Morphological Characteristics and Transcriptome Comparisons of the Shoot Buds from Flowering and Non-Flowering Pleioblastus pygmaeus

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Morphological and Anatomical Characterization
2.3. Sample and Library Preparation
2.4. Quality Control and Transcriptome Assembly
2.5. Gene Annotation and Differential Gene Expression
2.6. qRT-PCR Verification
3. Results
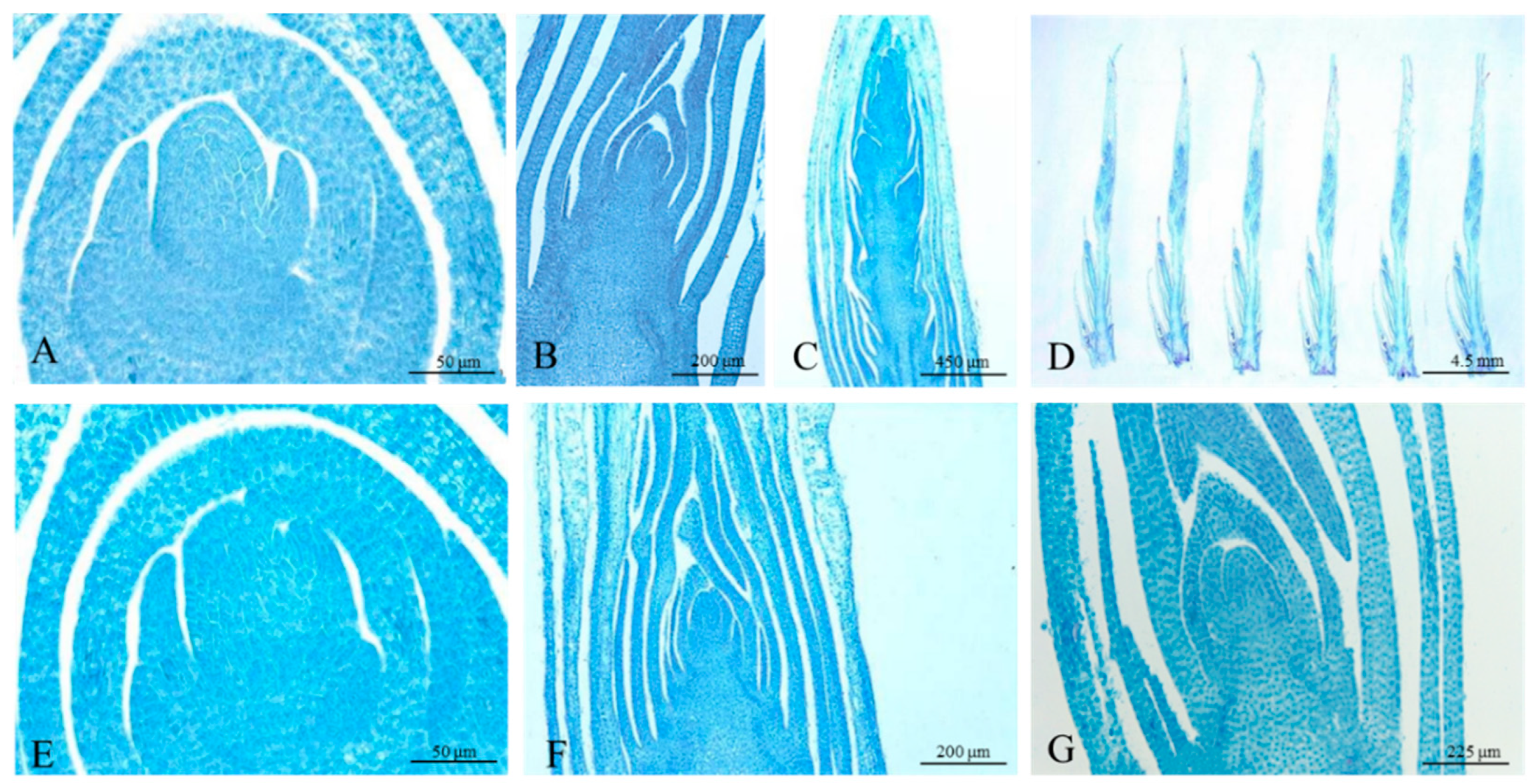
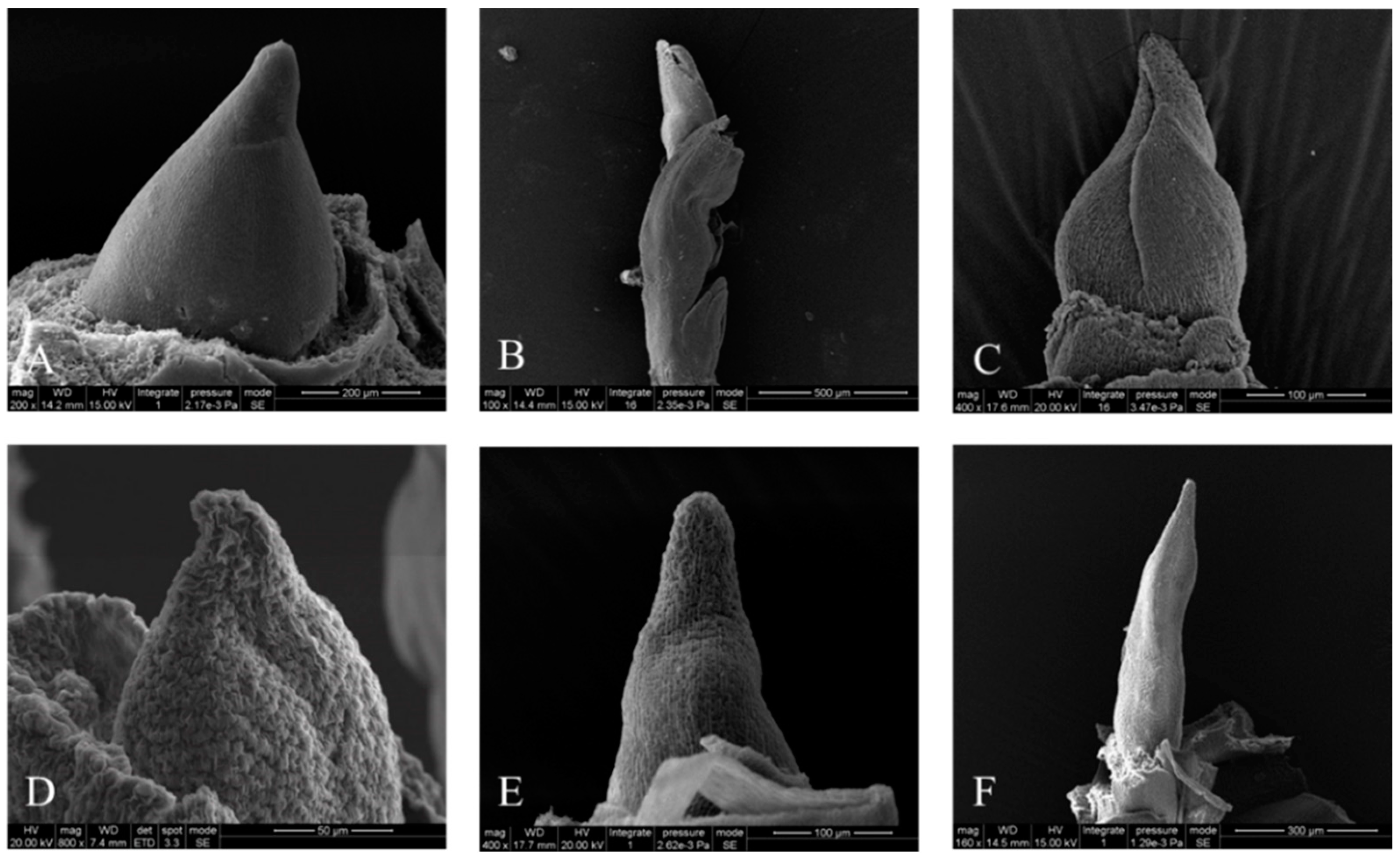
3.1. Morphological and Anatomical Characterization of the Samples
3.2. Data Filtering and Assembly
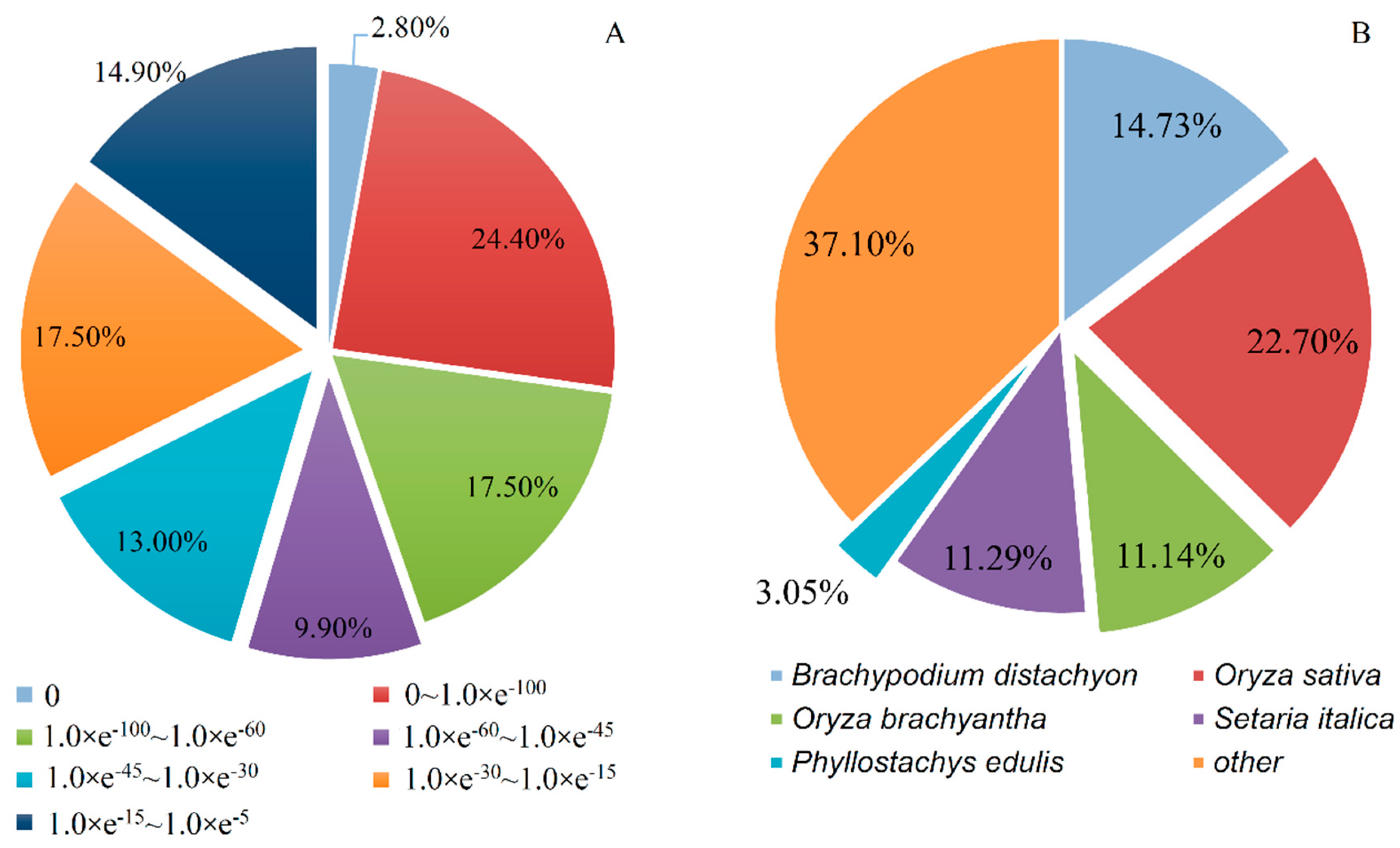
3.3. Gene Annotation
3.4. Differential Gene Enrichment Analysis
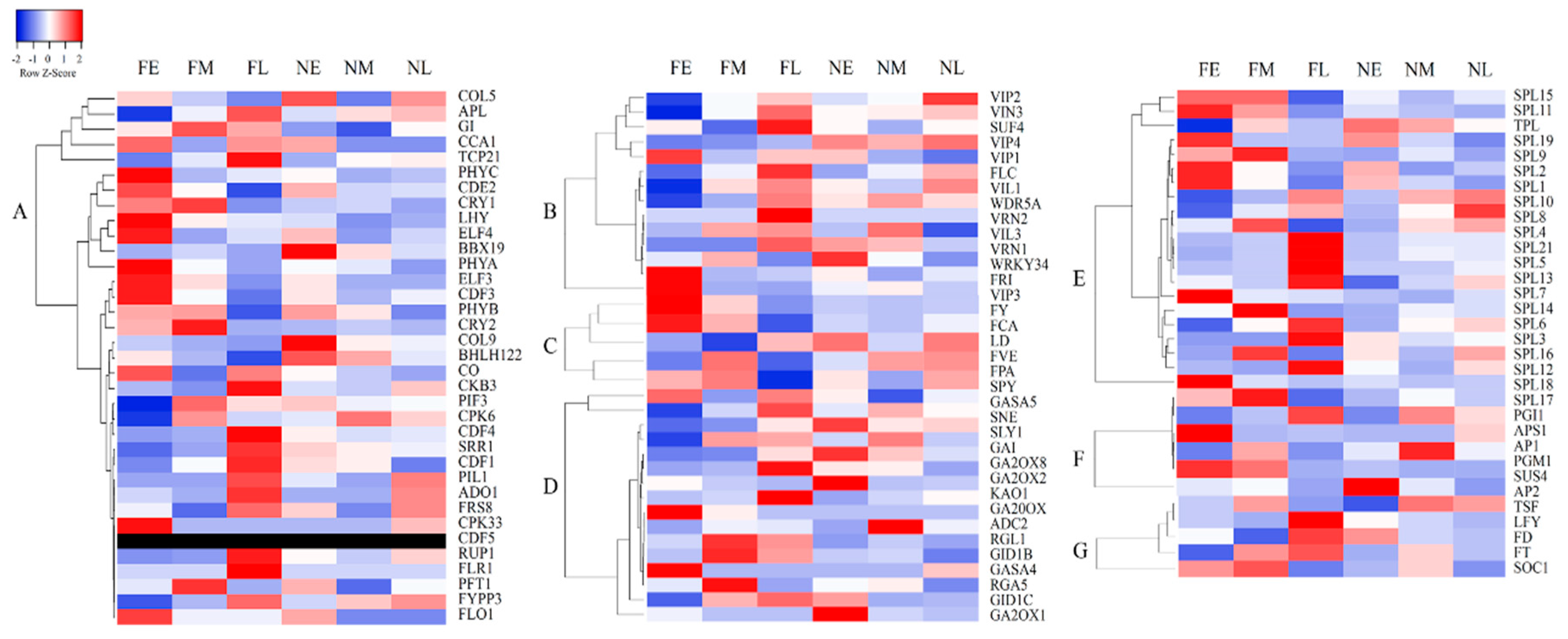
3.5. Expression Analysis of Key Genes Involved in Floral Development
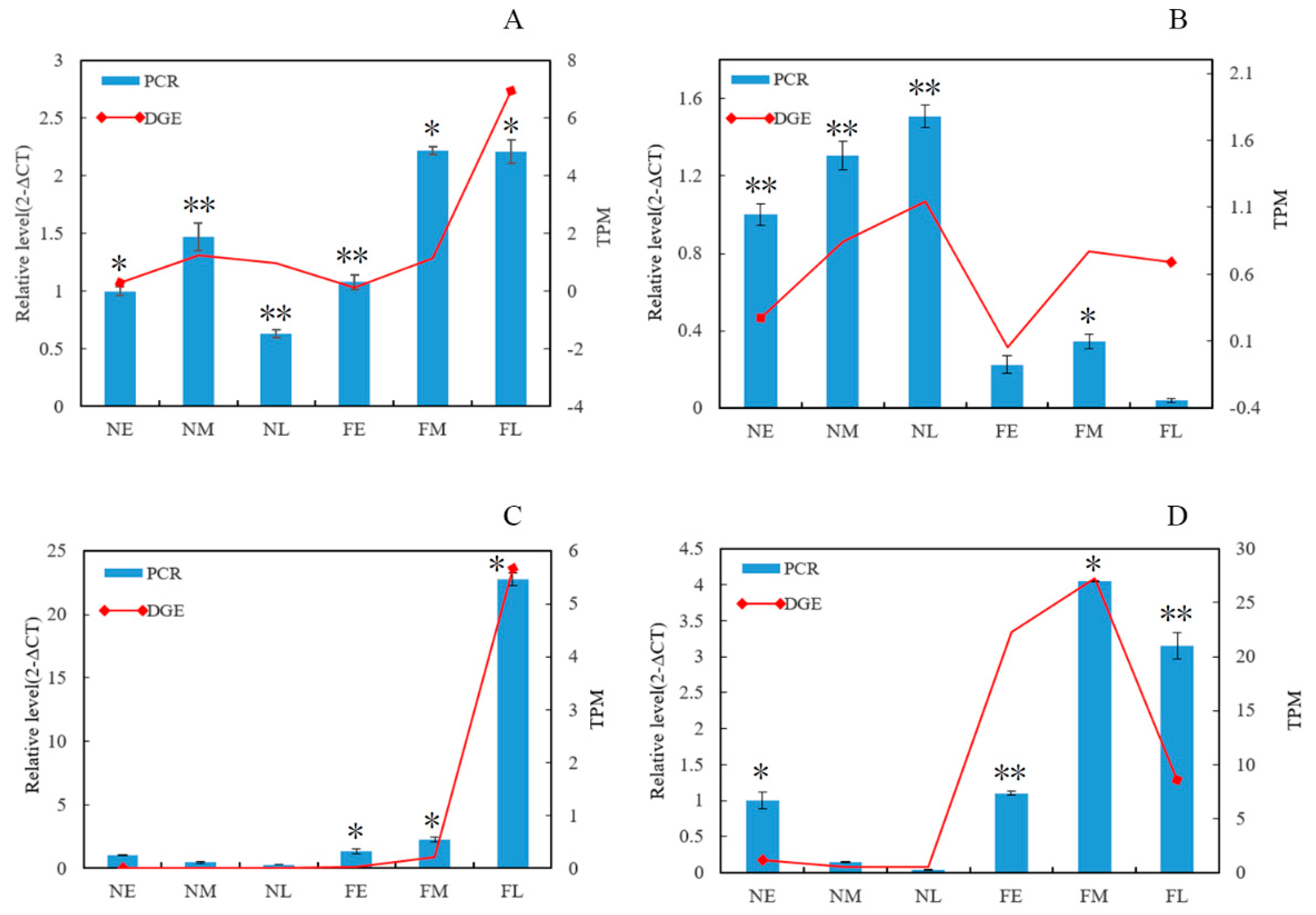
3.6. RNA-Seq Expression Validation of Key Genes Involved in Floral Development
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DEUs | differentially expressed unigenes |
| TF | Transcription factor |
| SEM | Scanning electron microscope |
| GA | Gibberellins |
| FE | Dormant shoot buds of flowering plants |
| FM | Germinated shoots of flowering plants |
| FL | Flower buds of flowering plants |
| NE | Dormant shoot buds of vegetative plants |
| NM | Germinated shoots of vegetative plants |
| NL | Leaf buds of vegetative plants |
| NR | NCBI non-redundant protein sequences |
| NT | NCBI non-redundant nucleotide sequences |
| Pfam | Protein family |
| KOG | Eukaryotic ortholog group |
| KEGG | Kyoto encyclopedia of genes and genomes |
| GO | Gene ontology |
| GC | guanine-cytosine |
| bp | base pair |
| RSEM | RNA-Seq by Expectation Maximization |
| FPKM | Fragments per Kilobase Million |
| TPM | Transcripts Perkilobase Million |
| RRMs | RNA recognition motifs |
References
- Liese, W.; Köhl, M. Bamboo: The Plant and Its Uses; Springer: Heidelberg/Berlin, Germany, 2015. [Google Scholar]
- Zheng, X.; Lin, S.Y.; Fu, H.J.; Wang, Y.W.; Ding, Y.L. The bamboo flowering cycle sheds light on flowering diversity. Front. Plant Sci. 2020, 11, 381. [Google Scholar] [PubMed]
- Jiao, Y.L.; Hu, Q.T.; Zhu, Y.; Zhu, L.F.; Ma, T.F.; Zeng, H.Y.; Zang, Q.L.; Li, L.; Lin, X.C. Comparative transcriptomic analysis of the flower induction and development of the Lei bamboo (Phyllostachys violascens). BMC Bioinfor. 2019, 20, 687. [Google Scholar]
- Rout, G.R.; Das, P. Somatic embryogenesis and in vitro flowering of 3 species of bamboo. Plant Cell Rep. 1994, 13, 683–686. [Google Scholar] [PubMed]
- Fan, T.T.; Guo, Z.H.; Fu, H.J.; Zhang, L.; Ding, Y.L. Germination and storage characteristics of Pleioblastus pygmaeus pollen. Forest Res. 2018, 31, 180–185. [Google Scholar]
- Lin, S.Y.; Li, J.; Zhao, R.; Dong, X.B.; Ding, Y.L. The development of flowering bud differentiation and male gametophyte of Bambusa multiplex. J. Nanjing For. Univ. Nat. Sci. 2015, 39, 51–56. [Google Scholar]
- Waikhom, S.D.; Louis, B.; Roy, P.; Singh, W.M.; Bharwaj, P.K.; Talukdar, N.C. Scanning electron microscopy of pollen structure throws light on resolving Bambusa–Dendrocalamus complex: Bamboo flowering evidence. Plant Syst. Evol. 2014, 300, 1261–1268. [Google Scholar]
- Saitoh, T.; Seiwa, K.; Nishiwaki, A. Importance of physiological integration of dwarf bamboo to persistence in forest understorey: A field experiment. J. Ecol. 2002, 90, 78–85. [Google Scholar]
- Prasun, B.; Sukanya, C.; Smritikana, D.; Amita, P.; Malay, D. Bamboo flowering from the perspective of comparative genomics and transcriptomics. Front. Plant Sci. 2016, 7, 1900. [Google Scholar]
- Zhao, H.S.; Gao, Z.M.; Wang, L.; Wang, J.L.; Wang, S.B.; Fei, B.H.; Chen, C.H.; Shi, C.C.; Liu, X.C.; Zhang, H.L.; et al. Chromosome-level reference genome and alternative splicing atlas of moso bamboo (Phyllostachys edulis). Gigascience 2018, 7, giy115. [Google Scholar]
- Li, Y.; Zhang, C.X.; Yang, K.B.; Shi, J.J.; Gao, Z.M. De novo sequencing of the transcriptome reveals regulators of the floral transition in Fargesia macclureana (Poaceae). BMC Genom. 2019, 20, 1035. [Google Scholar]
- Gao, J.; Zhang, Y.; Zhang, C.L.; Qi, F.Y.; Li, X.P.; Mu, S.H.; Peng, Z.H.; Gaquerel, E. Characterization of the floral transcriptome of Moso bamboo (Phyllostachys edulis) at different flowering developmental stages by transcriptome sequencing and RNA-Seq analysis. PLoS ONE 2014, 9, e98910. [Google Scholar]
- Zhang, X.M.; Zhao, L.; Larson-Rabin, Z.; Li, D.Z.; Guo, Z.H.; Nitabach, M.N. De novo sequencing and characterization of the floral transcriptome of Dendrocalamus latiflorus (Poaceae: Bambusoideae). PLoS ONE 2012, 7, e42082. [Google Scholar]
- Peng, Z.H.; Lu, Y.; Li, L.B.; Zhao, Q.; Jiang, Z.H. The draft genome of the fast-growing non-timber forest species moso bamboo (Phyllostachys heterocycla). Nat. Genet. 2013, 45, 456–461. [Google Scholar] [PubMed] [Green Version]
- Lin, S.Y.; Fan, T.T.; Jiang, M.Y.; Zhang, L.; Zheng, X.; Ding, Y.L. The revision of scientific names for three dwarf bamboo species (cultivar) based on the floral morphology. J. Nanjing For. Univ. Nat. Sci. 2017, 41, 189–193. [Google Scholar]
- Su, J.L.; Lin, S.Y.; Shi, W.S.; Wang, X.; Zheng, X.; Wan, Y.W.; Ding, Y.L. Anatomical observation and three-dimensional construction of leaf blades from six bamboos. J. Nanjing For. Univ. Nat. Sci. 2020, 44, 47–53. [Google Scholar]
- Wang, L.G.; Wang, S.Q.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar]
- Davidson, N.M.; Oshlack, A. Corset: Enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 2014, 15, 410. [Google Scholar]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar]
- Wang, L.K.; Feng, Z.X.; Wang, X.; Wang, X.W.; Zhang, X.G. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [PubMed] [Green Version]
- Mao, X.Z.; Cai, T.; Olyarchuk, J.G.; Wei, L.P. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [PubMed]
- Fan, C.J.; Ma, J.M.; Guo, Q.R.; Li, X.T.; Wang, H.; Lu, M.Z. Selection of Reference Genes for Quantitative Real-Time PCR in Bamboo (Phyllostachys edulis). PLoS ONE 2013, 8, e56573. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar]
- Ge, W.; Zhang, Y.; Cheng, Z.C.; Hou, D.; Gao, J. Main regulatory pathways, key genes and microRNAs involved in flower formation and development of Moso bamboo (Phyllostachys edulis). Plant Biotechnol. J. 2017, 15, 82–96. [Google Scholar]
- Putterill, J.; Laurie, R.; Macknight, R. It’s time to flower: The genetic control of flowering time. Bioessays 2004, 26, 363–373. [Google Scholar]
- Coupland, G. Regulation of flowering by photoperiod in Arabidopsis. Plant Cell Environ. 2010, 20, 785–789. [Google Scholar]
- Valverde, F.; Mouradov, A.; Soppe, W.; Ravenscroft, D.; Samach, A.; Coupland, G. Photoreceptor regulation of CONSTANS protein in photoperiodic flowering. Science 2004, 303, 1003–1006. [Google Scholar]
- An, H.; Roussot, C.; Suárez-López, P.; Corbesier, L.; Vincent, C.; Piñeiro, M.; Hepworth, S.; Mouradov, A.; Justin, S.; Turnbull, C.; et al. CONSTANS acts in the phloem to regulate a systemic signal that induces photoperiodic flowering of Arabidopsis. Development 2004, 131, 3615–3626. [Google Scholar]
- Castillejo, C.; Pelaz, S. The balance between CONSTANS and TEMPRANILLO activities determines FT expression to trigger flowering. Curr. Biol. 2008, 18, 1338–1343. [Google Scholar] [PubMed] [Green Version]
- Lee, J.E.; Lee, I. Regulation and function of SOC1, a flowering pathway integrator. J. Exp. Bot. 2010, 61, 2247–2254. [Google Scholar] [PubMed] [Green Version]
- Sheldon, C.C.; Finnegan, E.J.; Dennis, E.S.; Peacock, W.J. Quantitative effects of vernalization on FLC and SOC1 expression. Plant J. 2010, 45, 871–883. [Google Scholar]
- Dennis, E.S.; Peacock, W.J. Epigenetic regulation of flowering. Curr. Opin. Plant Biol. 2007, 10, 520–527. [Google Scholar] [PubMed]
- Bastow, R.; Mylne, J.S.; Lister, C.; Lippman, Z.; Martienssen, R.A.; Dean, C. Vernalization requires epigenetic silencing of FLC by histone methylation. Nature 2004, 427, 164–167. [Google Scholar]
- Aaron, G.; James, P.W.; Dennis, E.S.; Ben, T. The molecular biology of seasonal flowering-responses in Arabidopsis and the cereals. Ann. Bot. 2009, 103, 1165–1172. [Google Scholar]
- Yan, L.L.; Loukoianov, A.; Blechl, A.; Tranquilli, C.; Ramakrishna, W.; SanMiguel, P.; Bennetzen, J.L.; Echenique, V.; Dubcovsky, J. The wheat VRN2 gene is a flowering repressor down-regulated by vernalization. Science 2004, 303, 1640–1644. [Google Scholar]
- Finnegan, E.J.; Kovac, K.A.; Jaligot, E.; Sheldon, C.C.; Peacock, W.J.; Dennis, E.S. The downregulation of FLOWERING LOCUS C (FLC) expression in plants with low levels of DNA methylation and by vernalization occurs by distinct mechanisms. Plant J. 2010, 44, 420–432. [Google Scholar]
- Winichayakul, S.; Beswick, N.L.; Dean, C.; Macknight, R.C. Components of the Arabidopsis autonomous floral promotion pathway, FCA and FY, are conserved in monocots. Funct. Plant Biol. 2005, 32, 345–355. [Google Scholar]
- Jang, Y.H.; Park, H.Y.; Kim, S.K.; Hwan, L.J.; Chung, S.M.; Soo, C.Y.; Kyung-Hee, P.; Jeong-Kook, K. Survey of rice proteins interacting with OsFCA and OsFY proteins which are homologous to the Arabidopsis flowering time proteins, FCA and FY. Plant Cell Physiol. 2009, 50, 1479–1492. [Google Scholar]
- Lim, M.H.; Kim, J.; Kim, Y.S.; Chung, K.S.; Seo, Y.H.; Lee, I.; Kim, J.; Hong, C.B.; Kim, H.J.; Park, C.M. A new Arabidopsis gene, FLK, encodes an RNA binding protein with K homology motifs and regulates flowering time via FLOWERING LOCUS C. Plant Cell 2004, 16, 731–740. [Google Scholar] [PubMed] [Green Version]
- Simpson, G.G.; Quesada, V.; Henderson, I.R.; Dijkwel, P.P.; Dean, C. RNA processing and Arabidopsis flowering time control. Biochem. Soc. Trans. 2004, 32, 565–566. [Google Scholar] [PubMed]
- Lee, I.; Aukerman, M.J.; Gore, S.L.; Lohman, K.N.; Michaels, S.D.; Weaver, L.M.; John, M.C.; Feldmann, K.A.; Amasino, R.M. Isolation of LUMINIDEPENDENS: A gene involved in the control of flowering time in Arabidopsis. Plant Cell 1994, 6, 75–83. [Google Scholar] [PubMed] [Green Version]
- Marquardt, S.; Boss, P.K.; Hadfield, J.; Dean, C. Additional targets of the Arabidopsis autonomous pathway members, FCA and FY. J. Exp. Bot. 2006, 57, 3379–3386. [Google Scholar] [PubMed] [Green Version]
- Gocal, G.F.; Sheldon, C.C.; Gubler, F.; Moritz, T.; Bagnall, D.J.; MacMillan, C.P.; Li, S.F.; Parish, R.W.; Dennis, E.S.; Weigel, D.; et al. GAMYB-like genes, flowering, and gibberellin signaling in Arabidopsis. Plant Physiol. 2001, 127, 1682–1693. [Google Scholar] [PubMed]
- Peng, J.; Carol, P.; Richards, D.E.; King, K.E.; Cowling, R.J.; Murphy, G.P.; Harberd, N.P. The Arabidopsis GAI gene defines a signaling pathway that negatively regulates gibberellin responses. Genes Dev. 1997, 11, 3194–3205. [Google Scholar] [PubMed] [Green Version]
- Ueguchi-Tanaka, M.; Ashikari, M.; Nakajima, M.; Itoh, H.; Katoh, E.; Kobayashi, M.; Chow, T.Y.; Hsing, Y.C.; Kitano, H.; Yamaguchi, I. GIBBERELLIN INSENSITIVE DWARF1 encodes a soluble receptor for gibberellin. Nature 2005, 437, 693–698. [Google Scholar]
- Fornara, F.; Coupland, G. Plant phase transitions make a SPLash. Cell 2009, 138, 625–627. [Google Scholar]
- Khan, M.R.G.; Ai, X.Y.; Zhang, J.Z. Genetic regulation of flowering time in annual and perennial plants. Wiley Interdiscip. Rev. RNA 2014, 5, 347–359. [Google Scholar]
- Yamaguchi, A.; Wu, M.F.; Yang, L.; Wu, G.; Poethig, R.S.; Wagner, D. The microRNA-regulated SBP-Box transcription factor SPL3 is a direct upstream activator of LEAFY, FRUITFULL, and APETALA1. Dev. Cell 2009, 17, 268–278. [Google Scholar]
- Unte, U.S.; Sorensen, A.M.; Pesaresi, P.; Gandikota, M.; Leister, D.; Saedler, H.; Huijser, P. SPL8, an SBP-box gene that affects pollen sac development in Arabidopsis. Plant Cell 2003, 15, 1009–1019. [Google Scholar] [PubMed] [Green Version]
- Riese, M.; Zobell, O.; Saedler, H.; Huijser, P. SBP-domain transcription factors as possible effectors of cryptochrome-mediated blue light signalling in the moss Physcomitrella patens. Planta 2008, 227, 505–515. [Google Scholar] [PubMed] [Green Version]
- Komeda, Y. Genetic regulation of time to flower in Arabidopsis thaliana. Annu. Rev. Plant Biol. 2004, 55, 521–535. [Google Scholar] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, W.; Li, C.; Lin, S.; Ren, L.; Wan, Y.; Zhang, L.; Ding, Y. Morphological Characteristics and Transcriptome Comparisons of the Shoot Buds from Flowering and Non-Flowering Pleioblastus pygmaeus. Forests 2020, 11, 1229. https://doi.org/10.3390/f11111229
Yao W, Li C, Lin S, Ren L, Wan Y, Zhang L, Ding Y. Morphological Characteristics and Transcriptome Comparisons of the Shoot Buds from Flowering and Non-Flowering Pleioblastus pygmaeus. Forests. 2020; 11(11):1229. https://doi.org/10.3390/f11111229
Chicago/Turabian StyleYao, Wenjing, Chuanzhe Li, Shuyan Lin, Li Ren, Yawen Wan, Li Zhang, and Yulong Ding. 2020. "Morphological Characteristics and Transcriptome Comparisons of the Shoot Buds from Flowering and Non-Flowering Pleioblastus pygmaeus" Forests 11, no. 11: 1229. https://doi.org/10.3390/f11111229
APA StyleYao, W., Li, C., Lin, S., Ren, L., Wan, Y., Zhang, L., & Ding, Y. (2020). Morphological Characteristics and Transcriptome Comparisons of the Shoot Buds from Flowering and Non-Flowering Pleioblastus pygmaeus. Forests, 11(11), 1229. https://doi.org/10.3390/f11111229