Comparing the Effectiveness of Exome Capture Probes, Genotyping by Sequencing and Whole-Genome Re-Sequencing for Assessing Genetic Diversity in Natural and Managed Stands of Picea abies



Abstract
:1. Introduction
2. Method
2.1. Sampling
2.2. Whole-Genome Re-Sequencing Data
2.3. Targeted Capture Probe Data
2.4. Genotyping-by-Sequencing Data
2.5. Data Analysis
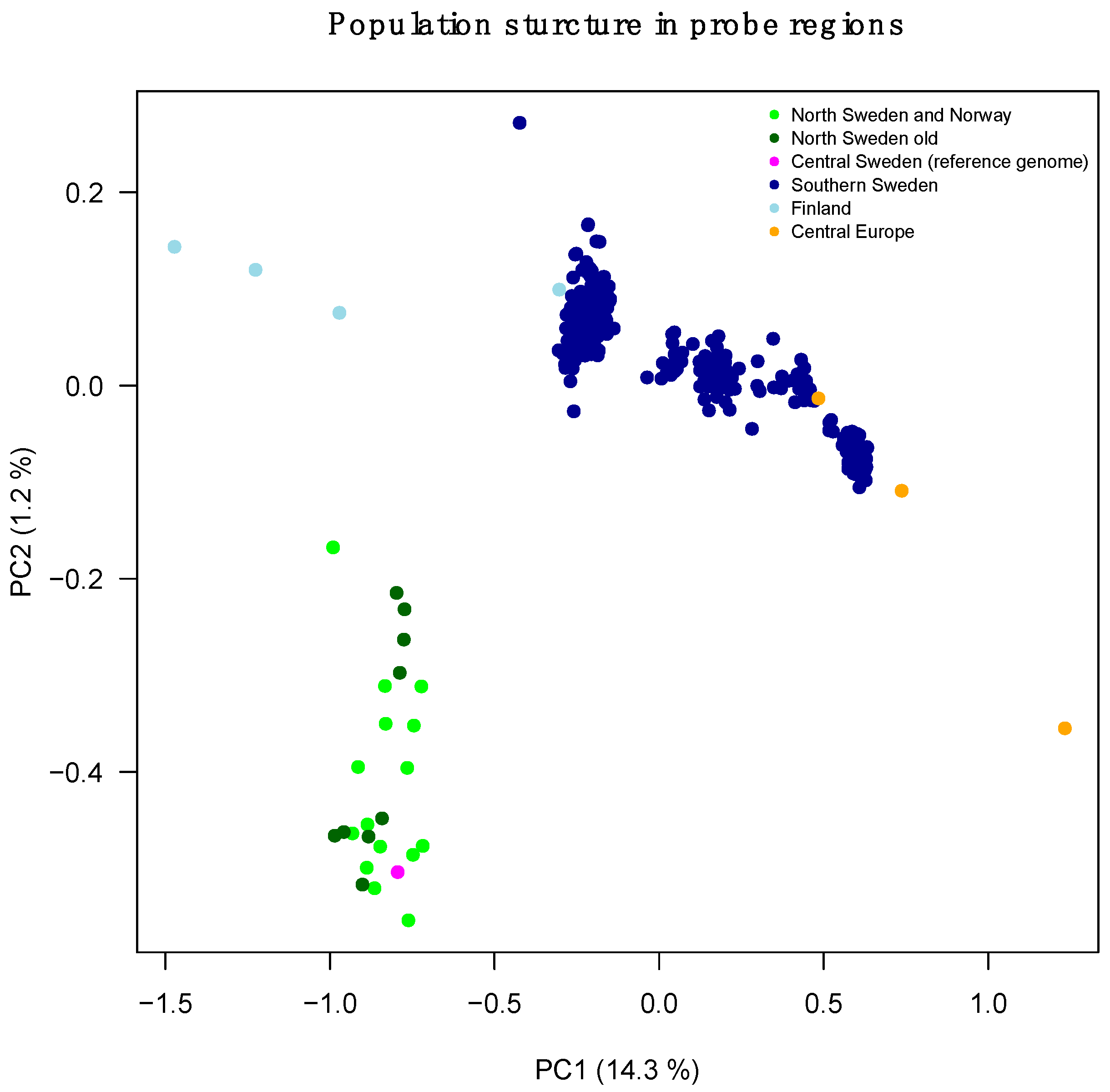
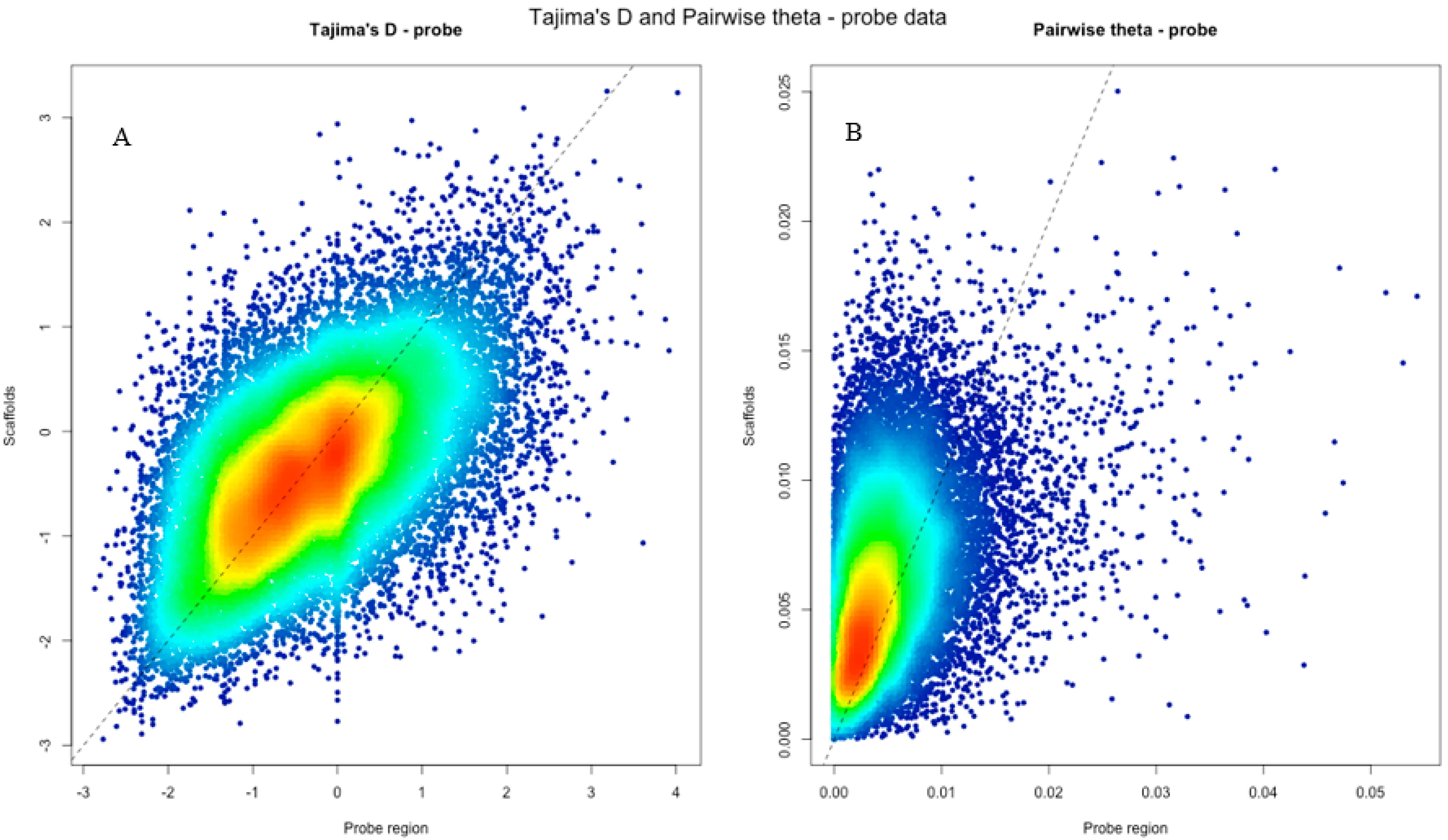
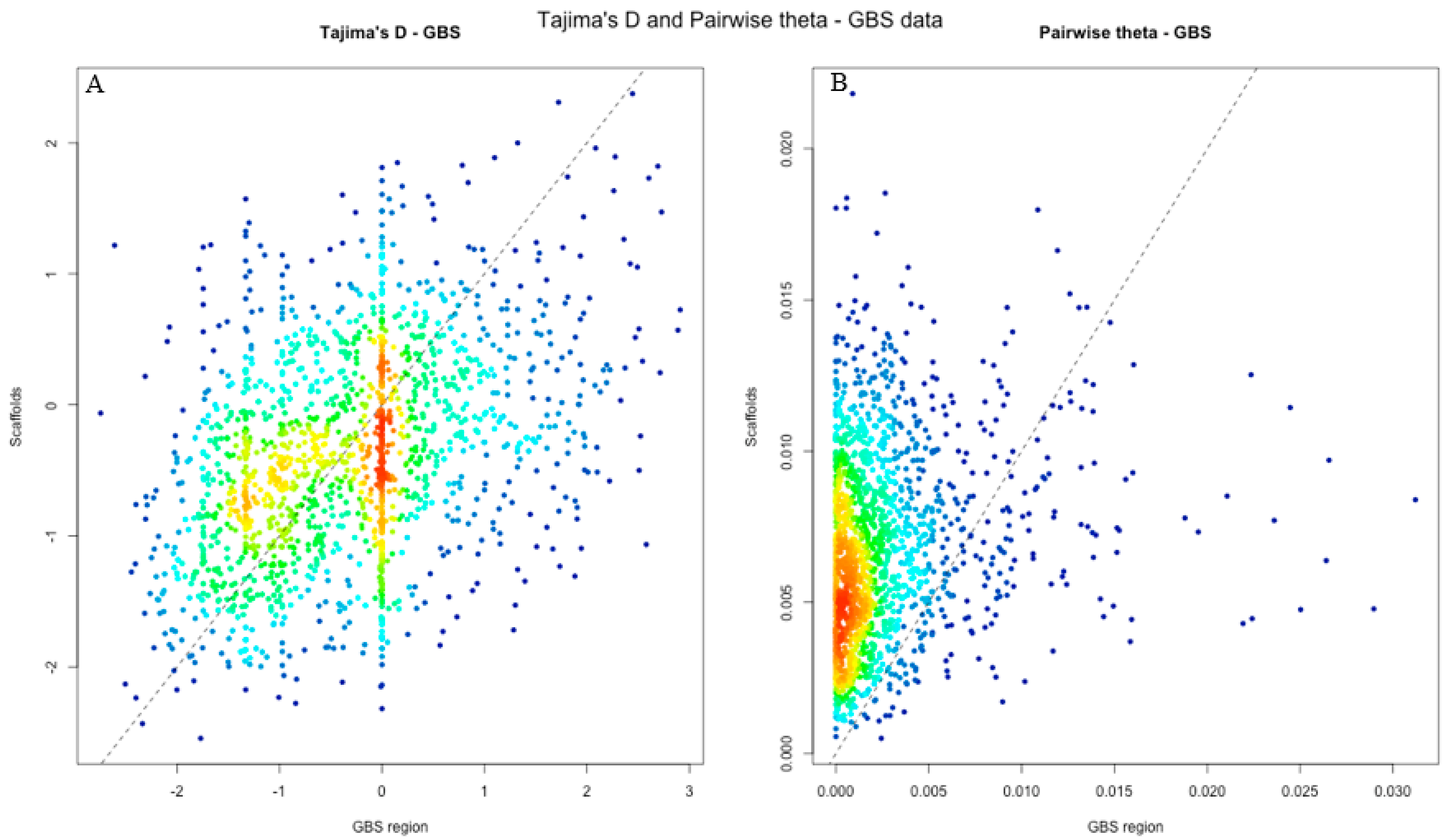
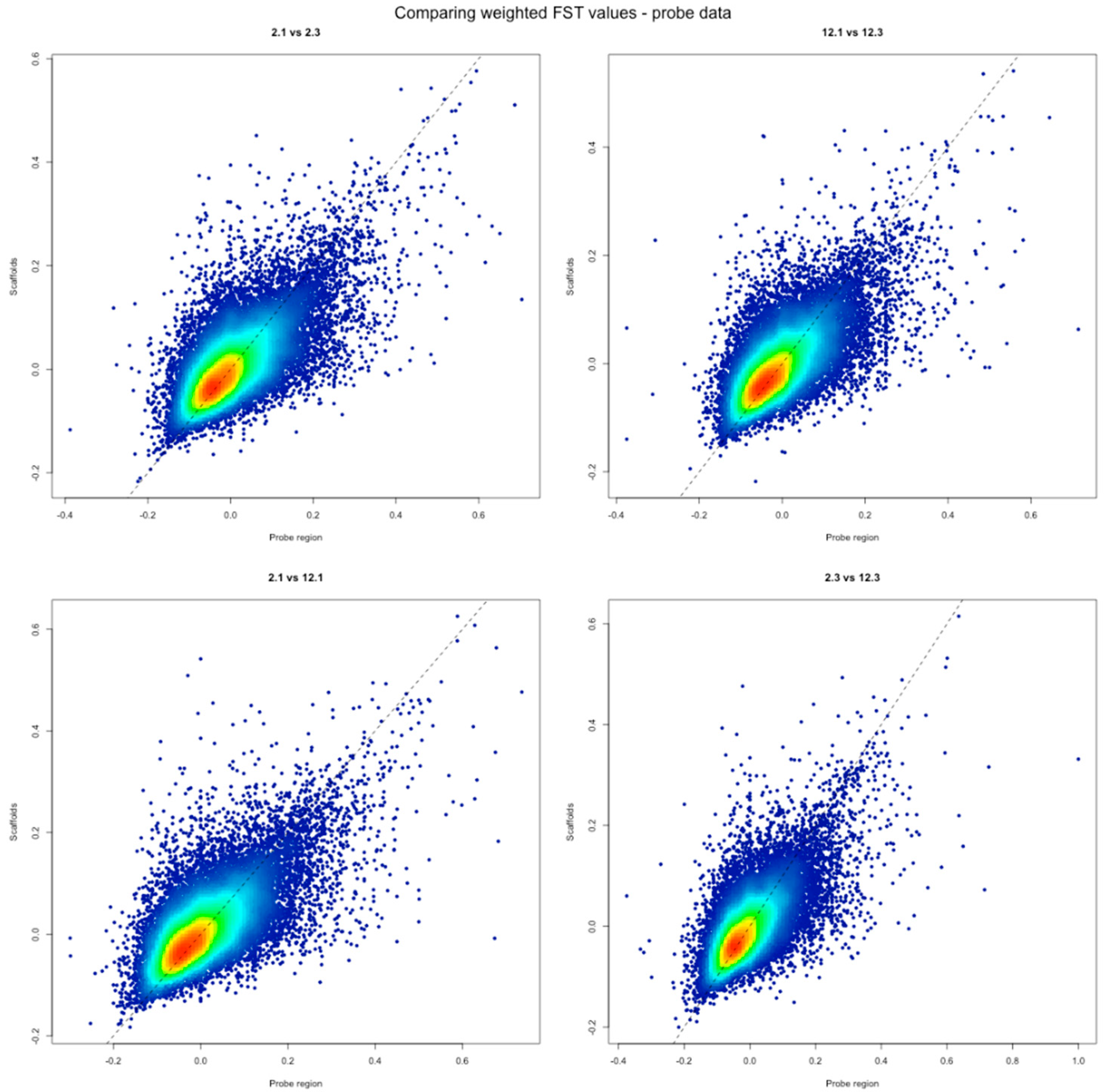
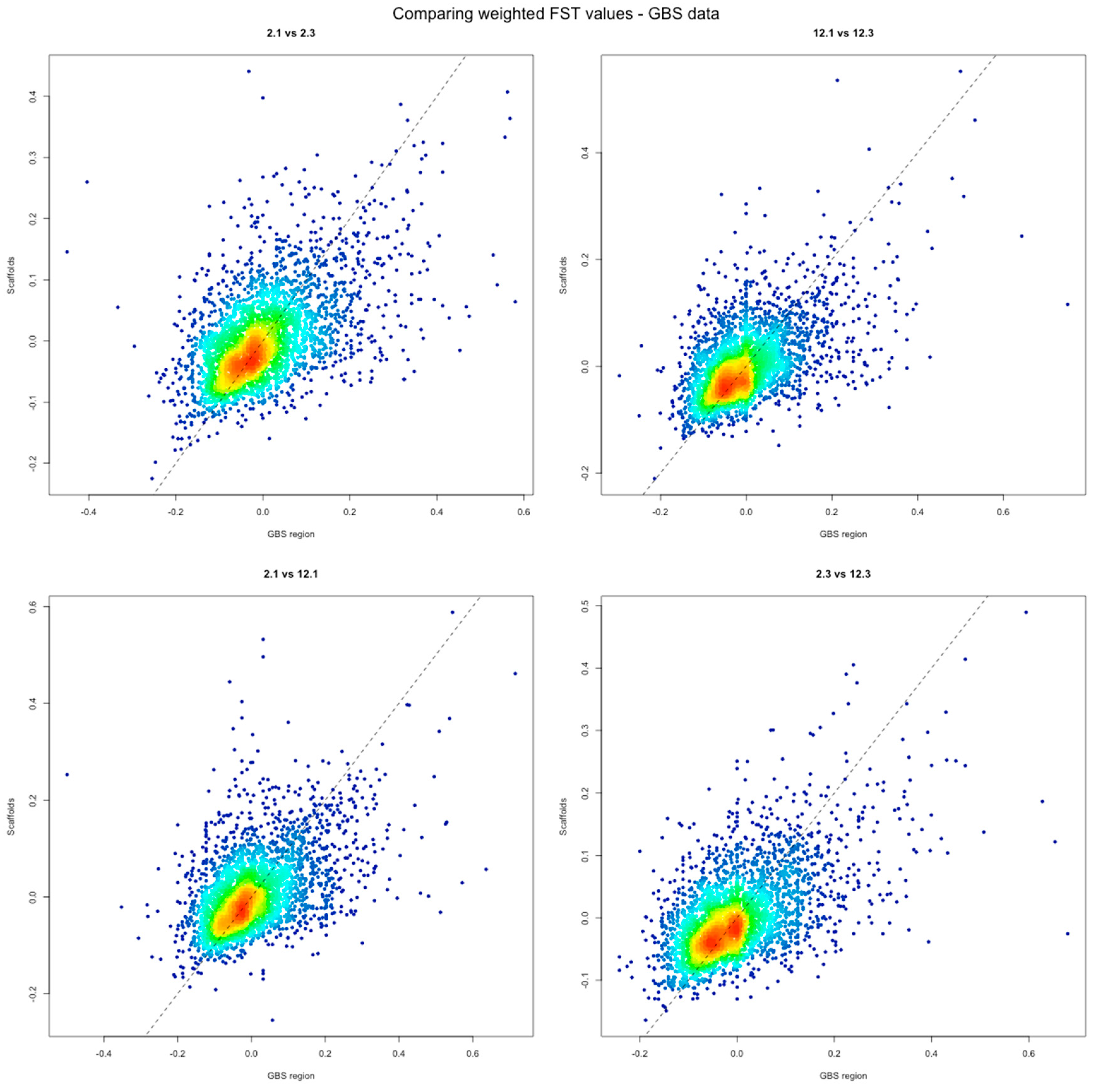
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Källman, T. Adaptive Evolution and Demographic History of Norway Spruce (Picea abies). Ph.D. Thesis, Uppsala University, Uppsala, Sweden, 2009. [Google Scholar]
- Farjón, A. Pinaceae: Drawings and Descriptions of the Genera Abies, Cedrus, Pseudolarix, Keteleeria, Nothotsuga, Tsuga, Cathaya, Pseudotsuga, Larix and Picea; Koeltz Scientific Books: Koönigstein, Germany, 1990. [Google Scholar]
- Burczyk, J.; Lewandowski, A.; Chalupka, W. Local pollen dispersal and distant gene flow in Norway spruce (Picea abies [L.] Karst.). For. Ecol. Manag. 2004, 197, 39–48. [Google Scholar] [CrossRef]
- Lindgren, D.; Karlsson, B.; Andersson, B.; Prescher, F. Swedish seed orchards for Scots pine and Norway spruce. In Proceedings of the a Seed Orchard Conference, Umeå, Sweden, 26–28 September 2007; pp. 26–28. Available online: http://daglindgren.upsc.se/Umea07/Umea07.htm (accessed on 6 November 2020).
- Lindgren, D.; Prescher, F. Optimal clone number for seed orchards with tested clones. Silvae Genet. 2005, 54, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Adams, W.T.; Burczyk, J. Magnitude and implications of gene flow in gene conservation reserves. In forest Conservation Genetics: Principles and Practice; Young, A., Boshier, D., Boyle, T., Eds.; CSIRO Publishing: Canberra, Australia, 2000; pp. 215–244. [Google Scholar]
- Pakkanen, A.; Nikkanen, T.; Pulkkinen, P. Annual variation in pollen contamination and outcrossing in a Picea abies seed orchard. Scand. J. For. Res. 2000, 15, 399–404. [Google Scholar] [CrossRef]
- Paule, L.; Lindgren, D.; Yazdani, R. Allozyme frequencies, outcrossing rate and pollen contamination in Picea abies seed orchards. Scand. J. For. Res. 1993, 8, 8–17. [Google Scholar] [CrossRef]
- Rosvall, O.; Almqvist, C.; Lindgren, D.; Hallander, J.; Berlin, M. Updates from Research on Selection and Mating Strategies. Review of the Swedish tree Breeding Programme. Skogforsk. 2011. Available online: https://www.skogforsk.se/kunskap/kunskapsbanken/2011/Utvardering-av-Skogforsks-foradlingsstrategi/ (accessed on 6 November 2020).
- Scotti, I.; Gugerli, F.; Pastorelli, R.; Sebastiani, F.; Vendramin, G.G. Maternally and paternally inherited molecular markers elucidate population patterns and inferred dispersal processes on a small scale within a subalpine stand of Norway spruce (Picea abies [L.] Karst.). For. Ecol. Manag. 2008, 255, 3806–3812. [Google Scholar] [CrossRef]
- Mehra, P.N.; Khoshoo, T.N. Cytology of conifers. I. J. Genet. 1956, 54, 165–180. [Google Scholar] [CrossRef]
- Morse, A.M.; Peterson, D.G.; Islam-Faridi, M.N.; Smith, K.E.; Magbanua, Z.; Garcia, S.A.; Kubisiak, T.L.; Amerson, H.V.; Carlson, J.E.; Nelson, C.D.; et al. Evolution of genome size and complexity in Pinus. PLoS ONE 2009, 4, e4332. [Google Scholar] [CrossRef] [Green Version]
- Neale, D.B.; Wheeler, N.C. The Conifers. In The Conifers: Genomes, Variation and Evolution; Springer: Cham, Switzerland, 2019; pp. 1–21. [Google Scholar]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Heuertz, M.; De Paoli, E.; Källman, T.; Larsson, H.; Jurman, I.; Morgante, M.; Lascoux, M.; Gyllenstrand, N. Multilocus patterns of nucleotide diversity, linkage disequilibrium and demographic history of Norway spruce [Picea abies (L.) Karst]. Genetics 2006, 174, 2095–2105. [Google Scholar] [CrossRef] [Green Version]
- Prunier, J.; Laroche, J.; Beaulieu, J.; Bousquet, J. Scanning the genome for gene SNPs related to climate adaptation and estimating selection at the molecular level in boreal black spruce. Mol. Ecol. 2011, 20, 1702–1716. [Google Scholar] [CrossRef]
- Acheré, V.; Favre, J.M.; Besnard, G.; Jeandroz, S. Genomic organization of molecular differentiation in Norway spruce (Picea abies). Mol. Ecol. 2005, 14, 3191–3201. [Google Scholar] [CrossRef] [PubMed]
- Gapare, W.J.; Aitken, S.N.; Ritland, C.E. Genetic diversity of core and peripheral Sitka spruce (Picea sitchensis (Bong.) Carr) populations: Implications for conservation of widespread species. Biol. Conserv. 2005, 123, 113–123. [Google Scholar] [CrossRef]
- Chen, C.; Mitchell, S.E.; Elshire, R.J.; Buckler, E.S.; El-Kassaby, Y.A. Mining conifers’ mega-genome using rapid and efficient multiplexed high-throughput genotyping-by-sequencing (GBS) SNP discovery platform. Tree Genet. Genomes 2013, 9, 1537–1544. [Google Scholar] [CrossRef]
- Karam, M.-J.; Lefèvre, F.; Dagher-Kharrat, M.B.; Pinosio, S.; Vendramin, G.G. Genomic exploration and molecular marker development in a large and complex conifer genome using RADseq and mRNAseq. Mol. Ecol. Resour. 2015, 15, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Wang, B.; Pei, Z.Y.; Zhao, W.; Gao, J.; Mao, J.F.; Wang, X.R. Optimization of the genotyping-by-sequencing strategy for population genomic analysis in conifers. Mol. Ecol. Resour. 2015, 15, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Vidalis, A.; Scofield, D.G.; Neves, L.G.; Bernhardsson, C.; García-Gil, M.R.; Ingvarsson, P.K. Design and evaluation of a large sequence-capture probe set and associated SNPs for diploid and haploid samples of Norway spruce (Picea abies). bioRxiv 2018, 291716. [Google Scholar] [CrossRef] [Green Version]
- De La Torre, A.R.; Puiu, D.; Crepeau, M.W.; Stevens, K.; Salzberg, S.L.; Langley, C.H.; Neale, D.B. Genomic architecture of complex traits in loblolly pine. New Phytol. 2019, 221, 1789–1801. [Google Scholar] [CrossRef]
- Wang, X.; Bernhardsson, C.; Ingvarsson, P.K. Demography and natural selection have shaped genetic variation in the widely distributed conifer Norway spruce (Picea abies). Genome Biol. Evol. 2020, 12, 3803–3817. [Google Scholar] [CrossRef]
- Bernhardsson, C.; Wang, X.; Eklöf, H.; Ingvarsson, P.K. Variant calling using whole genome resequencing and sequence capture for population and evolutionary genomic inferences in Norway Spruce (Picea abies). In The Spruce Genome. Compendium of Plant Genomes; Porth, I., De la Torre, A., Eds.; Springer: Cham, Switzerland, 2020; pp. 9–36. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Baison, J.; Vidalis, A.; Zhou, L.; Chen, Z.-Q.; Li, Z.; Sillanpää, M.J.; Bernhardsson, C.; Scofield, D.; Forsberg, N.; Grahn, T.; et al. Genome-wide association study (GWAS) identified novel candidate loci affecting wood formation in Norway spruce. Plant J. 2019, 100, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catchen, J.M.; Amores, A.; Hohenlohe, P.; Cresko, W.; Postlethwait, J.H. Stacks: Building and genotyping loci de novo from short-read sequences. G3 2011, 1, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Manichaikul, A.; Mychaleckyj, J.C.; Rich, S.S.; Daly, K.; Sale, M.; Chen, W.M. Robust relationship inference in genome-wide association studies. Bioinformatics 2010, 26, 2867–2873. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017; Available online: https://www.R-project.org/ (accessed on 6 November 2020).
- Korneliussen, T.S.; Albrechtsen, A.; Nielsen, R. ANGSD: Analysis of next generation sequencing data. BMC Bioinform. 2014, 15, 356. [Google Scholar] [CrossRef] [Green Version]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar]
- Korneliussen, T.S.; Moltke, I.; Albrechtsen, A.; Nielsen, R. Calculation of Tajima’s D and other neutrality test statistics from low hh next-generation sequencing data. BMC Bioinform. 2013, 14, 289. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Li, L.; Milesi, P.; Jansson, G.; Berlin, M.; Karlsson, B.; Aleksic, J.; Vendramin, G.G.; Lascoux, M. Genomic data provides new insights on the demographic history and the extent of recent material transfers in Norway spruce. Evol. Appl. 2019, 12, 1539–1551. [Google Scholar] [CrossRef] [Green Version]
- Shimono, A.; Wang, X.R.; Torimaru, T.; Lindgren, D.; Karlsson, B. Spatial variation in local pollen flow and mating success in a Picea abies clone archive and their implications for a novel “breeding without breeding” strategy. Tree Genet. Genomes 2011, 7, 499–509. [Google Scholar] [CrossRef]
- Fellers, J.P. Genome filtering using methylation-sensitive restriction enzymes with six base pair recognition sites. Plant Genome 2008, 1, 146–152. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Location | Population |
|---|---|---|
| Pab002 | Gettinge, Sweden | - |
| Pab003 | Vitebsk, Belarus | - |
| Pab004 | Blizyn, Poland | - |
| Pab005 | Toplita, Romania | - |
| Pab006 | Köttsjön, Sweden | - |
| Pab007 | Hatfjelldal, Norway | - |
| Pab008 | Rovaniemen, Finland | - |
| Pab009 | Kittilä, Finland | - |
| Pab010 | Suomussalmi, Finland | - |
| Pab011 | Hemnes, Norway | - |
| Pab012 | Levanger, Norway | - |
| Pab013 | Grane, Norway | - |
| Pab014 | Tyda, Norway | - |
| Pab015 | Loppi, Sweden | - |
| Pab016 | Marsfjället Planted | 12.3 |
| Pab017 | Marsfjället Planted | 12.3 |
| Pab018 | Marsfjället Planted | 12.3 |
| Pab019 | Marsfjället Planted | 12.3 |
| Pab020 | Marsfjället Planted | 12.3 |
| Pab021 | Marsfjället Old | 12.1 |
| Pab022 | Marsfjället Old | 12.1 |
| Pab023 | Marsfjället Old | 12.1 |
| Pab024 | Marsfjället Old | 12.1 |
| Pab025 | Marsfjället Old | 12.1 |
| Pab026 | Långrumpskogen Planted | 2.3 |
| Pab027 | Långrumpskogen Planted | 2.3 |
| Pab028 | Långrumpskogen Planted | 2.3 |
| Pab029 | Långrumpskogen Planted | 2.3 |
| Pab030 | Långrumpskogen Planted | 2.3 |
| Pab031 | Långrumpskogen Old | 2.1 |
| Pab032 | Långrumpskogen Old | 2.1 |
| Pab033 | Långrumpskogen Old | 2.1 |
| Pab034 | Långrumpskogen Old | 2.1 |
| Pab035 | Långrumpskogen Old | 2.1 |
| Probes | GBS | |||
|---|---|---|---|---|
| Target Regions | Scaffolds | Target Regions | Scaffolds | |
| Regions | 40,018 | - | 8731 | - |
| Mean size (bp) | 320 | 22,778 | 140 | 33,335 |
| Total size (bp) | 12,792,815 | 567,596,728 | 1,172,909 | 171,111,561 |
| Unique scaffolds | 24,919 | - | 5133 | - |
| Coding | 56.1% | 8.5% | 20.8% | 2.5% |
| Repeats | 1.8% | 27.5% | 19.0% | 38.2% |
| Subset | Pairwise Theta | Tajimas’D |
|---|---|---|
| Probe scaffolds | 0.0056 (0.0032) | −0.376 (0.84) |
| Probe regions | 0.0040 (0.0044) | −0.36 (1.017) |
| GBS scaffolds | 0.0063 (0.0031) | −0.393 (0.792) |
| GBS regions | 0.0014 (0.0034) | −0.311 (1.003) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eklöf, H.; Bernhardsson, C.; Ingvarsson, P.K. Comparing the Effectiveness of Exome Capture Probes, Genotyping by Sequencing and Whole-Genome Re-Sequencing for Assessing Genetic Diversity in Natural and Managed Stands of Picea abies. Forests 2020, 11, 1185. https://doi.org/10.3390/f11111185
Eklöf H, Bernhardsson C, Ingvarsson PK. Comparing the Effectiveness of Exome Capture Probes, Genotyping by Sequencing and Whole-Genome Re-Sequencing for Assessing Genetic Diversity in Natural and Managed Stands of Picea abies. Forests. 2020; 11(11):1185. https://doi.org/10.3390/f11111185
Chicago/Turabian StyleEklöf, Helena, Carolina Bernhardsson, and Pär K. Ingvarsson. 2020. "Comparing the Effectiveness of Exome Capture Probes, Genotyping by Sequencing and Whole-Genome Re-Sequencing for Assessing Genetic Diversity in Natural and Managed Stands of Picea abies" Forests 11, no. 11: 1185. https://doi.org/10.3390/f11111185
APA StyleEklöf, H., Bernhardsson, C., & Ingvarsson, P. K. (2020). Comparing the Effectiveness of Exome Capture Probes, Genotyping by Sequencing and Whole-Genome Re-Sequencing for Assessing Genetic Diversity in Natural and Managed Stands of Picea abies. Forests, 11(11), 1185. https://doi.org/10.3390/f11111185