Comparative Transcriptomic Response of Two Pinus Species to Infection with the Pine Wood Nematode Bursaphelenchus xylophilus

 , ,
, , 
Abstract
1. Introduction
2. Material and Methods
2.1. Biological Material, Pine Wood Nematode Inoculation, and Sampling
2.2. RNA Extraction, cDNA Synthesis, Library Preparation, and Sequencing
2.3. Pre-Processing RNA-Sequencing Data and Assembly
2.4. Prediction of Candidate Coding Regions
2.5. Mapping and Differential Expression Analysis
2.6. Quantitative Real-Time PCR Analysis
2.7. Transcriptome Annotation
2.8. Biological Networks Analysis
3. Results
3.1. Pre-Processing of RNA-Sequencing Data and Assembly
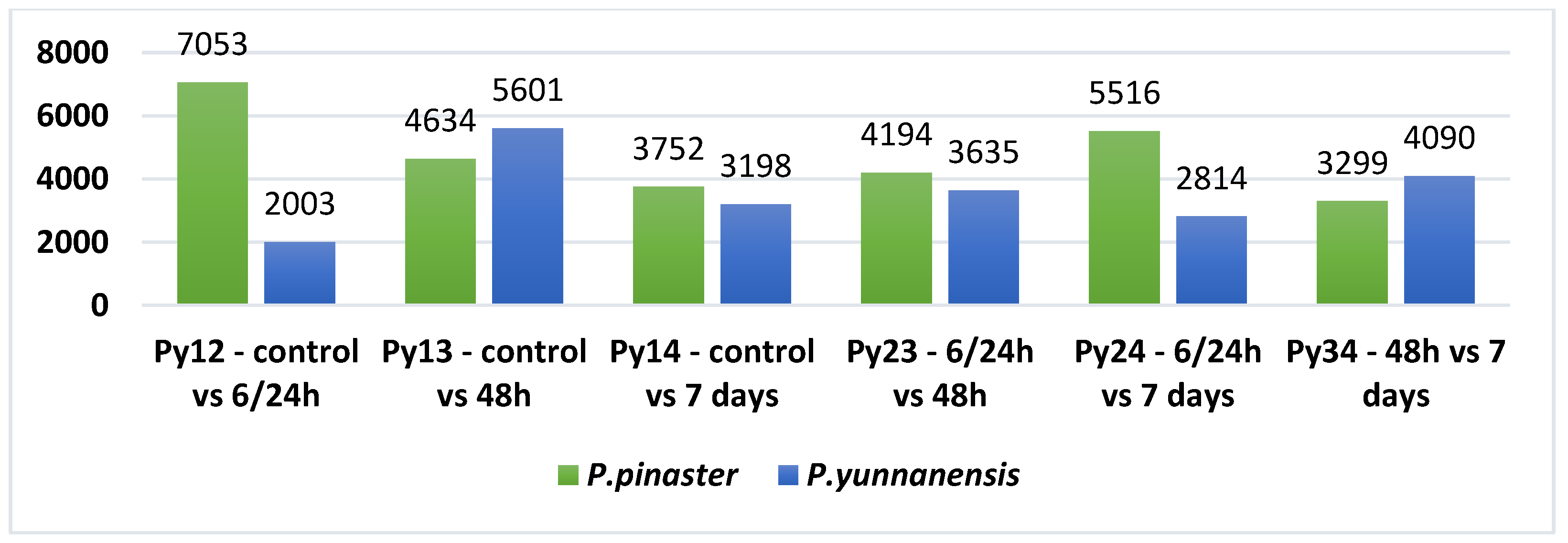
3.2. Mapping and Differential Expression Analysis
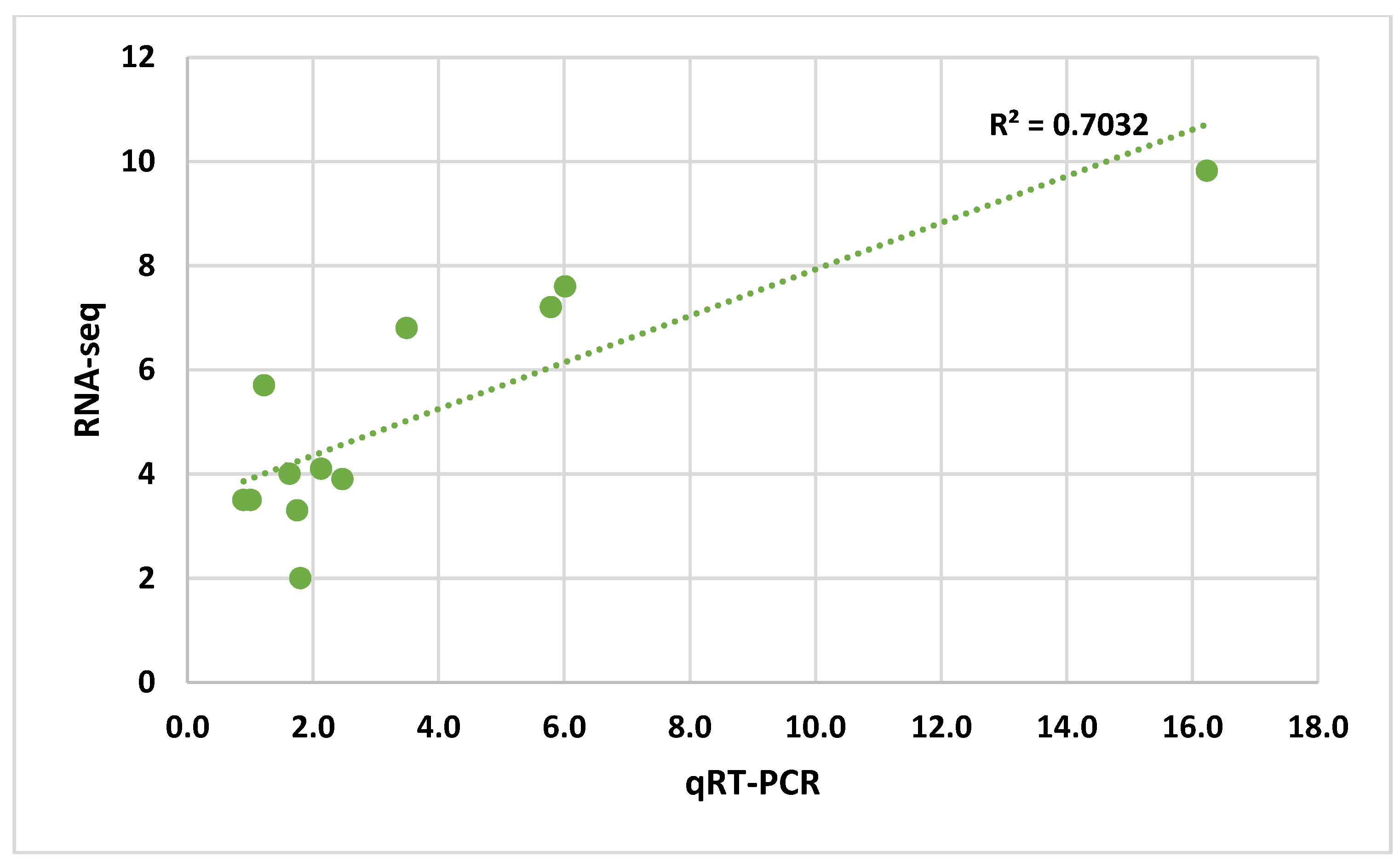
3.3. Quantitative Real-Time PCR Analysis
3.4. Transcriptome Annotation
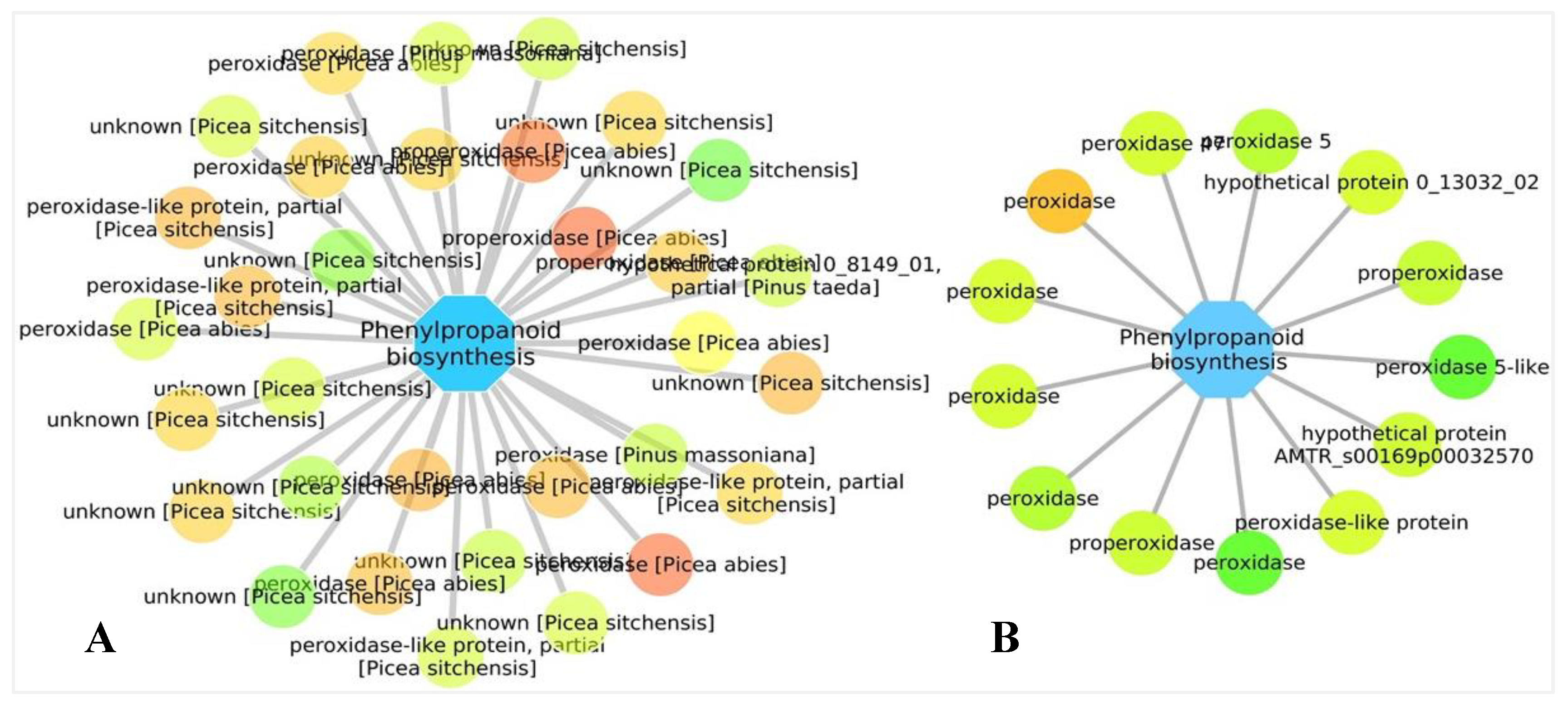
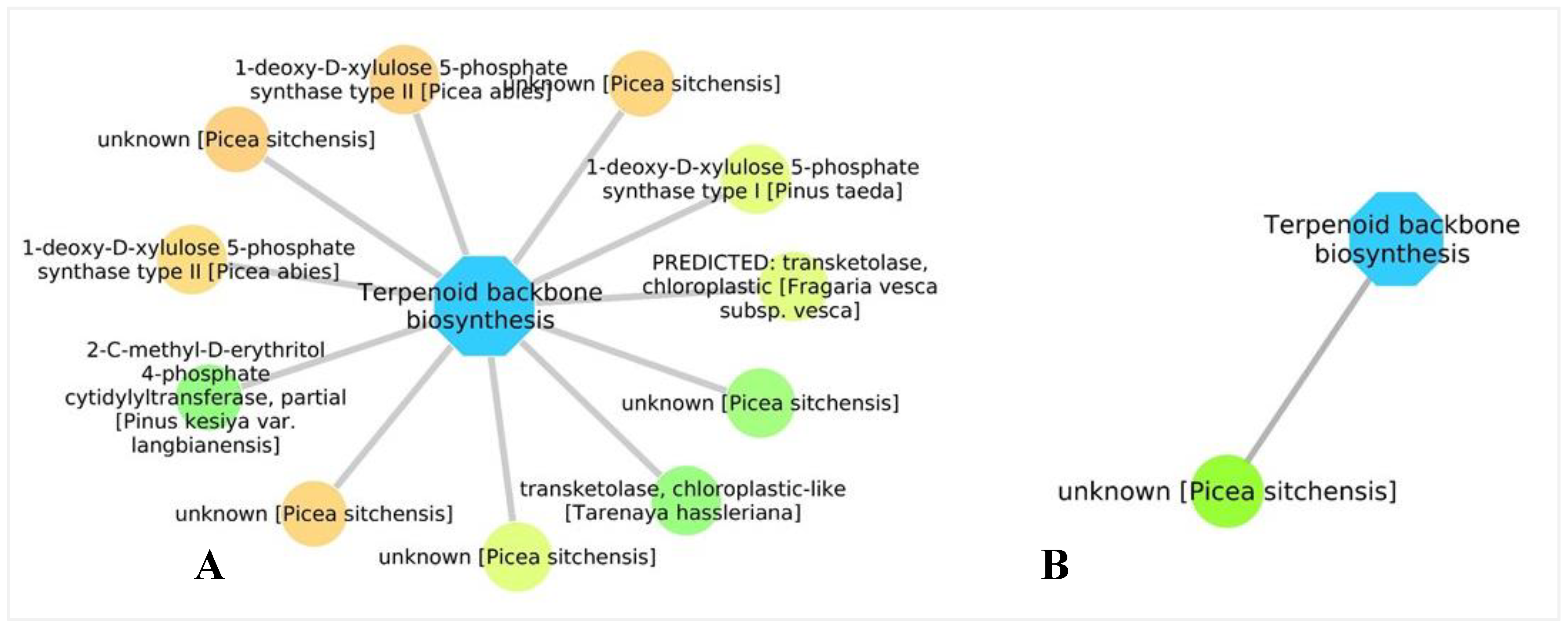
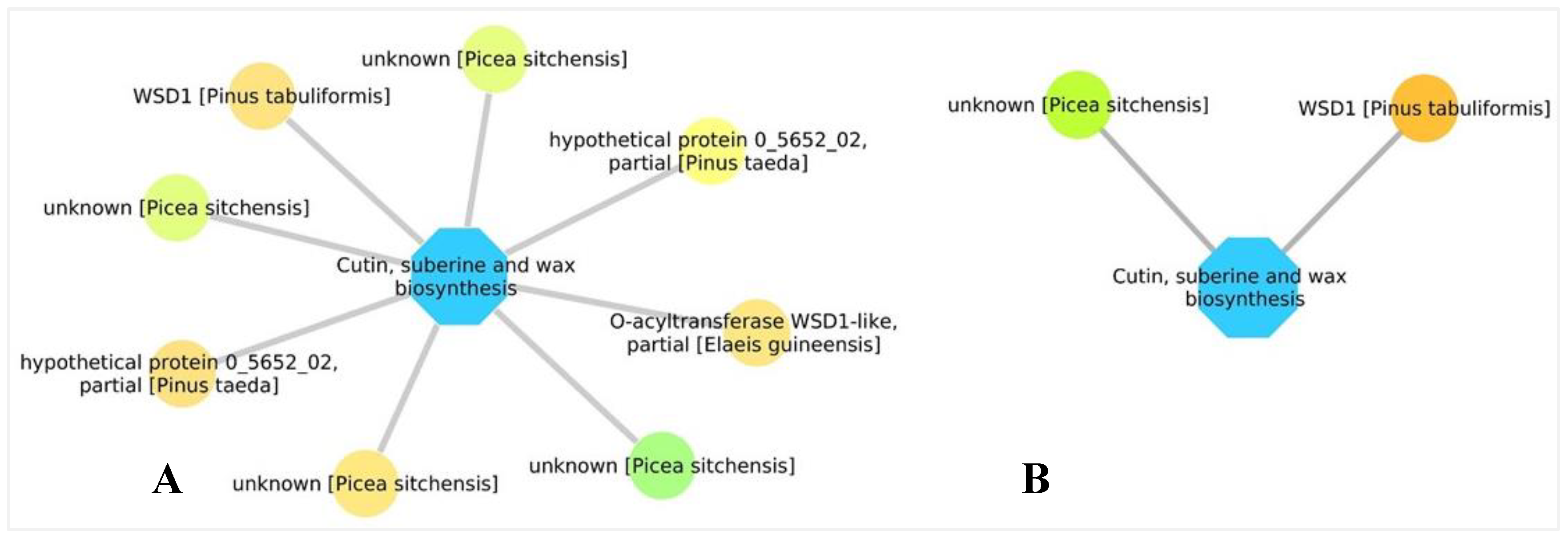
3.5. Biological Networks Analysis
4. Discussion
4.1. Response to PWN Infection Modulated by Time and Degree of Defense Mechanisms Following Pathogen Recognition
4.2. Production of Chemical Compounds and Physical Barriers as a Tool to Reduce Pathogen Growth and Proliferation
4.3. Overexpression of Defense-Related Genes Suggests a Continuum Reinforcement of the Immune System
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mota, M.M.; Vieira, P. Pine Wilt Disease: A Worldwide Threat to Forest Ecosystems; Springer: Dordrecht, The Netherlands, 2008; ISBN 978-1-4020-8454-6. [Google Scholar]
- Akbulut, S.; Stamps, W.T. Insect vectors of the pinewood nematode: A review of the biology and ecology of Monochamus species. For. Pathol. 2012, 42, 89–99. [Google Scholar] [CrossRef]
- Linit, M.J. Transmission of Pinewood Nematode Through Feeding Wounds of Monochamus carolinensis (Coleoptera: Cerambycidae). J. Nematol. 1990, 22, 231–236. [Google Scholar] [PubMed]
- Umebayashi, T.; Fukuda, K.; Haishi, T.; Sotooka, R.; Zuhair, S.; Otsuki, K. The developmental process of xylem embolisms in pine wilt disease monitored by multipoint imaging using compact magnetic resonance imaging. Plant Physiol. 2011, 156, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Hirao, T.; Fukatsu, E.; Watanabe, A.; Tokushige, Y.; Kiyohara, T.; Wang, Z.; Wang, C.; Fang, Z.; Zhang, D.; Liu, L.; et al. Characterization of resistance to pine wood nematode infection in Pinus thunbergii using suppression subtractive hybridization. BMC Plant Biol. 2012, 12, 13. [Google Scholar] [CrossRef]
- Xu, L.; Liu, Z.-Y.; Zhang, K.; Lu, Q.; Liang, J.; Zhang, X.-Y. Characterization of the Pinus massoniana transcriptional response to Bursaphelenchus xylophilus infection using suppression subtractive hybridization. Int. J. Mol. Sci. 2013, 14, 11356–11375. [Google Scholar] [CrossRef] [PubMed]
- Mota, M.; Braasch, H.; Bravo, M.A.; Penas, A.C.; Burgermeister, W.; Metge, K.; Sousa, E. First report of Bursaphelenchus xylophilus in Portugal and in Europe. Nematology 1999, 1, 727–734. [Google Scholar] [CrossRef]
- Gaspar, D.; Trindade, C.; Usié, A.; Meireles, B.; Barbosa, P.; Fortes, A.; Pesquita, C.; Costa, R.; Ramos, A. Expression Profiling in Pinus pinaster in Response to Infection with the Pine Wood Nematode Bursaphelenchus xylophilus. Forests 2017, 8, 279. [Google Scholar] [CrossRef]
- Santos, C.S.; Pinheiro, M.; Silva, A.I.; Egas, C.; Vasconcelos, M.W. Searching for resistance genes to Bursaphelenchus xylophilus using high throughput screening. BMC Genomics 2012, 13, 599. [Google Scholar] [CrossRef]
- Rassati, D.; Toffolo, E.P.; Battisti, A.; Faccoli, M. Monitoring of the pine sawyer beetle Monochamus galloprovincialis by pheromone traps in Italy. Phytoparasitica 2012, 40, 329–336. [Google Scholar] [CrossRef]
- Farjon, A.; Rushforth, K.; Christian, T. Pinus yunnanensis var. yunnanensis. The IUCN Red List of Threatened Species 2013: E.T191652A1991491. Available online: https://dx.doi.org/10.2305/IUCN.UK.2013-1.RLTS.T191652A1991491.en (accessed on 7 February 2018).
- C.A.B International. Pines of Silvicultural Importance, 1st ed.; CABI Pub: Oxon, UK, 2002. [Google Scholar]
- Wang, X.Z.; Wang, L.F.; Wang, Y.; Huang, Y.Q.; Ding, Z.G.; Zhou, J.; Gou, D.P. Identification and genetic analysis of the pinewood nematode Bursaphelenchus xylophilus from Pinus yunnanensis. For. Pathol. 2015, 45, 388–399. [Google Scholar] [CrossRef]
- Fielding, N.J.; Evans, H.F. The pine wood nematode Bursaphelenchus xylophilus (Steiner and Buhrer) Nickle (= B. lignicolus Mamiya and Kiyohara): An assessment of the current position. Forestry 1996, 69, 35–46. [Google Scholar] [CrossRef]
- Baermann, G. Ein einfache Methode zur Auffindung von Anklyostomum (Nematoden) Larven in Erdproben. Ned Tijdschr Geneeskd 1917, 57, 131–137. [Google Scholar]
- Futai, K.; Furuno, T. The variety of resistances among pine species to pine wood nematode, Bursaphelenchus lignicolus. Bull. Kyoto Univ. For. 1979, 51, 23–36. [Google Scholar]
- Le Provost, G.; Herrera, R.; Paiva, J.A.; Chaumeil, P.; Salin, F.; Plomion, C. A micromethod for high throughput RNA extraction in forest trees. Biol. Res. 2007, 40, 291–297. [Google Scholar] [CrossRef]
- Andrews, S. FastQC—A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics, Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Joshi, N.; Fass, J. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ files, Version 1.33. 2011.
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef]
- Haas, B. TransDecoder (Find Coding Regions within Transcripts). Available online: http://transdecoder.github.io (accessed on 16 May 2016).
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.-C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2015, 44, D279–D285. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Eddy, S.R. Multiple alignment using hidden Markov models. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1995, 3, 114–120. [Google Scholar]
- Srivastava, A.; Sarkar, H.; Patro, R. RapMap: A Rapid, Sensitive and Accurate Tool for Mapping RNA-seq Reads to Transcriptomes. bioRxiv 2015, 32, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [PubMed]
- Sebastiana, M.; Vieira, B.; Lino-Neto, T.; Monteiro, F.; Figueiredo, A.; Sousa, L.; Pais, M.S.; Tavares, R.; Paulo, O.S. Oak Root Response to Ectomycorrhizal Symbiosis Establishment: RNA-Seq Derived Transcript Identification and Expression Profiling. PLoS ONE 2014, 9, e98376. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A.M. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Zhi-Liang, H.; Jie, B.; James, M.R. CateGOrizer: A Web-Based Program to Batch Analyze Gene Ontology Classification Categories. Online J. Bioinform. 2008, 9, 108–112. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef]
- Chittoor, J.M.; Leach, J.E.; White, F.F. Differential Induction of a Peroxidase Gene Family During Infection of Rice by Xanthomonas oryzae pv. oryzae. Mol. Plant Microbe Interact. 1997, 10, 861–871. [Google Scholar] [CrossRef]
- Singh, B.; Sharma, R.A. Plant terpenes: Defense responses, phylogenetic analysis, regulation and clinical applications. 3 Biotech 2015, 5, 129–151. [Google Scholar] [CrossRef]
- Hunter, W.N. The non-mevalonate pathway of isoprenoid precursor biosynthesis. J. Biol. Chem. 2007, 282, 21573–21577. [Google Scholar] [CrossRef]
- Skryhan, K.; Gurrieri, L.; Sparla, F.; Trost, P.; Blennow, A. Redox regulation of starch metabolism. Front. Plant Sci. 2018, 9, 1–8. [Google Scholar] [CrossRef]
- Morkunas, I.; Ratajczak, L. The role of sugar signaling in plant defense responses against fungal pathogens. Acta Physiol. Plant. 2014, 36, 1607–1619. [Google Scholar] [CrossRef]
- Tauzin, A.S.; Giardina, T. Sucrose and invertases, a part of the plant defense response to the biotic stresses. Front. Plant Sci. 2014, 5, 293. [Google Scholar] [CrossRef]
- Ryss, A.Y.; Kulinich, O.A.; Sutherland, J.R. Pine wilt disease: A short review of worldwide research. For. Stud. China 2011, 13, 132–138. [Google Scholar] [CrossRef]
- Trindade, C. Avaliação da Expressão de Genes Relacionados com a Susceptibilidade a Bursaphelenchus xylophilus, Agente Causal da Doença da Murchidão dos Pinheiros (Pine Wilt Disease) em Pinus pinaster Ait e Pinus yunannensis Franch; Universidade de Lisboa: Lisboa, Portugal, 2012. [Google Scholar]
- War, A.R.; Paulraj, M.G.; Ahmad, T.; Buhroo, A.A.; Hussain, B.; Ignacimuthu, S.; Sharma, H.C. Mechanisms of plant defense against insect herbivores. Plant Signal. Behav. 2012, 7, 1306–1320. [Google Scholar] [CrossRef] [PubMed]
- Rejeb, I.B.; Pastor, V.; Mauch-Mani, B. Plant Responses to Simultaneous Biotic and Abiotic Stress: Molecular Mechanisms. Plants (Basel, Switz.) 2014, 3, 458–475. [Google Scholar] [CrossRef] [PubMed]
- Koornneef, A.; Pieterse, C.M.J. Cross talk in defense signaling. Plant Physiol. 2008, 146, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Ambawat, S.; Sharma, P.; Yadav, N.R.; Yadav, R.C. MYB transcription factor genes as regulators for plant responses: An overview. Physiol. Mol. Biol. Plants 2013, 19, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Van de Poel, B.; Van Der Straeten, D. 1-aminocyclopropane-1-carboxylic acid (ACC) in plants: More than just the precursor of ethylene! Front. Plant Sci. 2014, 5, 640. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.; Dixon, R.A. THE OXIDATIVE BURST IN PLANT DISEASE RESISTANCE. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1997, 48, 251–275. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Alscher, R.G.; Erturk, N.; Heath, L.S. Role of superoxide dismutases (SODs) in controlling oxidative stress in plants. J. Exp. Bot. 2002, 53, 1331–1341. [Google Scholar] [CrossRef]
- Kilili, K.G.; Atanassova, N.; Vardanyan, A.; Clatot, N.; Al-Sabarna, K.; Kanellopoulos, P.N.; Makris, A.M.; Kampranis, S.C. Differential roles of tau class glutathione S-transferases in oxidative stress. J. Biol. Chem. 2004, 279, 24540–24551. [Google Scholar] [CrossRef]
- Shahrtash, M. Plant glutathione s-transferases function during environmental stresses: A review article. Rom. J. Biol. Plant Biol. 2013, 58, 19–25. [Google Scholar]
- Sharma, R.; Sahoo, A.; Devendran, R.; Jain, M. Over-expression of a rice tau class glutathione s-transferase gene improves tolerance to salinity and oxidative stresses in Arabidopsis. PLoS ONE 2014, 9, e92900. [Google Scholar] [CrossRef] [PubMed]
- Nakai, Y.; Nakahira, Y.; Sumida, H.; Takebayashi, K.; Nagasawa, Y.; Yamasaki, K.; Akiyama, M.; Ohme-Takagi, M.; Fujiwara, S.; Shiina, T.; et al. Vascular plant one-zinc-finger protein 1/2 transcription factors regulate abiotic and biotic stress responses in Arabidopsis. Plant J. 2013, 73, 761–775. [Google Scholar] [CrossRef] [PubMed]
- Nawrot, R.; Barylski, J.; Nowicki, G.; Broniarczyk, J.; Buchwald, W.; Goździcka-Józefiak, A. Plant antimicrobial peptides. Folia Microbiol. (Praha) 2014, 59, 181–196. [Google Scholar] [CrossRef]
- Salas, C.E.; Badillo-Corona, J.A.; Ramírez-Sotelo, G.; Oliver-Salvador, C. Biologically active and antimicrobial peptides from plants. Biomed Res. Int. 2015, 2015, 102129. [Google Scholar] [CrossRef]
- Parage, C.; Tavares, R.; Réty, S.; Baltenweck-Guyot, R.; Poutaraud, A.; Renault, L.; Heintz, D.; Lugan, R.; Marais, G.A.B.; Aubourg, S.; et al. Structural, functional, and evolutionary analysis of the unusually large stilbene synthase gene family in grapevine. Plant Physiol. 2012, 160, 1407–1419. [Google Scholar] [CrossRef]
- Kodan, A.; Kuroda, H.; Sakai, F. A stilbene synthase from Japanese red pine (Pinus densiflora): Implications for phytoalexin accumulation and down-regulation of flavonoid biosynthesis. Proc. Natl. Acad. Sci. USA 2002, 99, 3335–3339. [Google Scholar] [CrossRef]
- Sharma, N.; Sharma, K.; Gaur, R.; Gupta, V. Role of Chitinase in Plant Defense. Asian J. Biochem. 2011, 29–37. [Google Scholar] [CrossRef]
- Miedes, E.; Vanholme, R.; Boerjan, W.; Molina, A. The role of the secondary cell wall in plant resistance to pathogens. Front. Plant Sci. 2014, 5, 358. [Google Scholar] [CrossRef]
- Malinovsky, F.G.; Fangel, J.U.; Willats, W.G.T. The role of the cell wall in plant immunity. Front. Plant Sci. 2014, 5, 178. [Google Scholar] [CrossRef]
- Lauvergeat, V.; Lacomme, C.; Lacombe, E.; Lasserre, E.; Roby, D.; Grima-Pettenati, J. Two cinnamoyl-CoA reductase (CCR) genes from Arabidopsis thaliana are differentially expressed during development and in response to infection with pathogenic bacteria. Phytochemistry 2001, 57, 1187–1195. [Google Scholar] [CrossRef]
- Waters, E.R.; Lee, G.J.; Vierling, E. Evolution, structure and function of the small heat shock proteins in plants. J. Exp. Bot. 1996, 47, 325–338. [Google Scholar] [CrossRef]
- Al-Whaibi, M.H. Plant heat-shock proteins: A mini review. J. King Saud. Univ. Sci. 2011, 23, 139–150. [Google Scholar] [CrossRef]
- Cao, J.; Lv, Y.; Hou, Z.; Li, X.; Ding, L. Expansion and evolution of thaumatin-like protein (TLP) gene family in six plants. Plant Growth Regul. 2016, 79, 299–307. [Google Scholar] [CrossRef]
- Shin, H.; Lee, H.; Woo, K.S.; Noh, E.W.; Koo, Y.B.; Lee, K.J. Identification of genes upregulated by pinewood nematode inoculation in Japanese red pine. Tree Physiol. 2009, 29, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Bommer, U.A.; Thiele, B.J. The translationally controlled tumour protein (TCTP). Int. J. Biochem. Cell Biol. 2004, 36, 379–385. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Han, Y.-J.; Hwang, O.-J.; Lee, S.-S.; Shin, A.-Y.; Kim, S.Y.; Kim, J.-I. Overexpression of Arabidopsis Translationally Controlled Tumor Protein Gene AtTCTP Enhances Drought Tolerance with Rapid ABA-Induced Stomatal Closure. Mol. Cells 2012, 33, 617–626. [Google Scholar] [CrossRef]
- Torii, K.U. Leucine-Rich Repeat Receptor Kinases in Plants: Structure, Function, and Signal Transduction Pathways. Int. Rev. Cytol. 2004, 234, 1–46. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Number of Sequenced Reads | Average Read Length (bp) | Number of Reads after QC | % Reads after QC |
|---|---|---|---|---|
| Py1—control | 43,708,074 | 113 | 35,593,607 | 81.4 |
| Py2—6 h + 24 h | 40,281,442 | 114 | 32,313,524 | 80.2 |
| Py3—48 h | 39,601,162 | 110 | 29,720,848 | 75.1 |
| Py4—7 days | 45,743,269 | 109 | 35,642,590 | 77.9 |
| Total | 169,333,947 | 111 | 133,270,569 | 78.7 |
| Sample | Number of Mapped Reads | Number of Uniquely Mapped Reads | % of Mapped Reads | % of Uniquely Mapped Reads |
|---|---|---|---|---|
| Py1—Control | 26,850,458 | 16,937,906 | 75.4% | 47.6% |
| Py2—6 h + 24 h | 24,124,055 | 15,319,554 | 74.7% | 47.4% |
| Py3—48 h | 23,420,583 | 14,894,121 | 78.8% | 50.1% |
| Py4—7 days | 28,578,528 | 18,221,649 | 80.2% | 51.1% |
| Total | 102,973,624 | 65,373,230 | 77.3% | 49.1% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaspar, D.; Trindade, C.; Usié, A.; Meireles, B.; Fortes, A.M.; Guimarães, J.B.; Simões, F.; Costa, R.L.; Ramos, A.M. Comparative Transcriptomic Response of Two Pinus Species to Infection with the Pine Wood Nematode Bursaphelenchus xylophilus. Forests 2020, 11, 204. https://doi.org/10.3390/f11020204
Gaspar D, Trindade C, Usié A, Meireles B, Fortes AM, Guimarães JB, Simões F, Costa RL, Ramos AM. Comparative Transcriptomic Response of Two Pinus Species to Infection with the Pine Wood Nematode Bursaphelenchus xylophilus. Forests. 2020; 11(2):204. https://doi.org/10.3390/f11020204
Chicago/Turabian StyleGaspar, Daniel, Cândida Trindade, Ana Usié, Brigida Meireles, Ana Margarida Fortes, Joana Bagoin Guimarães, Fernanda Simões, Rita Lourenço Costa, and António Marcos Ramos. 2020. "Comparative Transcriptomic Response of Two Pinus Species to Infection with the Pine Wood Nematode Bursaphelenchus xylophilus" Forests 11, no. 2: 204. https://doi.org/10.3390/f11020204
APA StyleGaspar, D., Trindade, C., Usié, A., Meireles, B., Fortes, A. M., Guimarães, J. B., Simões, F., Costa, R. L., & Ramos, A. M. (2020). Comparative Transcriptomic Response of Two Pinus Species to Infection with the Pine Wood Nematode Bursaphelenchus xylophilus. Forests, 11(2), 204. https://doi.org/10.3390/f11020204