Physiological Characterization and Transcriptome Analysis of Camellia oleifera Abel. during Leaf Senescence

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Growth Conditions
2.2. Physiological Analysis of C. oleifera Leaves
2.3. Total RNA Extraction
2.4. Construction of the cDNA Library and RNA Sequencing
2.5. Transcriptome Assembly and Annotation
2.6. Analysis and Enrichment of Differentially Expressed Genes (DEGs)
2.7. Screening of Key Senescence-Associated Genes (SAGs)
2.8. Analysis of Expression Correlation
2.9. Quantitative Real-Time PCR (qRT-PCR) Analysis of Gene Expression
3. Results
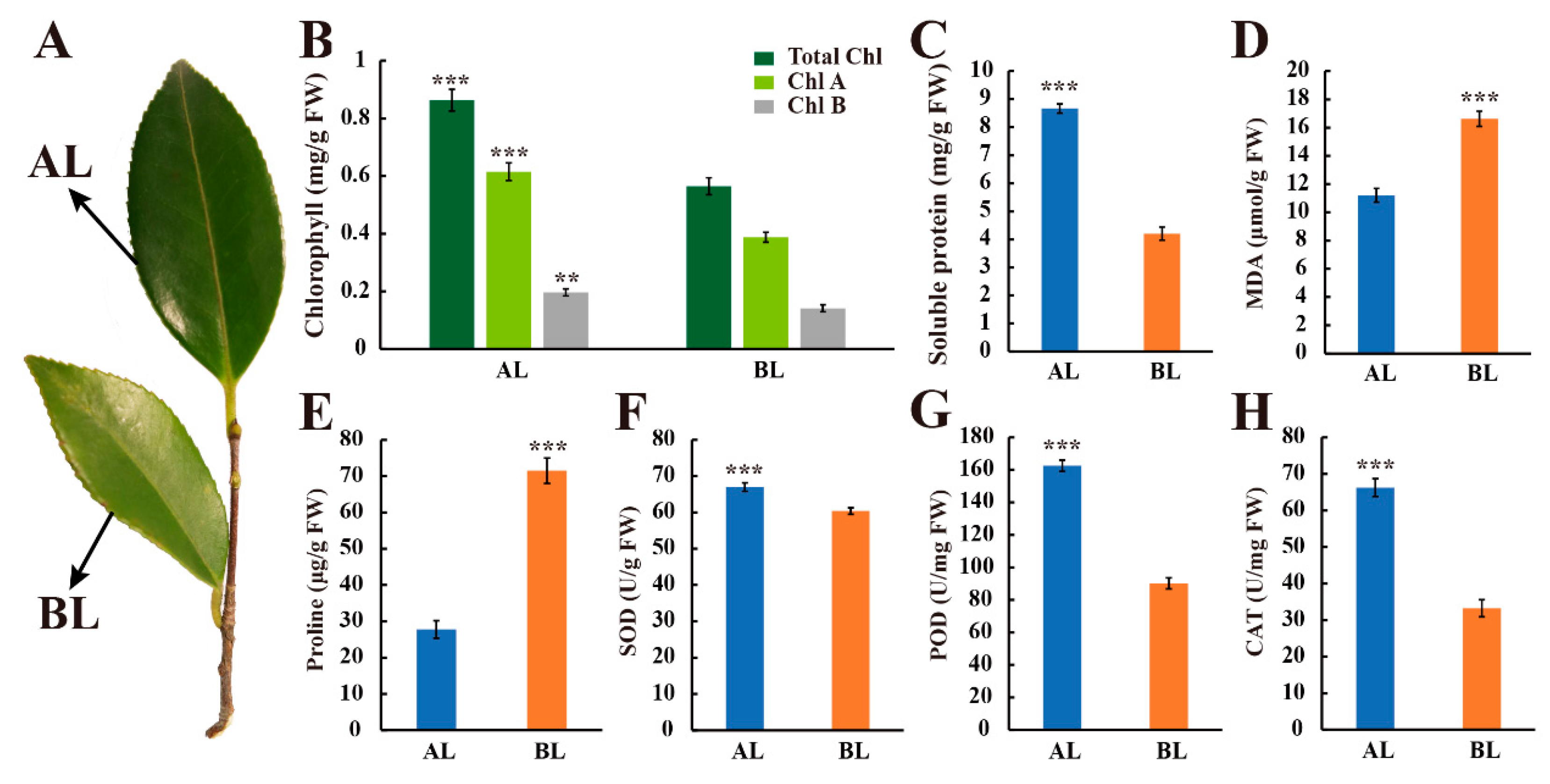
3.1. Morphological and Physiological Observation of Leaves at Different Leaf Age
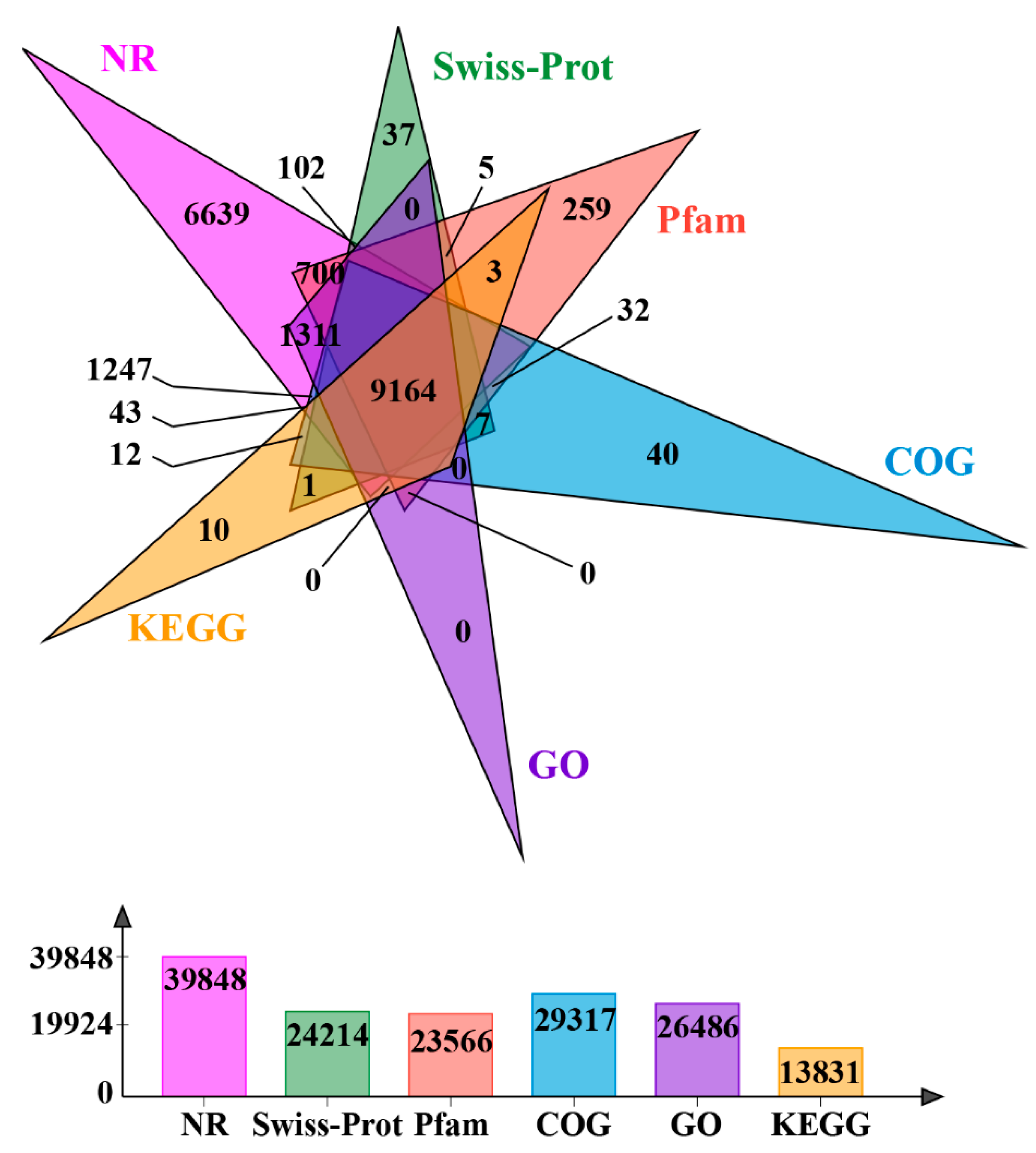
3.2. RNA Sequencing, De Novo Assembly, and Annotation
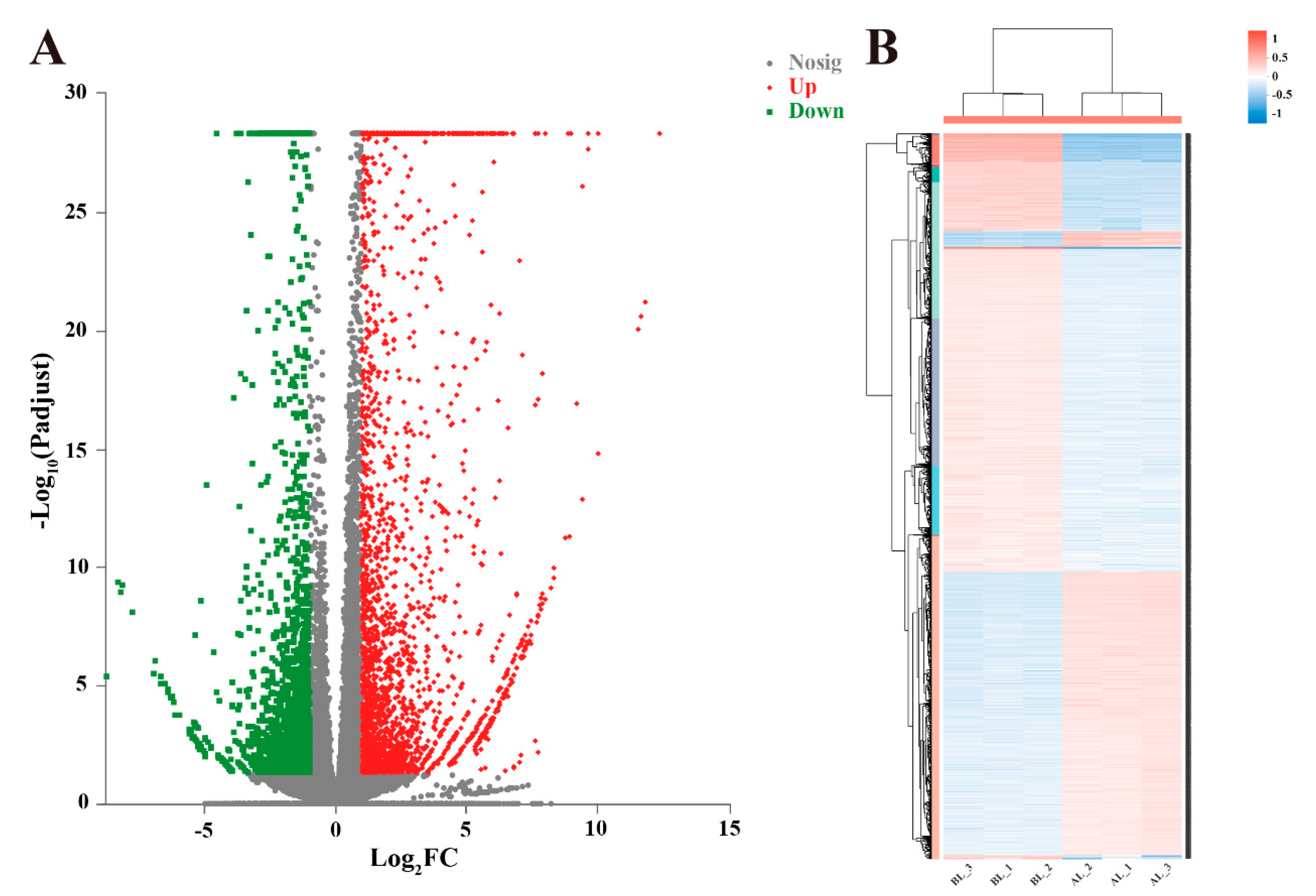
3.3. Analyses of Gene Expression and Identification of Differentially Expressed Genes (DEGs)
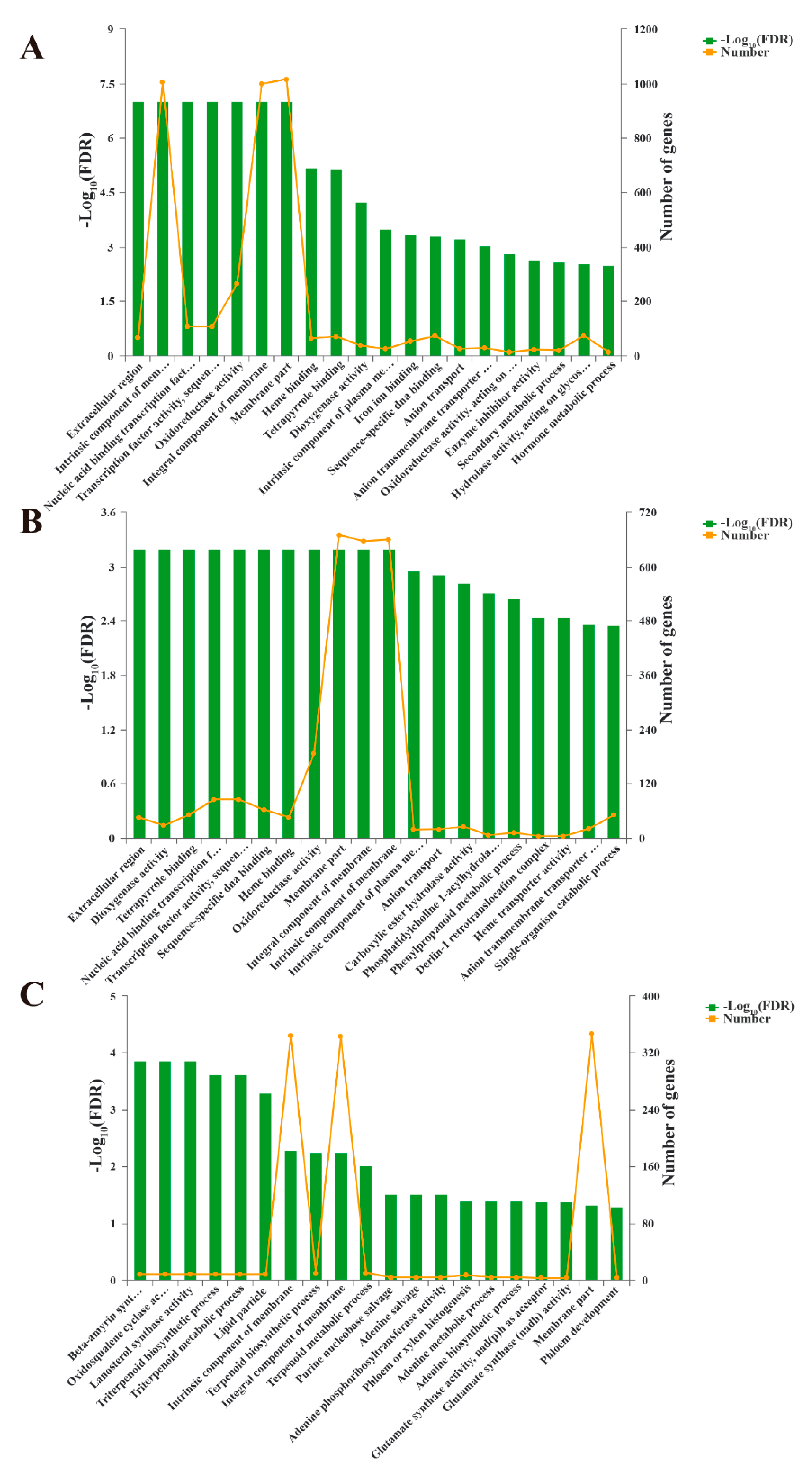
3.4. Gene Ontology (GO) Enrichment Analysis of DEGs
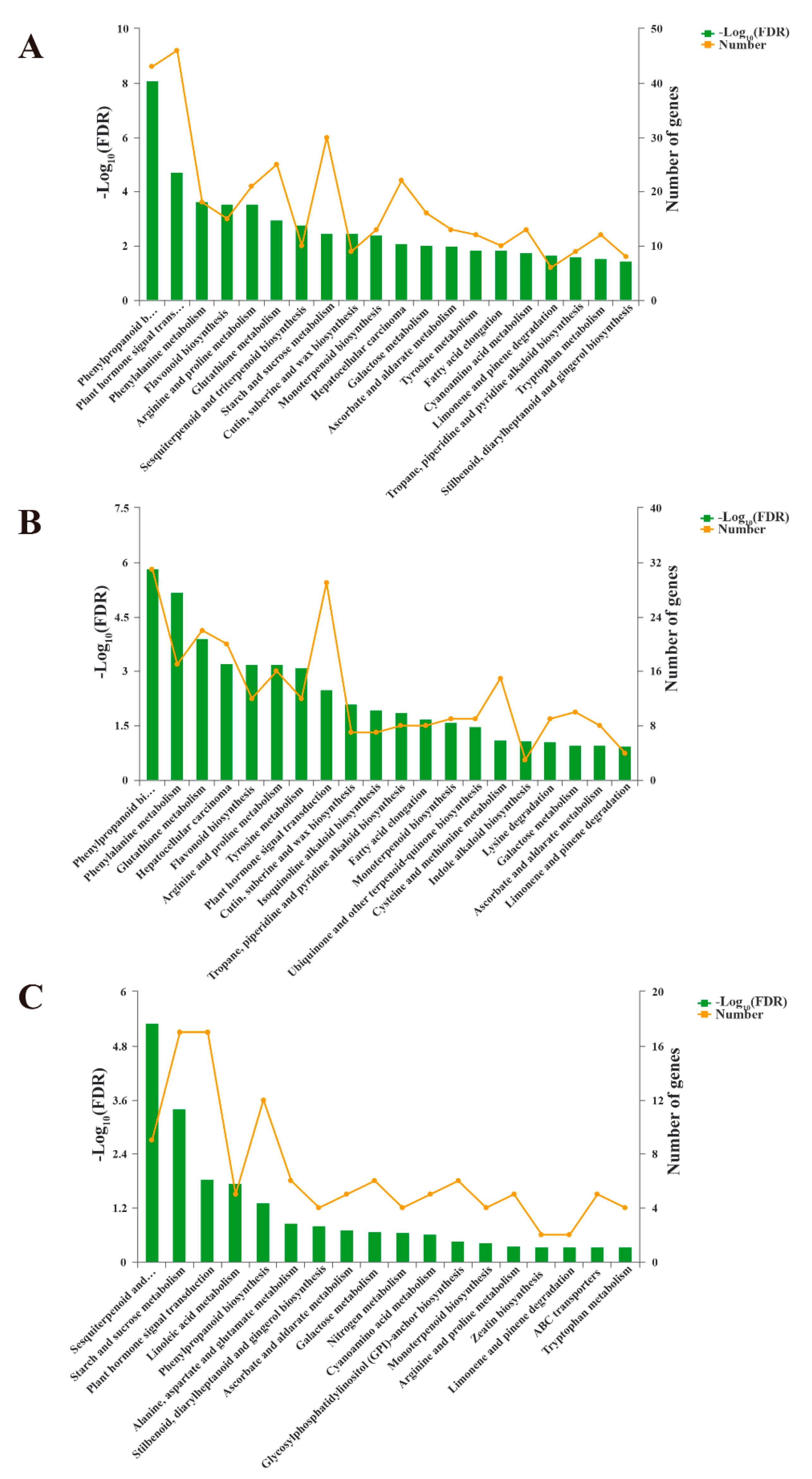
3.5. Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analysis of DEGs
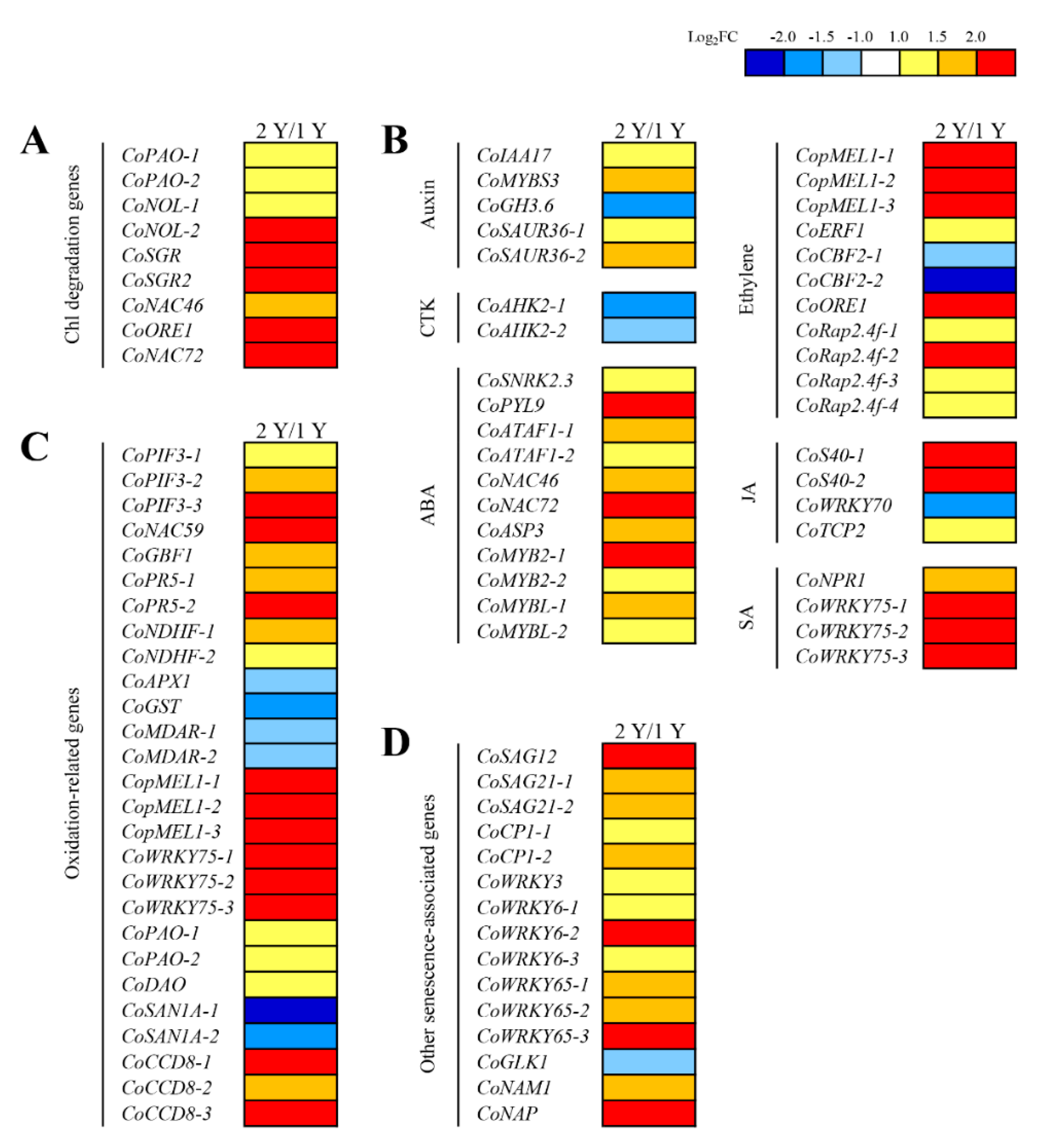
3.6. SAGs Differentially Expressed in Two Kinds of Leaves
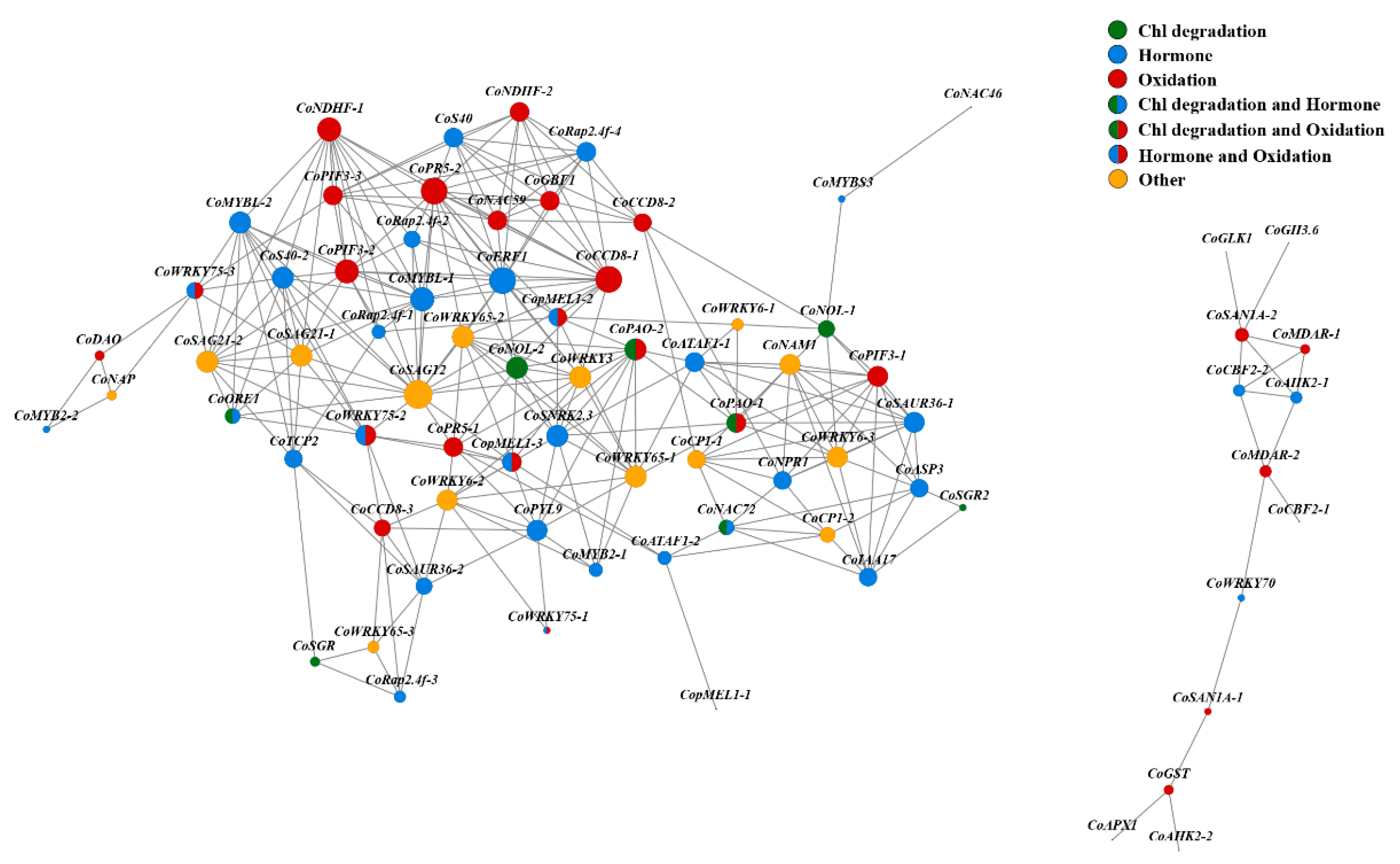
3.7. Correlation Analysis of SAGs
3.8. Transcription Factors (TFs) Responding to Leaf Senescence
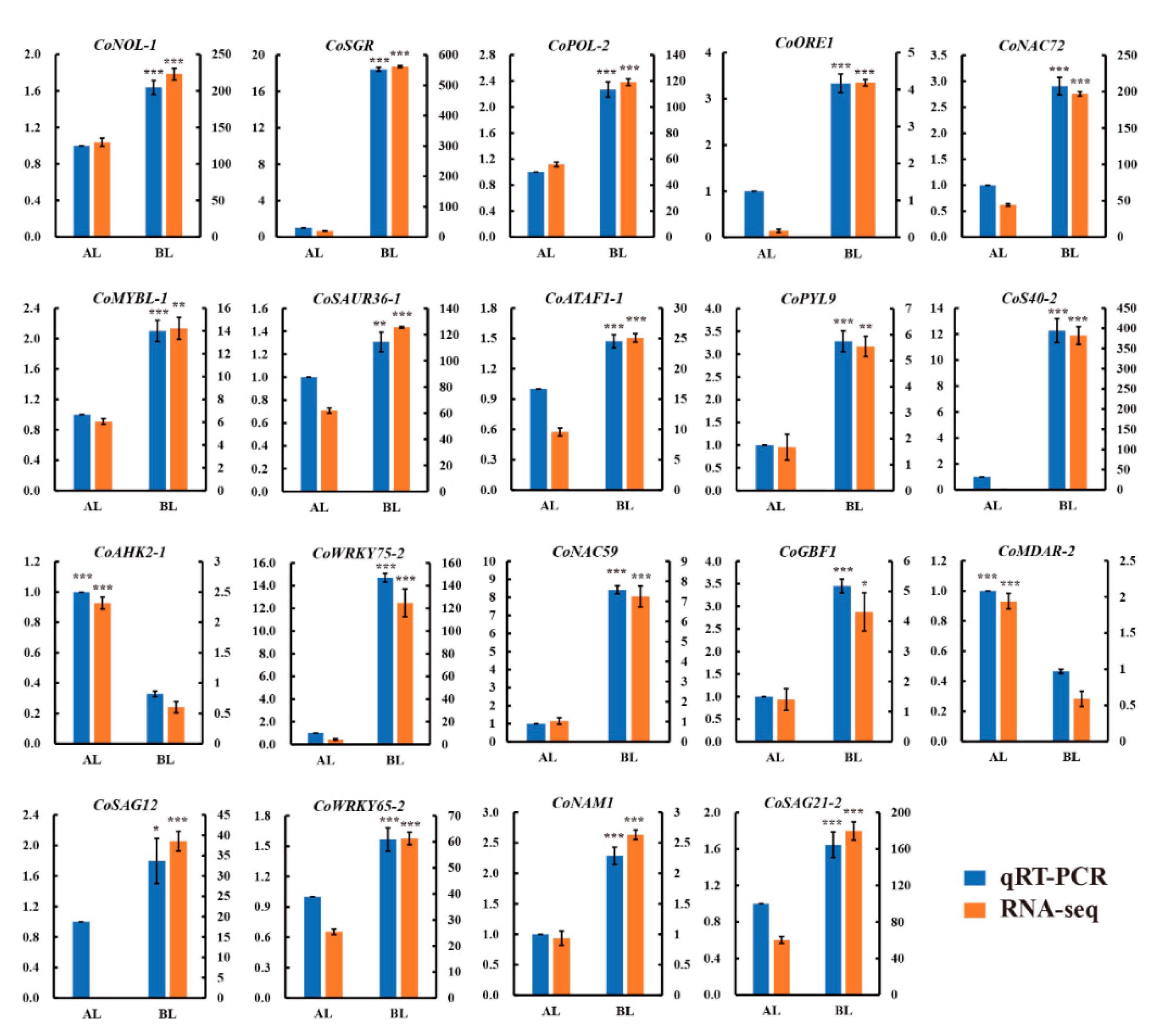
3.9. Validation of RNA-Seq Data by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhuang, R. Camellia Oleifera in China; China Forestry Press: Beijing, China, 2008; p. 366. [Google Scholar]
- Yang, C.; Liu, X.; Chen, Z.; Lin, Y.; Wang, S. Comparison of Oil Content and Fatty Acid Profile of Ten New Camellia oleifera Cultivars. J. Lipids 2016, 2016, 3982486. [Google Scholar] [CrossRef]
- Lee, C.P.; Yen, G.C. Antioxidant activity and bioactive compounds of tea seed (Camellia oleifera Abel.) oil. J. Agric. Food Chem. 2006, 54, 779–784. [Google Scholar] [CrossRef]
- Lee, C.P.; Shih, P.H.; Hsu, C.L.; Yen, G.C. Hepatoprotection of tea seed oil (Camellia oleifera Abel.) against CCl4-induced oxidative damage in rats. Food Chem. Toxicol. 2007, 45, 888–895. [Google Scholar] [CrossRef]
- Chen, Y.; Deng, S.; Chen, L.; Ma, L.; He, H.; Wang, X.; Peng, S.; Liu, C.; Wang, R.; Xu, Y.; et al. A new view on the development of oil tea camellia industry. J. Nanjing For. Univ. Nat. Sci. Edit. 2020, 44, 1–10. [Google Scholar] [CrossRef]
- Lin, P.; Wang, K.; Zhou, C.; Xie, Y.; Yao, X.; Yin, H. Seed Transcriptomics Analysis in Camellia oleifera Uncovers Genes Associated with Oil Content and Fatty Acid Composition. Int. J. Mol. Sci. 2018, 19, 118. [Google Scholar] [CrossRef]
- Guo, Y.; Gan, S.S. Translational researches on leaf senescence for enhancing plant productivity and quality. J. Exp. Bot. 2014, 65, 3901–3913. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.O.; Kim, H.J.; Nam, H.G. Leaf senescence. Annu. Rev. Plant Biol. 2007, 58, 115–136. [Google Scholar] [CrossRef] [PubMed]
- Podzimska-Sroka, D.; O’Shea, C.; Gregersen, P.L.; Skriver, K. NAC Transcription Factors in Senescence: From Molecular Structure to Function in Crops. Plants (Basel) 2015, 4, 412–448. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.R.; Kim, H.J.; Lim, P.O.; Nam, H.G. Leaf Senescence: Systems and Dynamics Aspects. Annu. Rev. Plant Biol. 2019, 70, 347–376. [Google Scholar] [CrossRef]
- Miller, J.D.; Arteca, R.N.; Pell, E.J. Senescence-associated gene expression during ozone-induced leaf senescence in Arabidopsis. Plant Physiol. 1999, 120, 1015–1024. [Google Scholar] [CrossRef]
- Pruzinska, A.; Tanner, G.; Anders, I.; Roca, M.; Hortensteiner, S. Chlorophyll breakdown: Pheophorbide a oxygenase is a Rieske-type iron-sulfur protein, encoded by the accelerated cell death 1 gene. Proc. Natl. Acad. Sci. USA 2003, 100, 15259–15264. [Google Scholar] [CrossRef] [PubMed]
- Jehanzeb, M.; Zheng, X.; Miao, Y. The Role of the S40 Gene Family in Leaf Senescence. Int. J. Mol. Sci. 2017, 18, 2152. [Google Scholar] [CrossRef] [PubMed]
- Garapati, P.; Xue, G.P.; Munne-Bosch, S.; Balazadeh, S. Transcription Factor ATAF1 in Arabidopsis Promotes Senescence by Direct Regulation of Key Chloroplast Maintenance and Senescence Transcriptional Cascades. Plant Physiol. 2015, 168, 1122–1139. [Google Scholar] [CrossRef] [PubMed]
- Smykowski, A.; Zimmermann, P.; Zentgraf, U. G-Box binding factor1 reduces CATALASE2 expression and regulates the onset of leaf senescence in Arabidopsis. Plant Physiol. 2010, 153, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Jehanzeb, M.; Habiba; Zhang, Y.; Li, L.; Miao, Y. Characterization of S40-like proteins and their roles in response to environmental cues and leaf senescence in rice. BMC Plant Biol. 2019, 19, 174. [Google Scholar] [CrossRef]
- Gombert, J.; Etienne, P.; Ourry, A.; Le Dily, F. The expression patterns of SAG12/Cab genes reveal the spatial and temporal progression of leaf senescence in Brassica napus L. with sensitivity to the environment. J. Exp. Bot. 2006, 57, 1949–1956. [Google Scholar] [CrossRef]
- Kuai, B.; Chen, J.; Hortensteiner, S. The biochemistry and molecular biology of chlorophyll breakdown. J. Exp. Bot. 2018, 69, 751–767. [Google Scholar] [CrossRef]
- Ramkumar, M.K.; Senthil Kumar, S.; Gaikwad, K.; Pandey, R.; Chinnusamy, V.; Singh, N.K.; Singh, A.K.; Mohapatra, T.; Sevanthi, A.M. A Novel Stay-Green Mutant of Rice with Delayed Leaf Senescence and Better Harvest Index Confers Drought Tolerance. Plants (Basel) 2019, 8, 375. [Google Scholar] [CrossRef]
- Sato, Y.; Morita, R.; Katsuma, S.; Nishimura, M.; Tanaka, A.; Kusaba, M. Two short-chain dehydrogenase/reductases, NON-YELLOW COLORING 1 and NYC1-LIKE, are required for chlorophyll b and light-harvesting complex II degradation during senescence in rice. Plant J. 2009, 57, 120–131. [Google Scholar] [CrossRef]
- Jan, S.; Abbas, N.; Ashraf, M.; Ahmad, P. Roles of potential plant hormones and transcription factors in controlling leaf senescence and drought tolerance. Protoplasma 2019, 256, 313–329. [Google Scholar] [CrossRef]
- Zhao, Y.; Chan, Z.; Gao, J.; Xing, L.; Cao, M.; Yu, C.; Hu, Y.; You, J.; Shi, H.; Zhu, Y.; et al. ABA receptor PYL9 promotes drought resistance and leaf senescence. Proc. Natl. Acad. Sci. USA 2016, 113, 1949–1954. [Google Scholar] [CrossRef] [PubMed]
- Riefler, M.; Novak, O.; Strnad, M.; Schmulling, T. Arabidopsis cytokinin receptor mutants reveal functions in shoot growth, leaf senescence, seed size, germination, root development, and cytokinin metabolism. Plant Cell 2006, 18, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Rogers, H.; Munne-Bosch, S. Production and Scavenging of Reactive Oxygen Species and Redox Signaling during Leaf and Flower Senescence: Similar but Different. Plant Physiol. 2016, 171, 1560–1568. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Shen, Y.; Ni, Z.; Wang, Q.; Lei, Z.; Xu, N.; Deng, Q.; Lin, L.; Wang, J.; Lv, X.; et al. Exogenous Melatonin Application Delays Senescence of Kiwifruit Leaves by Regulating the Antioxidant Capacity and Biosynthesis of Flavonoids. Front Plant Sci. 2018, 9, 426. [Google Scholar] [CrossRef]
- Bengoa Luoni, S.; Astigueta, F.H.; Nicosia, S.; Moschen, S.; Fernandez, P.; Heinz, R. Transcription Factors Associated with Leaf Senescence in Crops. Plants (Basel) 2019, 8, 411. [Google Scholar] [CrossRef]
- Guo, P.; Li, Z.; Huang, P.; Li, B.; Fang, S.; Chu, J.; Guo, H. A Tripartite Amplification Loop Involving the Transcription Factor WRKY75, Salicylic Acid, and Reactive Oxygen Species Accelerates Leaf Senescence. Plant Cell 2017, 29, 2854–2870. [Google Scholar] [CrossRef]
- Qiu, K.; Li, Z.; Yang, Z.; Chen, J.; Wu, S.; Zhu, X.; Gao, S.; Gao, J.; Ren, G.; Kuai, B.; et al. EIN3 and ORE1 Accelerate Degreening during Ethylene-Mediated Leaf Senescence by Directly Activating Chlorophyll Catabolic Genes in Arabidopsis. PLoS Genet. 2015, 11, e1005399. [Google Scholar] [CrossRef]
- Zhang, X. Determination of chlorophyll content in plants -- Acetone ethanol mixture method. Liaoning Agric. Sci. 1986, 3, 28–30. [Google Scholar]
- Sedmak, J.J.; Grossberg, S.E. A rapid, sensitive, and versatile assay for protein using Coomassie brilliant blue G250. Anal. Biochem. 1977, 79, 544–552. [Google Scholar] [CrossRef]
- Dhindsa, R.S.; Plumb-Dhindsa, P.; Thorpe, T.A. Leaf senescence: Correlation with increased levels of membrane permeability and lipid peroxidation and increased levels of superoxide dismutase and catalase. J. Exp. Bot. 1981, 32, 93–101. [Google Scholar] [CrossRef]
- Bates, L.S.; Waldren, R.P.; Teare, I.D. Rapid Determination of Free Proline for Water-Stress Studies. Plant & Soil 1973, 39, 205–207. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Xian, A.; Lin, F.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotech. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Smith-Unna, R.; Boursnell, C.; Patro, R.; Hibberd, J.M.; Kelly, S. TransRate: Reference-free quality assessment of de novo transcriptome assemblies. Genome Res. 2016, 26, 1134–1144. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol. Biol. 2019, 1962, 227. [Google Scholar]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; He, F.C. Integrated nr Database in Protein Annotation System and Its Localization. Comput. Eng. 2006, 32, 71–72. [Google Scholar]
- Rolf, A.; Amos, B.; Wu, C.H.; Barker, W.C.; Brigitte, B.; Serenella, F.; Elisabeth, G.; Huang, H.; Rodrigo, L.; Michele, M. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar]
- Finn, R.D. Pfam: The Protein Families Database; American Cancer Society: Atlanta, GA, USA, 2005. [Google Scholar]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Minoru, K.; Yoko, S.; Masayuki, K.; Miho, F.; Mao, T. KEGG as a reference resource for gene and protein annotation. Nuclc Acids Res. 2016, 44, D457–D462. [Google Scholar]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Klopfenstein, D.V.; Zhang, L.; Pedersen, B.S.; Fidel, R.; Alex, W.V.; Aurélien, N.; Mungall, C.J.; Yunes, J.M.; Olga, B.; Mark, W. GOATOOLS: A Python library for Gene Ontology analyses. Sci. Rep. 2018, 8, 10872. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nuclc Acids Res. 2011, 39, 316–322. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Zou, D.; Zhao, Y.; Wang, H.; Zhang, Y.; Xia, X.; Luo, J.; Guo, H.; Zhang, Z. LSD 3.0: A comprehensive resource for the leaf senescence research community. Nucleic Acids Res. 2019, 48, D1069–D1075. [Google Scholar] [CrossRef]
- Liang, K.; Wang, A.; Sun, Y.; Yu, M.; Zhang, L. Identification and Expression of NAC Transcription Factors of Vaccinium corymbosum L. in Response to Drought Stress. Forests 2019, 10, 1088. [Google Scholar] [CrossRef]
- Jeongsik, K.; Hee, K.J.; Il, L.J.; Ryun, W.H.; Ok, L.P. New insights into the regulation of leaf senescence in Arabidopsis. J. Exp. Bot. 2017, 4, 787–799. [Google Scholar]
- Landis, J.B.; Soltis, D.E.; Soltis, P.S. Comparative transcriptomic analysis of the evolution and development of flower size in Saltugilia (Polemoniaceae). BMC Genom. 2017, 18, 475. [Google Scholar] [CrossRef]
- Lin, M.; Pang, C.; Fan, S.; Song, M.; Wei, H.; Yu, S. Global analysis of the Gossypium hirsutum L. Transcriptome during leaf senescence by RNA-Seq. BMC Plant Biol. 2015, 15, 43. [Google Scholar] [CrossRef]
- Srivalli, S.; Khanna-Chopra, R. Delayed wheat flag leaf senescence due to removal of spikelets is associated with increased activities of leaf antioxidant enzymes, reduced glutathione/oxidized glutathione ratio and oxidative damage to mitochondrial proteins. Plant Physiol. Biochem. 2009, 47, 663–670. [Google Scholar] [CrossRef]
- Moore, K.; Roberts, L.J. Measurement of Lipid Peroxidation. Free Radic. Res. Commun. 1998, 28, 659–671. [Google Scholar] [CrossRef]
- Wu, X.; Hu, W.; Luo, H.; Xia, Y.; Zhao, Y.; Wang, L.; Zhang, L.; Luo, J.; Jing, H. Transcriptome profiling of developmental leaf senescence in sorghum (Sorghum bicolor). Plant Mol. Biol. 2016, 92, 555–580. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Z.; Chen, Y.; He, J.; Bi, Y. PHYTOCHROME-INTERACTING FACTOR 5 (PIF5) positively regulates dark-induced senescence and chlorophyll degradation in Arabidopsis. Plant Sci. 2015, 237, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.Q.; Tan, X.L.; Chen, J.W.; Liu, Z.L.; Kuang, J.F.; Lu, W.J.; Shan, W.; Chen, J.Y. BrNAC055, a Novel Transcriptional Activator, Regulates Leaf Senescence in Chinese Flowering Cabbage by Modulating Reactive Oxygen Species Production and Chlorophyll Degradation. J. Agric. Food Chem. 2018, 66, 9399–9408. [Google Scholar] [CrossRef]
- Sakuraba, Y.; Kim, D.; Han, S.-H.; Kim, S.-H.; Paek, N.-C. Multilayered Regulation of Membrane-Bound ONAC054 Is Essential for Abscisic Acid-Induced Leaf Senescence in Rice. Plant Cell 2020, 32, 630–649. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Zhang, Z.; Wang, C.; Li, X.; Guo, C.; Yang, L.; Guo, Y. Identification of a Novel Melon Transcription Factor CmNAC60 as a Potential Regulator of Leaf Senescence. Genes 2019, 10, 584. [Google Scholar] [CrossRef]
- Jin, S.J.; Park, Y.T.; Jung, H.; Park, S.H.; Kim, J.K. Rice NAC proteins act as homodimers and heterodimers. Plant Biotechnol. Rep. 2009, 3, 127–134. [Google Scholar]
- Wang, X.; Culver, J.N. DNA binding specificity of ATAF2, a NAC domain transcription factor targeted for degradation by Tobacco mosaic virus. BMC Plant Biol. 2012, 12, 1–14. [Google Scholar] [CrossRef]
- Mathew, I.E.; Das, S.; Mahto, A.; Agarwal, P. Three Rice NAC Transcription Factors Heteromerize and Are Associated with Seed Size. Front Plant Sci. 2016, 7, 1638. [Google Scholar] [CrossRef]
- Besseau, S.; Li, J.; Palva, E.T. WRKY54 and WRKY70 co-operate as negative regulators of leaf senescence in Arabidopsis thaliana. J. Exp. Bot. 2012, 63, 2667–2679. [Google Scholar] [CrossRef]
- Liu, C.; Sun, Q.; Zhao, L.; Li, Z.; Peng, Z.; Zhang, J. Heterologous Expression of the Transcription Factor EsNAC1 in Arabidopsis Enhances Abiotic Stress Resistance and Retards Growth by Regulating the Expression of Different Target Genes. Front Plant Sci. 2018, 9, 1495. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Galiano, M.J.; Gonzalez-Hernandez, A.I.; Crespo-Salvador, O.; Rausell, C.; Real, M.D.; Escamilla, M.; Camanes, G.; Garcia-Agustin, P.; Gonzalez-Bosch, C.; Garcia-Robles, I. Epigenetic regulation of the expression of WRKY75 transcription factor in response to biotic and abiotic stresses in Solanaceae plants. Plant Cell Rep. 2018, 37, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Deng, Z.; Lai, J.; Zhang, Y.; Yang, C.; Yin, B.; Zhao, Q.; Zhang, L.; Li, Y.; Yang, C.; et al. Dual function of Arabidopsis ATAF1 in abiotic and biotic stress responses. Cell Res. 2009, 19, 1279–1290. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Unigene |
|---|---|
| Total number | 78,860 |
| Total base | 68,289,653 |
| Average length (bp) | 865.96 |
| N50 length (bp) | 1374 |
| GC percent (%) | 39.19 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.; Liang, K.; Wang, A.; Zhang, M.; Qiu, J.; Zhang, L. Physiological Characterization and Transcriptome Analysis of Camellia oleifera Abel. during Leaf Senescence. Forests 2020, 11, 812. https://doi.org/10.3390/f11080812
Yang S, Liang K, Wang A, Zhang M, Qiu J, Zhang L. Physiological Characterization and Transcriptome Analysis of Camellia oleifera Abel. during Leaf Senescence. Forests. 2020; 11(8):812. https://doi.org/10.3390/f11080812
Chicago/Turabian StyleYang, Shiwen, Kehao Liang, Aibin Wang, Ming Zhang, Jiangming Qiu, and Lingyun Zhang. 2020. "Physiological Characterization and Transcriptome Analysis of Camellia oleifera Abel. during Leaf Senescence" Forests 11, no. 8: 812. https://doi.org/10.3390/f11080812
APA StyleYang, S., Liang, K., Wang, A., Zhang, M., Qiu, J., & Zhang, L. (2020). Physiological Characterization and Transcriptome Analysis of Camellia oleifera Abel. during Leaf Senescence. Forests, 11(8), 812. https://doi.org/10.3390/f11080812