Assessment of Genetic Diversity of Tea Germplasm for Its Management and Sustainable Use in Korea Genebank

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA Extraction
2.3. SSR Genotyping
2.4. Population Structure and Genetic Diversity
2.5. Estimation of Reproduction Mode among 410 Korean Tea Accessions
3. Results
3.1. Regional Distribution of 410 Tea Accessions in South Korea
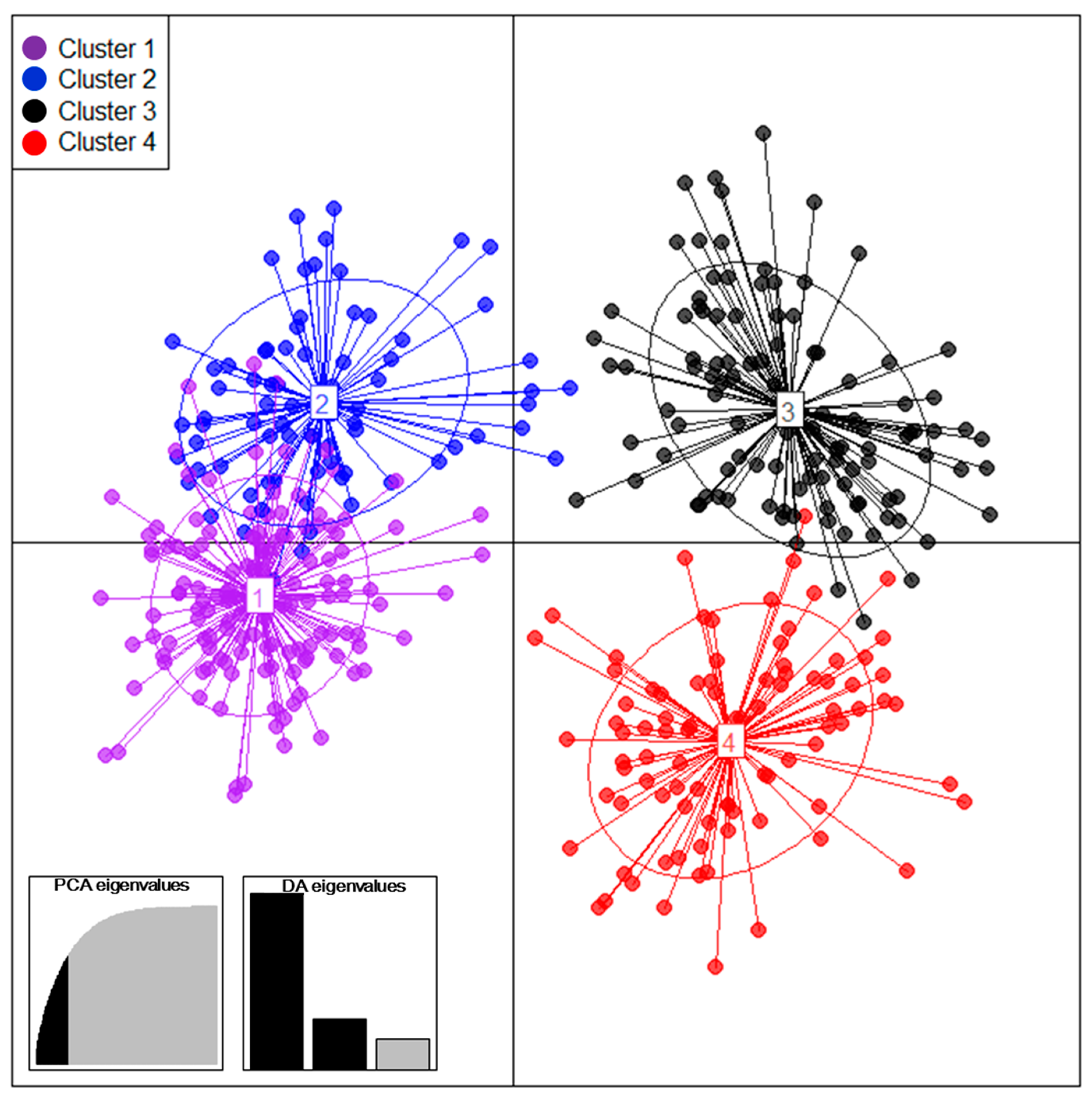
3.2. Population Structure and Mode of Reproduction of 410 Tea Accessions
3.3. Genetic Diversity
3.4. Gene Flow
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Karak, T.; Bhagat, R.M. Trace Elements in Tea Leaves, Made Tea and Tea Infusion: A Review. Food Res. Int. 2010, 43, 2234–2252. [Google Scholar] [CrossRef]
- Wambulwa, M.C.; Meegahakumbura, M.K.; Kamunya, S.; Muchugi, A.; Möller, M.; Liu, J.; Xu, J.-C.; Li, D.-Z.; Gao, L.-M. Multiple origins and a narrow genepool characterise the African tea germplasm: Concordant patterns revealed by nuclear and plastid DNA markers. Sci. Rep. 2017, 7, 4053. [Google Scholar] [CrossRef] [PubMed]
- Karunarathna, K.H.T.; Mewan, K.M.; Weerasena, O.V.D.S.J.; Perera, S.A.C.N.; Edirisinghe, E.N.U.; Jayasoma, A.A. Understanding the genetic relationships and breeding patterns of Sri Lankan tea cultivars with genomic and EST-SSR markers. Sci. Hortic. 2018, 240, 72–80. [Google Scholar] [CrossRef]
- Wight, W. Nomenclature and Classification of the Tea Plant. Nature 1959, 183, 1726–1728. [Google Scholar] [CrossRef]
- Matsumoto, S.; Kiriiwa, Y.; Yamaguchi, S. The Korean tea plant (Camellia sinensis): RFLP analysis of genetic diversity and relationship to Japanese tea. Breed. Sci. 2004, 54, 231–237. [Google Scholar] [CrossRef]
- Thomas, T.A.; Mathur, P.N. Germplasm Evaluation and Utilzation. In Plant Genetic Resources: Conservation and Management. Concepts and Approaches; Paroda, R.S., Arora, R.K., Eds.; IBPGR Regional Office: New Delhi, India, 1991. [Google Scholar]
- Wambulwa, M.C.; Meegahakumbura, M.K.; Samson, K.; Alice, M.; Michael, M.; Liu, J.; Xu, J.C.; Sailesh, R.; Li, D.Z.; Gao, L.M.; et al. Insights into the genetic relationships and breeding patterns of the african tea germplasm based on nSSR markers and cpDNA sequences. Front. Plant Sci. 2016, 7, 1244. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, C.; Li, Y.; Ding, Z.; Shen, J.; Wang, Y.; Zhao, L.; Xu, M.; Li, Y.; Chen, C.; et al. The identification and evaluation of two different color variations of tea. J. Sci. Food Agric. 2016, 96, 4951–4961. [Google Scholar] [CrossRef]
- Feng, L.; Gao, M.J.; Hou, R.Y.; Hu, X.Y.; Zhang, L.; Wan, X.C.; Wei, S.; Feng, L.; Gao, M.J.; Hou, R.Y.; et al. Determination of quality constituents in the young leaves of albino tea cultivars. Food Chem. 2014, 155, 98–104. [Google Scholar] [CrossRef]
- Gunasekare, M.T.K. Applications of molecular markers to the genetic improvement of Camellia sinensis L. (tea)—A review. J. Pomol. Hort. Sci. 2007, 82, 161–169. [Google Scholar]
- Chen, L.; Zhou, Z.X.; Chen, L.; Zhou, Z.X. Variations of main quality components of tea genetic resources [Camellia sinensis (L.) O. Kuntze] preserved in the China National Germplasm Tea Repository. Plant Foods Hum. Nutr. 2005, 60, 31–35. [Google Scholar] [CrossRef]
- Chen, L.; Yamaguchi, S.; Chen, L.; Yamaguchi, S. RAPD markers for discriminating tea germplasms at the inter-specific level in China. Plant Breed. 2010, 124, 404–409. [Google Scholar] [CrossRef]
- Fang, W.; Cheng, H.; Duan, Y.; Jiang, X.; Li, X. Genetic diversity and relationship of clonal tea (Camellia sinensis) cultivars in China as revealed by SSR markers. Plant Syst. Evol. 2012, 298, 469–483. [Google Scholar] [CrossRef]
- Park, Y.-G.; Kaundun, S.S.; Zhyvoloup, A. Use of the bulked genomic DNA-based RAPD methodology to assess the genetic diversity among abandoned Korean tea plantations. Genet. Resour. Crop Evol. 2002, 49, 159–165. [Google Scholar]
- Lai, J.A.; Yang, W.C.; Hsiao, J.Y. An assessment of genetic relationships in cultivated tea clones and native wild tea in Taiwan using RAPD and ISSR markers. Bot. Bull. Acad. Sin. 2001, 42, 93–100. [Google Scholar]
- Oh, C.J.; Lee, S.; You, H.C.; Chae, J.G.; Han, S.S. Genetic Diversity of Wild Tea (Camellia sinensis L.) in Korea. Korean J. Plant Resour. 2008, 21, 41–46. [Google Scholar]
- Korir, N.K.; Han, J.; Shangguan, L.; Wang, C.; Kayesh, E.; Zhang, Y.; Fang, J. Plant variety and cultivar identification: Advances and prospects. Crit. Rev. Biotechnol. 2013, 33, 111–125. [Google Scholar] [CrossRef]
- Jeong, B.-C.; Park, Y.-G. Tea Plant (Camellia sinensis) Breeding in Korea. In Global Tea Breeding: Achievements, Challenges and Perspectives; Springer: Berlin/Heidelberg, Germany, 2012; pp. 263–288. [Google Scholar]
- Gai, Z.; Wang, Y.; Jiang, J.; Xie, H.; Ding, Z.; Ding, S.; Wang, H. The quality evaluation of tea (Camellia sinensis) varieties based on the metabolomics. Horticulture 2019, 54, 409. [Google Scholar] [CrossRef]
- Chen, L.; Yao, M.; Zhao, L.P.; Wang, X. Recent Research Progresses on Molecular Biology of Tea Plant (Camellia sinensis). In Floriculture, Ornamental and Plant Biotechnology: Advances and Topical Issues, 1st ed.; da Silva, J.A.T., Ed.; Global Science Books: London, UK, 2006; Volume 4, pp. 425–436. [Google Scholar]
- Kaundun, S.S.; Zhyvoloup, A.; Park, Y.-G. Evaluation of the genetic diversity among elite tea (Camellia sinensis var. sinensis) accessions using RAPD markers. Euphytica 2000, 115, 7–16. [Google Scholar] [CrossRef]
- Yao, M.-Z.; Ma, C.-L.; Qiao, T.-T.; Jin, J.-Q.; Chen, L. Diversity distribution and population structure of tea germplasms in China revealed by EST-SSR markers. Tree Genet. Genomes 2012, 8, 205–220. [Google Scholar] [CrossRef]
- Jeong, B.-C.; Song, Y.S.; Moon, Y.H.; Han, S.K.; Bang, J.K. Collection and Multiplication of Superior Germplasm for Development of the New Tea Tree Cultivar; Joint Research Report for Tea germplasm in Korea; RDA: Mokpo, Korea, 2007; pp. 1–46. [Google Scholar]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ. 2014, 2, e281. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research±an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Ivandic, V.; Hackett, C.A.; Nevo, E.; Keith, R.; Thomas, W.T.B.; Forster, B.P. Analysis of simple sequence repeats (SSRs) in wild barley from the Fertile Crescent: Associations with ecology, geography and flowering time. Plant Mol. Biol. 2002, 48, 511–527. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T.; Collins, C. A tutorial for Discriminant Analysis of Principal Components (DAPC) using adegenet 2.0.0. Available online: http://adegenet.r-forge.r-project.org/files/tutorial-dapc.pdf (accessed on 6 September 2019).
- Grunwald, N.J.; Kamvar, Z.N.; Everhart, S.E.; Tabima, J.F.; Knaus, B.J. Population Genetic and Genomics in R. Available online: http://grunwaldlab.github.io/Population_Genetics_in_R/ (accessed on 3 April 2019).
- Agapow, P.-M.; Burt, A. Indices of multilocus linkage disequilibrium. Mol. Ecol. Notes 2001, 1, 101–102. [Google Scholar] [CrossRef]
- van de Wouw, M.; Kik, C.; van Hintum, T.; van Treuren, R.; Visser, B. Genetic erosion in crops: Concept, research results and challenges. Plant Genet. Resour. 2010, 8, 1–15. [Google Scholar] [CrossRef]
- van Heerwaarden, J.; Hellin, J.; Visser, R.F.; van Eeuwijk, F.A. Estimating maize genetic erosion in modernized smallholder agriculture. Theor. Appl. Genet. 2009, 119, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.G.; Sa, K.J.; Lee, J.R.; Lee, J.K. Genetic analysis of maize germplasm in the Korean Genebank and association with agronomic traits and simple sequence repeat markers. Genes Genom. 2017, 39, 843–853. [Google Scholar] [CrossRef]
- Gepts, P. Plant genetic resources conservation and utilization: The accomplishments and future of a societal insurance policy. Crop Sci. 2006, 46, 2278–2292. [Google Scholar] [CrossRef]
- Eom, B.-C.; Kim, J.-W. Phytocoenosen and Distribution of a Wild Tea (Camellia sinensis (L.) Kuntze) Population in South Korea. Korean J. Plant Resour. 2017, 30, 176–190. [Google Scholar] [CrossRef][Green Version]
- Harris, A.M.; DeGiorgio, M. An Unbiased Estimator of Gene Diversity with Improved Variance for Samples Containing Related and Inbred Individuals of any Ploidy. G3 Genes Genom. Genet. 2017, 7, 671–691. [Google Scholar] [CrossRef] [PubMed]
- Balloux, F.; Lehmann, L.; de Meeûs, T. The Population Genetics of Clonal and Partially Clonal Diploids. Genetics 2003, 164, 1635–1644. [Google Scholar] [PubMed]
- Birky, C.W., Jr. Heterozygosity, heteromorphy, and phylogenetic trees in asexual eukaryotes. Genetics 1996, 144, 427–437. [Google Scholar] [PubMed]
- Halkett, F.; Simon, J.C.; Balloux, F. Tackling the population genetics of clonal and partially clonal organisms. Trends Ecol. Evol. 2005, 20, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.S.; Moon, Y.H.; Han, S.K.; Jeong, B.C.; Bang, J.K. Morphological characteristics of progeny population in collected wild tea (Camellia sinensis). J. Korean Tea Soc. 2005, 11, 93–105. [Google Scholar]
- Campoy, J.A.; Lerigoleur-Balsemin, E.; Christmann, H.; Beauvieux, R.; Girollet, N.; Quero-García, J.; Dirlewanger, E.; Barreneche, T. Genetic diversity, linkage disequilibrium, population structure and construction of a core collection of Prunus avium L. landraces and bred cultivars. BMC Plant Biol. 2016, 16, 49. [Google Scholar] [CrossRef] [PubMed]
- Deperi, S.I.; Tagliotti, M.E.; Bedogni, M.C.; Manrique-Carpintero, N.C.; Coombs, J.; Zhang, R.; Douches, D.; Huarte, M.A. Discriminant analysis of principal components and pedigree assessment of genetic diversity and population structure in a tetraploid potato panel using SNPs. PLoS ONE 2018, 13, e0194398. [Google Scholar] [CrossRef]
- Rosyara, U.R.; De Jong, W.S.; Douches, D.S.; Endelman, J.B. Software for Genome-Wide Association Studies in Autopolyploids and Its Application to Potato. Plant Genome 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Choi, S. Studies of the aroma of Korean green tea. In Proceedings of the International Symposium of Tea Science; ISTS: Shizuoka, Japan, 1991; pp. 130–134. [Google Scholar]
- Hamrick, J. Isozymes and the analysis of genetic structure in plant populations. In Isozymes in Plant Biology; Soltis, D.E., Soltis, P.S., Dudley, T.R., Eds.; Springer: Dordrecht, The Netherlands, 1989; pp. 87–105. [Google Scholar]


{kind=link}
| Locus | Sequence (5′→ 3′) | Dye |
|---|---|---|
| MSG0258 | F: ACTCATCACCATGCCTTCTCCATC | FAM |
| R: GTTTAGCTCAACTGGTGGAACCTCAACT | ||
| MSE0173 | F: GTGTTCACCAACAACTCACCAAGG | FAM |
| R: TGTCGAACAAAGATACACCCCAAA | ||
| MSE0313 | F: TGCTATGCCGCCTAACAAAAACTT | FAM |
| R: ACCACCAACAACAATTCCCACTCT | ||
| MSG0403 | F: ATGATCGCCGGTTTAGAGATGAAT | FAM |
| R: GTTTAAGCTGGCTAACCTACACGGAGC | ||
| MSE0083 | F: GAGGAAAGAGATTATGCGGTGTGG | FAM |
| R: GTGAGCCTTCAAAAGACAGCAACG | ||
| TM604 | F: TCTCAATCAAGGTTCCGAGG | FAM |
| R: TGACATATTCGCCCAACAAA | ||
| TM530 | F: CCGTGTTTACCACCACCCT | FAM |
| R: CCCTGGGAACAAGAAAGTGA | ||
| MSG0361 | F: AGATGGAGGTAGAGAGAGAGGCAG | HEX |
| R: GTTTGTCCCTCTCATTTTCAACGC | ||
| MSE0029 | F: ATAGCCAATCAAGCTCCTCCTCCT | HEX |
| R: AGTCTGTTCCTCCCTTGATGATCG | ||
| MSG0610 | F: ACAGAGGAGGAAGATGATCGGTAA | HEX |
| R: GTTTGAAGAAGAAGAAAACTCCCGCCAT | ||
| MSE0291 | F: AATCAAATAACACTTGCACCCGC | HEX |
| R: AAAAAGAGAAAGTCACGTCCACGG | ||
| MSG0681 | F: AGGGTTTGCGTCTTCAAAGAGAGA | HEX |
| R: GTTTGTAACACTTGCCACGTTTCG | ||
| MSG0470 | F: ATAGGGTTCGAAAATGGCAGG | HEX |
| R: GTTTGAGGTGGCAAGTTTGTGACTGT | ||
| MSG0699 | F: ATGCGACAGTGTTGCTGAGATTTT | HEX |
| R: GTTTCAAAAATGGGGTGTCTACAGAGGG | ||
| MSG0429 | F: AGGACCGTTCTTCCCTACCTGTAA | NED |
| R: GTTTGAGATTGAGGATGTGGTCGTTGT | ||
| MSG0380 | F: ACAGACCTTCACCCTCTCCATTTC | NED |
| R: GTTTACCTCTGCCTTCGTTCTTCAGC | ||
| MSG0423 | F: ACTCCATGTGCTGCTCTGTAGTTC | NED |
| R: GTTTGCAGGAAGTTGAGCCAGAC | ||
| MSE0237 | F: CTCTCCTTCTTCACACCCTCCAAA | NED |
| R: TTGTTCTCAAAGAACCTCCTTCGC | ||
| MSE0113 | F: TACCTTCTGCAACTCCAGCAATCC | NED |
| R: TGAGATTGACCATCTTTCATCGGA | ||
| MSE0107 | F: TCTCTCTACTCCTGCGCAATCTCA | NED |
| R: TCAAAGATGTTGCTCTCGTCAACC | ||
| TM382 | F: TCTCAAAACCAAATAGGCTCAA | NED |
| R: TTGCGTTATGATTTCTGGGA |
| Location | Cluster 1 | Total (%) | |||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | ||
| GN 2 | |||||
| Gimhae | 1 | 1 | 3 | 1 | 6 (1.5) |
| Namhae | 1 | 2 | 3 (0.7) | ||
| Sacheon | 2 | 1 | 3 (0.7) | ||
| Sancheong | 2 | 1 | 3 (0.7) | ||
| Yangsan | 2 | 2 | 1 | 1 | 6 (1.5) |
| Jinju | 2 | 1 | 1 | 4 (1.0) | |
| Hadong | 2 | 1 | 1 | 4 (1.0) | |
| Subtotal | 6 | 7 | 9 | 7 | 29 (7.1) |
| JN | |||||
| Gangjin | 6 | 2 | 5 | 11 | 24 (5.9) |
| Goheung | 2 | 1 | 3 (0.7) | ||
| Gokseong | 1 | 6 | 2 | 9 (2.2) | |
| Gwangyang | 2 | 1 | 2 | 5 (1.2) | |
| Gwangju | 11 | 3 | 14 (3.4) | ||
| Gurye | 6 | 7 | 2 | 3 | 18(4.4) |
| Naju | 10 | 3 | 10 | 6 | 29 (7.1) |
| Damyang | 2 | 3 | 5 (1.2) | ||
| Mu’an | 7 | 1 | 1 | 2 | 11 (2.7) |
| Boseong | 28 | 17 | 16 | 16 | 77 (18.8) |
| Suncheon | 17 | 7 | 19 | 14 | 57 (13.9) |
| Yeosu | 1 | 3 | 3 | 3 | 10 (2.4) |
| Yeong’am | 4 | 5 | 1 | 2 | 12 (2.9) |
| Jangseong | 3 | 3 | 9 | 5 | 20 (4.9) |
| Jangheung | 3 | 2 | 3 | 8 (2.0) | |
| Jindo | 1 | 1 | 2 (0.5) | ||
| Hampyeong | 3 | 1 | 2 | 2 | 8 (2.0) |
| Haenam | 11 | 1 | 3 | 2 | 17 (4.1) |
| Hwasun | 11 | 4 | 4 | 2 | 21 (5.1) |
| Subtotal | 126 | 60 | 86 | 78 | 350 (85.4) |
| JB | |||||
| Gochang | 2 | 1 | 1 | 4 (1.0) | |
| Gimje | 1 | 1 | 1 | 3 (0.7) | |
| Sunchang | 2 | 3 | 5 (1.2) | ||
| Iksan | 1 | 1 (0.2) | |||
| Jeongeup | 1 | 3 | 3 | 1 | 8 (2.0) |
| Subtotal | 5 | 5 | 8 | 3 | 21 (5.1) |
| Unknown | 1 | 3 | 5 | 1 | 10 (2.4) |
| Total | 138 | 75 | 108 | 89 | 410 |
| Cluster | N a | MLG | λ | GD | Ia | rbarD | p-Value (rbarD) |
|---|---|---|---|---|---|---|---|
| C1 | 138 | 138 | 0.993 | 0.778 | 0.123 | 0.0062 | 0.018 |
| C2 | 75 | 75 | 0.987 | 0.805 | 0.554 | 0.0282 | 0.001 |
| C3 | 108 | 108 | 0.991 | 0.724 | 0.462 | 0.0235 | 0.001 |
| C4 | 89 | 88 | 0.989 | 0.695 | 0.999 | 0.0508 | 0.001 |
| Total | 410 | 409 | 0.998 | 0.792 | 1.154 | 0.0583 | 0.001 |
| Locus | C1 | C2 | C3 | C4 | Total |
|---|---|---|---|---|---|
| MSG0258 | 2 (2) | 1 (1) | 1 (1) | 1 (1) | 3 (5) |
| MSE0173 | 0 (0) | 2 (2) | 1 (1) | 0 (0) | 2 (3) |
| MSE0313 | 5 (5) | 1 (1) | 0 (0) | 1 (1) | 5 (7) |
| MSE0403 | 3 (3) | 2(3) | 1 (2) | 1 (1) | 6 (9) |
| MSE0083 | 6 (9) | 3 (4) | 1 (2) | 0 (0) | 6 (15) |
| TM604 | 1 (1) | 0 (0) | 0 (0) | 0 (0) | 1 (1) |
| TM530 | 0 (0) | 0 (0) | 0 (0) | 1 (1) | 1 (1) |
| MSG0361 | 0 (0) | 0 (0) | 0 (0) | 0(0) | 6 (0) |
| MSE0029 | 1 (3) | 1 (1) | 0 (0) | 0 (0) | 1 (4) |
| MSE0291 | 1 (4) | 1 (2) | 1 (1) | 1 (1) | 4 (8) |
| MSG0681 | 2 (2) | 1 (1) | 2 (2) | 0 (0) | 5 (5) |
| MSG0470 | 5 (7) | 2 (2) | 0 (0) | 0 (0) | 6 (9) |
| MSG0699 | 1 (1) | 1 (2) | 4 (4) | 1 (1) | 6 (2) |
| MSG0429 | 1 (3) | 0 (0) | 0 (0) | 1 (1) | 1 (4) |
| MSG0380 | 2 (2) | 2 (2) | 1 (1) | 0 (0) | 2 (5) |
| MSG0423 | 2 (3) | 1 (2) | 1 (1) | 1 (1) | 4 (7) |
| MSE0237 | 0 (0) | 1 (2) | 2 (4) | 0 (0) | 3 (6) |
| MSE0113 | 3 (5) | 4 (6) | 2 (2) | 2 (2) | 6 (15) |
| MSE0107 | 3 (3) | 4 (5) | 0 (0) | 0 (0) | 5 (8) |
| TM382 | 2 (2) | 1 (1) | 0 (0) | 2 (2) | 3 (5) |
| Total | 41 (56) | 30 (39) | 20 (24) | 16 (16) | 76 (119) |
| Source | df | SS | MS | Est. Var. | % | Value | p Value | |
|---|---|---|---|---|---|---|---|---|
| Among Pops | 3 | 201.375 | 67.125 | 0.388 | 1% | PhiPT | 0.014 | 0.001 |
| Within Pops | 406 | 11391.250 | 28.057 | 28.057 | 99% | Nm | 36.156 | |
| Total | 409 | 11592.624 | 28.445 | 100% |
| C1 | C2 | C3 | C4 | |
|---|---|---|---|---|
| C1 | - | 45.120 | 33.525 | 40.760 |
| C2 | 0.011 | - | 23.717 | 47.872 |
| C3 | 0.015 | 0.021 | - | 37.797 |
| C4 | 0.012 | 0.010 | 0.013 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.J.; Lee, J.-R.; Sebastin, R.; Shin, M.-J.; Kim, S.-H.; Cho, G.-T.; Hyun, D.Y. Assessment of Genetic Diversity of Tea Germplasm for Its Management and Sustainable Use in Korea Genebank. Forests 2019, 10, 780. https://doi.org/10.3390/f10090780
Lee KJ, Lee J-R, Sebastin R, Shin M-J, Kim S-H, Cho G-T, Hyun DY. Assessment of Genetic Diversity of Tea Germplasm for Its Management and Sustainable Use in Korea Genebank. Forests. 2019; 10(9):780. https://doi.org/10.3390/f10090780
Chicago/Turabian StyleLee, Kyung Jun, Jung-Ro Lee, Raveendar Sebastin, Myoung-Jae Shin, Seong-Hoon Kim, Gyu-Taek Cho, and Do Yoon Hyun. 2019. "Assessment of Genetic Diversity of Tea Germplasm for Its Management and Sustainable Use in Korea Genebank" Forests 10, no. 9: 780. https://doi.org/10.3390/f10090780
APA StyleLee, K. J., Lee, J.-R., Sebastin, R., Shin, M.-J., Kim, S.-H., Cho, G.-T., & Hyun, D. Y. (2019). Assessment of Genetic Diversity of Tea Germplasm for Its Management and Sustainable Use in Korea Genebank. Forests, 10(9), 780. https://doi.org/10.3390/f10090780