google Scholar]
- McKenna, D.D.; Scully, E.D.; Pauchet, Y.; Hoover, K.; Kirsch, R.; Geib, S.M.; Mitchell, R.F.; Waterhouse, R.M.; Ahn, S.J.; Arsala, D.; et al. Genome of the Asian longhorned beetle (Anoplophora glabripennis), a globally significant invasive species, reveals key functional and evolutionary innovations at the beetle-plant interface. Genome Biol. 2016, 17, 227. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C. Horizontal Gene Transfer and Gene Duplication of Plant Cell Wall Degrading Enzyme Genes in an Invasive Insect Pest. Ohio State Univ. 2014, 1, 1. [Google Scholar]
- Eyun, S.I.; Wang, H.; Pauchet, Y.; Ffrench-Constant, R.H.; Benson, A.K.; Valencia-Jimenez, A.; Moriyama, E.N.; Siegfried, B.D. Molecular evolution of glycoside hydrolase genes in the Western corn rootworm (Diabrotica virgifera virgifera). PLoS ONE 2014, 9, e94052. [Google Scholar] [CrossRef]
- Zhang, L.L.; Liang, G.M.; Cao, G.C.; Gao, X.W.; Guo, Y.Y. Approaches for enhancing the insecticidal activity of Bacillus thuringiensis Cry toxins: Application of synergistic factors and genetic improvement of crystal protein. J. Environ. Entomol. 2010, 32, 525–531. [Google Scholar]
- Moses, E.; Maureen, S.; Samuel, K.; Mwanga, R.; Benson, O.; Marc, G.; Moar, W.J.J.J. Toxicity of Seven Bacillus thuringiensis Cry Proteins Against Cylas puncticollis and Cylas brunneus (Coleoptera: Brentidae) Using a Novel Artificial Diet. J. Econ. Entomol. 2010, 4, 4. [Google Scholar]
- Bergamasco, V.B.; Mendes, D.R.P.; Fernandes, O.A.; Desidério, J.A.; Lemos, M.V. Bacillus thuringiensis Cry1Ia10 and Vip3Aa protein interactions and their toxicity in Spodoptera spp. Lepidoptera 2013, 112, 152–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravo, A.; Gill, S.S.; Soberon, M. Mode of action of Bacillus thuringiensis Cry and Cyt toxins and their potential for insect control. Toxicon 2007, 49, 423–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y. Detection and Mechanisms of Resistance Evolved in Insects to Cry Toxins from Bacillus thuringiensis. Adv. Insect Physiol. 2014, 47, 297–342. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schffer, A.A.; Zhang, J.; Zhang, Z.; Webb, M.; Lipman, D.J.J.N. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389. [Google Scholar] [CrossRef] [Green Version]
- Nastou, K.C.; Tsaousis, G.N.; Papandreou, N.C.; Hamodrakas, S.J. MBPpred: Proteome-wide detection of membrane lipid-binding proteins using profile Hidden Markov Models. Biochim. Biophys. Acta 2016, 1864, 747–754. [Google Scholar] [CrossRef]
- Deng, Y.; Jianqi, L.I.; Songfeng, W.U.; Zhu, Y.; Chen, Y.; Fuchu, H.E.J.C.E. Integrated nr Database in Protein Annotation System and Its Localization. Comput. Eng. 2006, 32, 71–72. [Google Scholar]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Biology, A.J.J.G. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef] [Green Version]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [Green Version]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, G. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Thissen, D.; Steinberg, L.; Kuang, D. Quick and Easy Implementation of the Benjamini-Hochberg Procedure for Controlling the False Positive Rate in Multiple Comparisons. J. Educ. Behav. Stat. 2016, 27, 77–83. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeling, C.I.; Yuen, M.M.; Liao, N.Y.; Docking, T.R.; Chan, S.K.; Taylor, G.A.; Palmquist, D.L.; Jackman, S.D.; Nguyen, A.; Li, M.; et al. Draft genome of the mountain pine beetle, Dendroctonus ponderosae Hopkins, a major forest pest. J. Genome Biol. 2013, 14, R27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Song, W.; Zhang, Z.; Wang, H.; Yang, M.; Guo, R.; Li, M. Transcriptome analysis of Dastarcus helophoroides (Coleoptera: Bothrideridae) using Illumina HiSeq sequencing. PLoS ONE 2014, 9, e100673. [Google Scholar] [CrossRef]
- Wu, S.; Zhu, X.; Liu, Z.; Shao, E.; Rebeca, C.L.; Guo, Y.; Xiong, Y.; Mou, Y.; Xu, R.; Hu, X.; et al. Identification of Genes Relevant to Pesticides and Biology from Global Transcriptome Data of Monochamus alternatus Hope (Coleoptera: Cerambycidae) Larvae. PLoS ONE 2016, 11, e0147855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Ren, S.; Ji, M.A.; Liu, X.J.J. Expression Profile of Alcohol Dehydrogenase from Helicoverpa Armigera. J. Xinjiang Univ. (Nat. Sci. Ed.) 2017, 34, 267–272. [Google Scholar]
- Fernandez, L.E.; Aimanova, K.G.; Gill, S.S.; Bravo, A.; Soberon, M. A GPI-anchored alkaline phosphatase is a functional midgut receptor of Cry11Aa toxin in Aedes aegypti larvae. Biochem. J. 2006, 394 Pt 1, 77–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, N.; Malhotra, P.; Bhatnagar, R.K. Interaction of gene-cloned and insect cell-expressed aminopeptidase N of Spodoptera litura with insecticidal crystal protein Cry1C. Appl. Environ. Microbiol. 2002, 68, 4583–4592. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Zhang, X.; Zhang, J. Molecular characterization of four midgut aminopeptidase N isozymes from the cabbage looper, Trichoplusia ni. Insect Biochem. Mol. Biol. 2005, 35, 611–620. [Google Scholar] [CrossRef]
- Luo, K.; Sangadala, S.; Masson, L.; Mazza, A.; Brousseau, R.; Adang, M.J.J. The heliothis virescens 170 kDa aminopeptidase functions as “receptor A” by mediating specific Bacillus thuringiensis Cry1A delta-endotoxin binding and pore formation. Insect Biochem. Mol. Biol. 1997, 27, 735–743. [Google Scholar] [CrossRef]
- Midboe, E. Expression of a midgut-specific cadherin BT-R1 during the development of Manduca sexta larva. Comp. Biochem. Physiol. Part. B Biochem. Mol. Biol. 2003, 135, 125–137. [Google Scholar] [CrossRef]
- Flagel, L.; Lee, Y.W.; Wanjugi, H.; Swarup, S.; Brown, A.; Wang, J.; Kraft, E.; Greenplate, J.; Simmons, J.; Adams, N.; et al. Mutational disruption of the ABCC2 gene in fall armyworm, Spodoptera frugiperda, confers resistance to the Cry1Fa and Cry1A.105 insecticidal proteins. Sci. Rep. 2018, 8, 7255. [Google Scholar] [CrossRef] [Green Version]
- Oppert, B. Physiology, Protease interactions with Bacillus thuringiensis Insecticidal toxins. Arch. Insect Biochem. Physiol. 1999, 42, 1–12. [Google Scholar] [CrossRef]
- Brennan, Y.; Callen, W.N.; Christoffersen, L.; Dupree, P.; Goubet, F.; Healey, S.; Hernandez, M.; Keller, M.; Li, K.; Palackal, N.; et al. Unusual microbial xylanases from insect guts. Appl. Environ. Microbiol. 2004, 70, 3609–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knorr, E.; Schmidtberg, H.; Vilcinskas, A.; Altincicek, B. MMPs regulate both development and immunity in the tribolium model insect. PLoS ONE 2009, 4, e4751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauchet, Y.; Wilkinson, P.; van Munster, M.; Augustin, S.; Pauron, D.; ffrench-Constant, R.H. Pyrosequencing of the midgut transcriptome of the poplar leaf beetle Chrysomela tremulae reveals new gene families in Coleoptera. Insect Biochem. Mol. Biol. 2009, 39, 403–413. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Length Range | Transcript | Unigene |
|---|---|---|
| Total number | 95,899 | 67,456 |
| Total length | 106,767,745 | 52,950,876 |
| N50 length | 2233 | 1527 |
| Mean length | 1113.34 | 784.97 |
| Group | All_DEG | Up-Regulated | Down-Regulated |
|---|---|---|---|
| L1_vs_L2 | 1213 | 447 | 766 |
| L1_vs_L3 | 4732 | 2185 | 2547 |
| L1_vs_L4 | 5310 | 2727 | 2583 |
| L2_vs_L3 | 4314 | 2248 | 2066 |
| L2_vs_L4 | 4695 | 2569 | 2126 |
| L3_vs_L4 | 2707 | 1331 | 1376 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.; Guo, Y.; Weng, X.; Sun, Y.; Carballar-Lejarazú, R.; Hu, X.; Wang, R.; Liang, G.; Zhang, F.; Wu, S. Comparative Transcriptome Analysis of Stage-Specific Changes in Gene Expression during Larval Development in Monochamus alternatus Hope. Forests 2021, 12, 1312. https://doi.org/10.3390/f12101312
Huang J, Guo Y, Weng X, Sun Y, Carballar-Lejarazú R, Hu X, Wang R, Liang G, Zhang F, Wu S. Comparative Transcriptome Analysis of Stage-Specific Changes in Gene Expression during Larval Development in Monochamus alternatus Hope. Forests. 2021; 12(10):1312. https://doi.org/10.3390/f12101312
Chicago/Turabian StyleHuang, Jing, Yajie Guo, Xiaoqian Weng, Yunzhu Sun, Rebeca Carballar-Lejarazú, Xia Hu, Rong Wang, Guanghong Liang, Feiping Zhang, and Songqing Wu. 2021. "Comparative Transcriptome Analysis of Stage-Specific Changes in Gene Expression during Larval Development in Monochamus alternatus Hope" Forests 12, no. 10: 1312. https://doi.org/10.3390/f12101312
APA StyleHuang, J., Guo, Y., Weng, X., Sun, Y., Carballar-Lejarazú, R., Hu, X., Wang, R., Liang, G., Zhang, F., & Wu, S. (2021). Comparative Transcriptome Analysis of Stage-Specific Changes in Gene Expression during Larval Development in Monochamus alternatus Hope. Forests, 12(10), 1312. https://doi.org/10.3390/f12101312