Deep Sequencing and Analysis of Transcriptomes of Pinus koraiensis Sieb. & Zucc.

Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. RNA Sequencing and De Novo Transcriptome Assembly
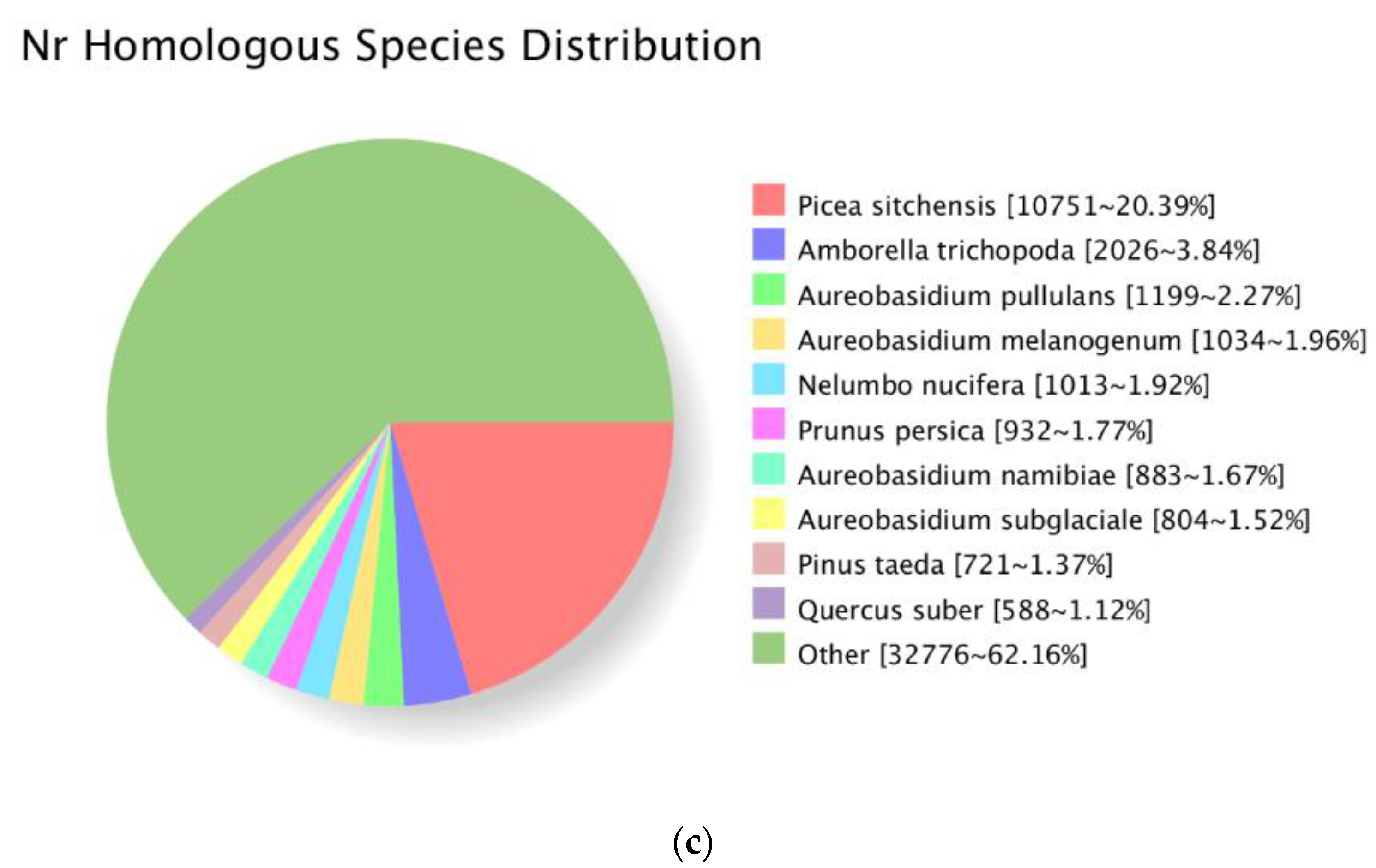
3.2. Functional Annotation of the P. Koraiensis Unigenes
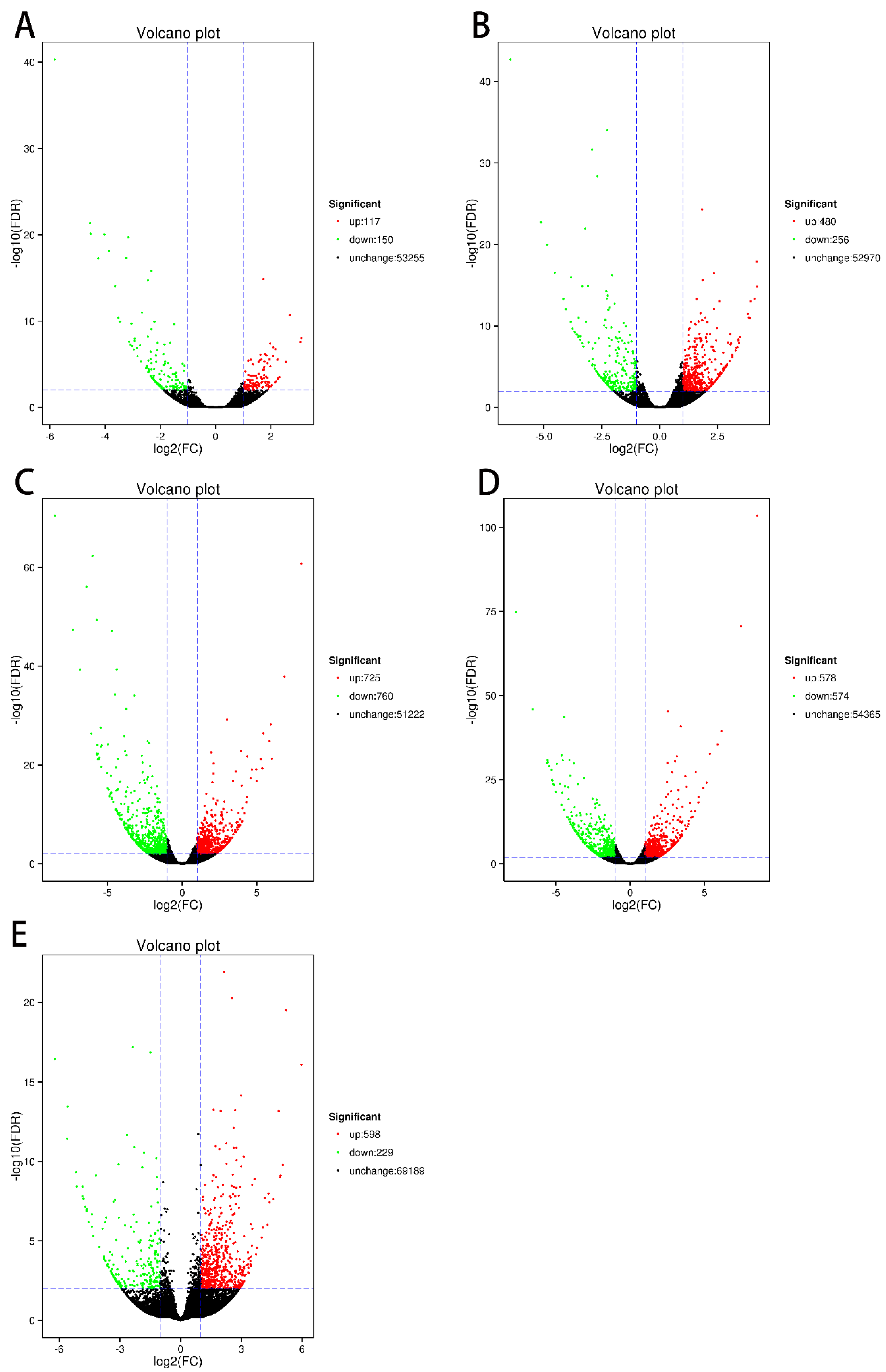
3.3. Identification of Differentially Expressed Genes (DEGs)
3.4. Gene Ontology (GO) Enrichment of the DEGs
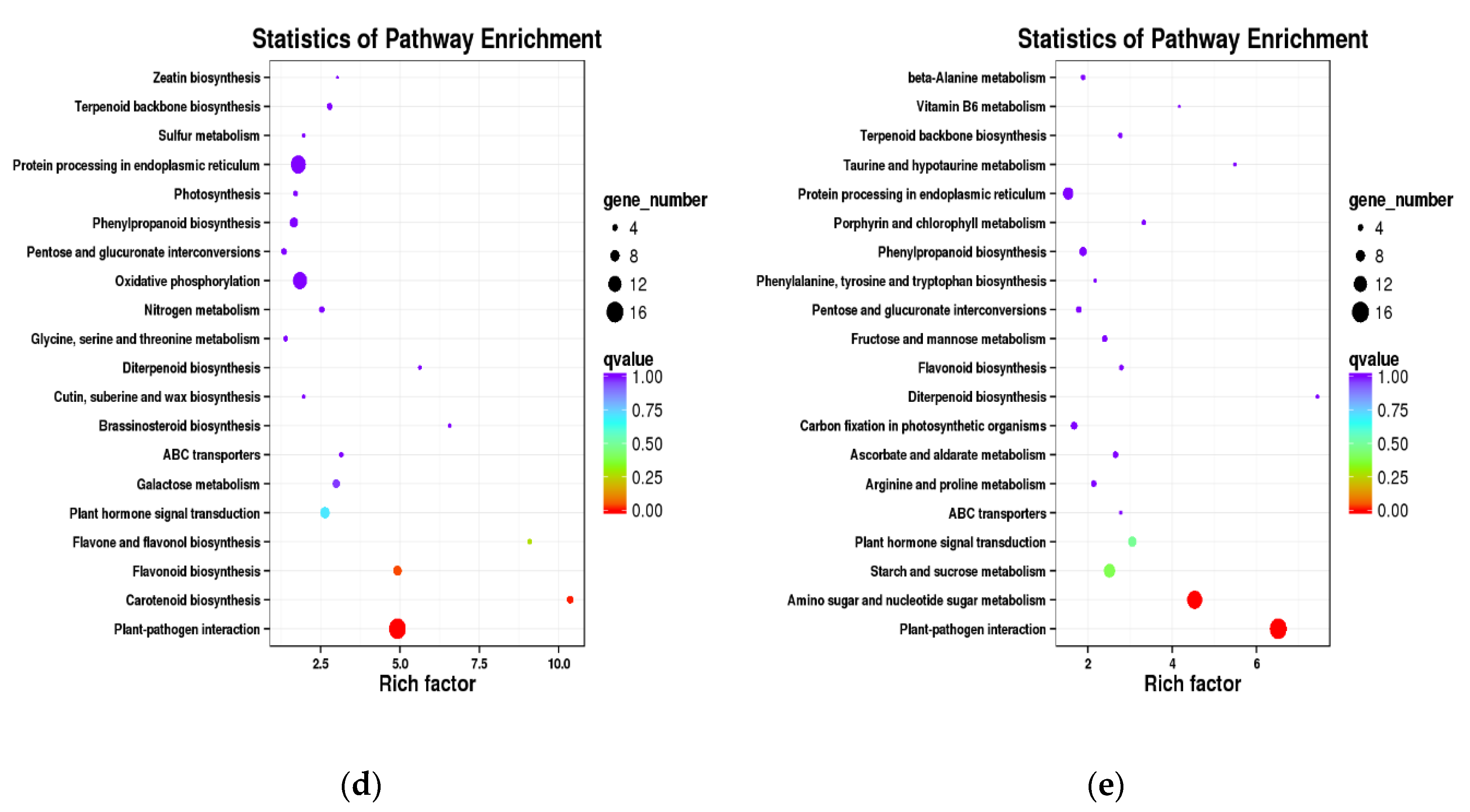
3.5. KEGG Pathway Enrichment Analysis of the DEGs
3.6. The Expression Level of Genes Related to Flowers and Hormones
4. Discussion
4.1. Analysis of DEGs that Show Changes in Expression during Reproductive Development
4.2. Tree Age Affects Flowering Time
4.3. Plant Hormones Related to Flowering
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Santelices, B.; Varela, D. Intra-clonal variation in the red seaweed Gracilaria chilensis. Marine Biol. 1993, 116, 543–552. [Google Scholar] [CrossRef]
- Heide, O.M. Primary and secondary induction requirements for flowering in Alopecurus pratensis. Physiol. Plant. 2006, 66, 251–256. [Google Scholar] [CrossRef]
- Summerfield, R.J.; Roberts, E.H.; Erskine, W.; Ellis, R.H. Effects of temperature and photoperiod on flowering in Lentils (Lens culinaris Medic.). Ann. Bot. 1985, 56, 659–671. [Google Scholar] [CrossRef]
- Sessions, A.; Nemhauser, J.L.; Mccoll, A.; Roe, J.L.; Feldmann, K.A.; Zambryski, P.C. ETTIN patterns the Arabidopsis floral meristem and reproductive organs. Development 1997, 124, 4481–4491. [Google Scholar] [PubMed]
- Weigel, D.; Meyerowitz, E.M. Activation of Floral Homeotic Genes in Arabidopsis. Science 1993, 261, 1723–1726. [Google Scholar] [CrossRef] [PubMed]
- Okamuro, J.K.; Caster, B.; Villarroel, R.; Van Montagu, M.; Jofuku, K.D. The AP2 domain of APETALA2 defines a large new family of DNA binding proteins in Arabidopsis. Proc. Natl. Acad. Sci. USA 1997, 94, 7076–7081. [Google Scholar] [CrossRef]
- Weigel, D.; Alvarez, J.; Smyth, D.R.; Yanofsky, M.F.; Meyerowitz, E.M. LEAFY controls floral meristem identity in Arabidopsis. Cell 1993, 69, 843–859. [Google Scholar] [CrossRef]
- Stinchcombe, J.R.; Weinig, C.; Ungerer, M.; Olsen, K.M.; Schmitt, J. A latitudinal cline in flowering time in Arabidopsis thaliana modulated by the flowering time gene FRIGIDA. Proc. Natl. Acad. Sci. USA 2004, 101, 4712–4717. [Google Scholar] [CrossRef]
- Martinez-Garcia, J.F.; Virgos-Soler, A.; Prat, S. Control of photoperiod-regulated tuberization in potato by the Arabidopsis flowering-time gene CONSTANS. Proc. Natl. Acad. Sci. USA 2002, 99, 15211–15216. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Haughn, G.W. UNUSUAL FLORAL ORGANS controls meristem identity and organ primordia fate in Arabidopsis. Plant Cell 1995, 7, 1485–1499. [Google Scholar] [CrossRef]
- Mouradov, A.; Glassick, T.; Hamdorf, B.; Murphy, L.; Fowler, B.; Marla, S.; Teasdale, R.D. NEEDLY, a Pinus radiata ortholog of FLORICAULA/LEAFY genes, expressed in both reproductive and vegetative meristems. Proc. Natl. Acad. Sci. USA 1998, 95, 6537–6542. [Google Scholar] [CrossRef] [PubMed]
- Skriabin, K.G.; Alekseev, D.V.; Ezhova, T.A.; Kozlov, V.N.; Kudriavtsev, V.B.; Nosov, M.V.; Penin, A.A.; Chub, V.V.; Shestakov, S.V.; Shul’Ga, O.A. Determination of type and spatial pattern formation of flower organs: Dynamic model of development. Izvestiâ Akademii Nauk.rossijskaâ Akademiâ Nauk.seriâ Biologiceskaâ 2006, 645–659. [Google Scholar]
- Ferrandiz, C.; Navarro, C.; Gomez, M.D.; Canas, L.A.; Beltran, J.P. Flower development in Pisum sativum: From the war of the whorls to the battle of the common primordia. Dev. Genet. 1999, 25, 280–290. [Google Scholar] [CrossRef]
- Goto, K. Molecular and genetic analyses of flower homeotic genes of Arabidopsis. J. Biosci. 1996, 21, 369–378. [Google Scholar] [CrossRef]
- Blazquez, M.A.; Soowal, L.N.; Lee, I.; Weigel, D. LEAFY expression and flower initiation in Arabidopsis. Development 1997, 124, 3835–3844. [Google Scholar]
- Yuan, Z.; Zhou, A.; Liu, Y.; Chengzhong, H. The Polar Transport and Regulatory Mechanism of Auxin in Plant. J. Yunnan. Agric. University 2013, 28, 878–884. [Google Scholar]
- Cheng, Y. Auxin biosynthesis by the YUCCA flavin monooxygenases controls the formation of floral organs and vascular tissues in Arabidopsis. Gene. Dev. 2006, 20, 1790–1799. [Google Scholar] [CrossRef]
- Křeček, P.; Skůpa, P.; Libus, J.; Naramoto, S.; Tejos, R.; Friml, J.; Zažímalová, E. The PIN-FORMED (PIN) protein family of auxin transporters. Genome Biol. 2009, 10. [Google Scholar]
- Balla, J.; Medved’ová, Z.; Kalousek, P.; Matiješčuková, N.; Friml, J.; Reinöhl, V.; Procházka, S. Auxin flow-mediated competition between axillary buds to restore apical dominance. Sci. Rep-UK 2016, 6, 35955. [Google Scholar] [CrossRef]
- Müller, C.J.; Larsson, E.; Spíchal, L.; Sundberg, E. Cytokinin-auxin crosstalk in the gynoecial primordium ensures correct domain patterning. Plant Physiol. 2017, 175, 1144–1157. [Google Scholar] [CrossRef] [PubMed]
- Lagoze, C.; Hunter, J. The ABC Ontology and Model. In Proceedings of the International Conference on Dublin Core and Metadata Applications, Toyota, Japan, 24–26 October 2001. [Google Scholar]
- Litt, A.; Kramer, E.M. The ABC model and the diversification of floral organ identity. Semin. Cell Dev. Biol. 2010, 21, 0–137. [Google Scholar] [CrossRef] [PubMed]
- Sussex, I.I.M. Function of the apetala-1 Gene during Arabidopsis Floral Development. Plant Cell 1990, 2, 741–753. [Google Scholar]
- Kotoda, N.; Wada, M.; Kusaba, S.; Kano-Murakami, Y.; Masuda, T.; Soejima, J. Overexpression of MdMADS5, an APETALA1-like gene of apple, causes early flowering in transgenic Arabidopsis. Plant Sci. 2002, 162, 679–687. [Google Scholar] [CrossRef]
- Goto, K.; Meyerowitz, E.M. Function and regulation of the Arabidopsis floral homeotic gene PISTILLATA. Gene Dev. 1994, 8, 1548–1560. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.S.; Hill, T.A.; Tan, K.G.; Irish, V.F. Regulation of APETALA3 floral homeotic gene expression by meristem identity genes. Development 2002, 129, 2079–2086. [Google Scholar]
- Pelaz, S.; Tapia-López, R.; Alvarez-Buylla, E.R.; Yanofsky, M.F. Conversion of leaves into petals in Arabidopsis. Curr. Biol. 2001, 11, 182–184. [Google Scholar] [CrossRef]
- Pelaz, S.; Ditta, G.S.; Baumann, E.; Wisman, E.; Yanofsky, M.F. B and C floral organ identity functions require SEPALLATA MADS-box genes. Nature 2000, 405, 200–203. [Google Scholar] [CrossRef]
- Plomion, C.; Chagne, D.; Pot, D.; Kumar, S.; Wilcox, P.L.; Burdon, R.D.; Prat, D.; Peterson, D.G.; Paiva, J.; Chaumeil, P.; et al. Pines. In Genome Mapping & Molecular Breeding in Plants; USDA: Washington, DC, USA, 2007; Volume 7, pp. 213–226. [Google Scholar]
- Chen, M.M.; Feng, F.; Sui, X.; Li, M.-H.; Zhao, D.; Han, S. Construction of a framework map forPinus koraiensisSieb. et Zucc. using SRAP, SSR and ISSR markers. Trees 2010, 24, 685–693. [Google Scholar] [CrossRef]
- Feng, F.J.; Han, S.J.; Wang, H.M. Genetic diversity and genetic differentiation of natural Pinus koraiensis population. J. For. Res. 2006, 17, 21–24. [Google Scholar] [CrossRef]
- Tandre, K.; Svenson, M.; Svensson, M.E.; Engström, P. Conservation of gene structure and activity regulation of reproductive organ development of conifers and angiosperms. Plant J. 1998, 15, 615–623. [Google Scholar] [CrossRef]
- Mouradov, A.; Glassick, T.; Hamdorf, B.; Teasdale, R.D. Molecular control of early cone development in Pinus radiata. Protoplasma 1999, 208, 3–12. [Google Scholar] [CrossRef]
- Müller, B.M.; Saedler, H.; Zachgo, S. The MADS-box gene DEFH28 from Antirrhinum is involved in the regulation of floral meristem identity and fruit development. Plant. J. 2001, 28, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.W.; Li-Hua, M.A. Extracting DNA from Edible Fungus by Combined Method of CTAB and DNA Gel Purification Kit. Biotechnology 2009, 19, 32–34. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- ALTSCHUL, S.F. Gapped BLAST and PSI-BLAST: A new generation of protein detabase search programs. Nucleic Acids. Res. 1997, 25. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatiana, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids. Res. 2005, 33 (Suppl. 1), D501–D504. [Google Scholar] [CrossRef] [PubMed]
- Amos Bairoch, R.A. The SWISS-PROT protein sequence data bank and its supplement TrEMBL in 1999. Nucleic Acids. Res. 1999, 28, 49–54. [Google Scholar] [CrossRef]
- Gene, O.C.; Mulder, N. The Gene Ontology (GO) project in 2006. Nucleic Acids. Res. 2005, 34, D322–D326. [Google Scholar]
- Koonin, E.V.; Tatusov, R.L.; Galperin, M.Y.; Rosanov, M.N. Genome analysis using clusters of orthologous groups (COGs). In Proceedings of the International Conference on Computational Molecular Biology, Bethesda, MD, USA, March 1998; pp. 135–139. [Google Scholar]
- Sander, C.; Schneider, R. Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins 1991, 9, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Sean, P.; Damian, S.; Kalliopi, T.; Alexander, R.; Michael, K.; Jean, M.; Roland, A.; Thomas, R.; Ivica, L.; Tobias, D. eggNOG v3.0: Orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 2011, 40, D284–D289. [Google Scholar]
- Minoru, K.; Susumu, G. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar]
- Nastou, K.C.; Tsaousis, G.N.; Papandreou, N.C.; Hamodrakas, S.J. MBPpred: Proteome-wide detection of membrane lipid-binding proteins using profile Hidden Markov Models. BBA 2016, 1864, 747–754. [Google Scholar] [CrossRef]
- Holmes, S.; Alekseyenko, A.; Timme, A.; Nelson, T.; Pasricha, P.J.; Spormann, A. Visualization and Statistical Comparisons of Microbial Communities Using R Packages on Phylochip Data. Pac. Symp. Biocomput. 2011, 20, 142–153. [Google Scholar]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome. Biol. 2010, 1. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. Bioinformatics 2011, 12, 323. [Google Scholar] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and abundance estimation from RNA-Seq reveals thousands of new transcripts and switching among isoforms. Nat. Biotechnol. 2010, 28, 511. [Google Scholar] [CrossRef] [PubMed]
- Lopes, R.H.C. Kolmogorov–Smirnov test. Int. Encycl. Stat. Sci. 2014, 10, 718–720. [Google Scholar]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- von, M.C.; Martijn, H.; Daniel, J.; Steffen, S.; Peer, B.; Berend, S. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 2003, 31, 258–261. [Google Scholar]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef]
- Boenn, M. Hypergea: Hypergeometric Tests. 2016. Available online: https://cran.r-project.org/web/packages/hypergea/hypergea.pdf (accessed on 5 February 2020).
- Jin, C. The Development of The Gametophytes of Pinus koraiensis. Bull. Bot. Res. 1985, 5, 113–126. [Google Scholar]
- Bari, R.; Jones, J.D.G. Role of plant hormones in plant defence responses. Plant Mol. Biol. 2009, 69, 473–488. [Google Scholar] [CrossRef]
- Folkerts, G.W.; Folkerts, D.R. Unique Capsule Dehiscence in Sarracenia leucophylla Raf. and a Hypothesis concerning Post-Anthesis Tilting in Sarracenia Flowers. Castanea 1989, 54, 111–114. [Google Scholar]
- Hillman, W.S. The physiology of flowering. Q. Rev. Biol. 1963, 109. [Google Scholar]
- Charles, E.D.A. A hypothesis about the control of flowering. Ann. Bot-London 1983, 52, 105–107. [Google Scholar] [CrossRef]
- Gang, C.; Zhong, W.; Zhang, M.; Li, W. The development of embryo and endosperm in tobacco seed and its nutrients accumulation. J. Yangzhou Uni. Nat. Sci. Ed. 1998, 1, 25–29. [Google Scholar]
- Hirschberg, K.; Hubner, G.; Borriss, H. Cytokinin-induzierte de novo-Synthese der Nitratreductase im Embryonen von Agrostemma githago. Planta 1972, 108, 333–337. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Related Gene | Function |
|---|---|
| APETALA1 (AP1) | control the characteristics of sepals and petals, be related to the meristem of flowers, a transcriptional activator that regulates AP3 and PI |
| APETALA2 (AP2) | class A genes, control the characteristics of sepals and petals |
| APETALA3 (AP3) | class B genes, specify the development of petals and stamens |
| PISTILLATA (PI) | class B genes, specify the development of petals and stamens |
| AGAMOUS (AG) | class C genes, control the characteristics of stamens and carpel |
| SEP1, SEP2, and SEP3 | were found to be essential for petal, stamen, and carpel development, that is the SEP gene product was required for B and C gene activity |
| CAULIflower (CAL) | confer floral identity to emerging floral primordia, a paralogous genes of APETALA1 (AP1), act redundantly to control inflorescence architecture by affecting the domains of LFY expression as well as the relative levels of its activities |
| LEAFY (LFY) | is expressed throughout the flower, participates in the activation of homeotic genes, are expressed in specific regions of the flower, encodes a transcription factor that determines a meristem will generate flowers, is a direct upstream regulator of floral homeotic genes |
| FRIGIDA (FRI) | the flowering time gene, alleles were associated with accelerated flowering |
| Arabidopsis CONSTANS (AtCO) | regulate flowering time, accelerates flowering in response to long days, exerts its inhibitory effect on tuber formation by acting in the leaves |
| UNUSUAL FLORAL ORGANS (UFO) | controls meristem identity and organ primordia fate, related to floral-organ type |
| YUCCA (YUC) | spatially and temporally regulated auxin biosynthesis, is essential for the formation of floral organs and vascular tissues |
| PIN (PIN) | auxin efflux transport proteins, controlling auxin polar transport |
| Anno_Database | Annotated_Number |
|---|---|
| COG_Annotation | 18,577 |
| GO_Annotation | 33,277 |
| KEGG_Annotation | 19,726 |
| KOG_Annotation | 32,241 |
| Pfam_Annotation | 38,956 |
| Swissprot_Annotation | 30,528 |
| eggNOG_Annotation | 51,957 |
| nr_Annotation | 52,872 |
| All_Annotated | 58,706 |
| DEG_Comparison | All_DEGs | Up-Regulated | Down-Regulated |
|---|---|---|---|
| Y2_vs_Y5 | 267 | 117 | 150 |
| Y2_vs_Y10 | 736 | 480 | 256 |
| Y2_vs_Y15 | 1485 | 725 | 760 |
| Y2_vs_Y30 | 1152 | 578 | 574 |
| Y2_vs_grafted | 827 | 598 | 229 |
| Gene_ID | Description |
|---|---|
| c64070.graph_c0 | Hypothetical protein GOBAR_AA39818, related to pollen germination and regulates flower development |
| c68641.graph_c0 | The SRF-type transcription factor and agamous-like MADS-box protein AGL15, and related to somatic embryogenesis. |
| c65543.graph_c1 | It has a K-box region and is a SRF-type and MADS-box transcription factor |
| c77350.graph_c2 | It has a K-box region and is a MADS-box transcription factor DAL1 (Picea abies) |
| c72714.graph_c0 | It is SRF-type and MADS-box transcription factor with transcription factor activity |
| c76037.graph_c0 | CKX3 [Pinus tabuliformis], which is involved in the metabolism of cytokinins and has oxidoreductase activity. |
| c55708.graph_c0 | The abscisic acid receptor PYR/PYL family, involved in the abscisic acid-activated signaling pathway. |
| c61855.graph_c0 | It participates in abscisic acid catabolic process and has the activity of abscisic acid 8’ hydroxylase |
| c73235.graph_c0 | It is related to the signaling mechanism and responds to abscisic acid |
| c82790.graph_c0 | Related to hormone-mediated signaling pathway, histidine kinase 5 (Herrania umbratica) |
| c55558.graph_c0 | Predicted as CYP85A1(Musa acuminata), which is a brassinosteroid-6-oxidase and involved in the synthesis of sterol hormones. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, S.; Yan, S.; Zhao, C.; Zhang, P.; Yang, L.; Wang, C.; Shen, H. Deep Sequencing and Analysis of Transcriptomes of Pinus koraiensis Sieb. & Zucc. Forests 2020, 11, 350. https://doi.org/10.3390/f11030350
Shi S, Yan S, Zhao C, Zhang P, Yang L, Wang C, Shen H. Deep Sequencing and Analysis of Transcriptomes of Pinus koraiensis Sieb. & Zucc. Forests. 2020; 11(3):350. https://doi.org/10.3390/f11030350
Chicago/Turabian StyleShi, Shaolin, Siyu Yan, Chao Zhao, Peng Zhang, Ling Yang, Chao Wang, and Hailong Shen. 2020. "Deep Sequencing and Analysis of Transcriptomes of Pinus koraiensis Sieb. & Zucc." Forests 11, no. 3: 350. https://doi.org/10.3390/f11030350
APA StyleShi, S., Yan, S., Zhao, C., Zhang, P., Yang, L., Wang, C., & Shen, H. (2020). Deep Sequencing and Analysis of Transcriptomes of Pinus koraiensis Sieb. & Zucc. Forests, 11(3), 350. https://doi.org/10.3390/f11030350