Abstract
The dove tree (Davidia involucrata Baill.), a tertiary relic species, is adapted to cool climates. With the progression of global warming, high-temperature stress has become the primary environmental factor restricting geographic distribution, ex situ conservation, and landscape application for D. involucrata resources. However, the detailed molecular events underlying D. involucrata responses to heat stress are poorly understood. Here, we conducted RNA-Seq-based gene expression profiling in D. involucrata seedlings during the time course of a 42 °C heat treatment (0, 1, 6, and 12 h). After de novo assembly, we obtained 138,923 unigenes, of which 69,743 were annotated in public databases. Furthermore, 19,532, 20,497 and 27,716 differentially expressed genes (DEGs) were identified after 1 h (HS1), 6 h (HS6), and 12 h (HS12) of heat treatment in comparison to 0 h (HS0), respectively. Based on a KEGG enrichment analysis, the two pathways “protein processing in endoplasmic reticulum” and “plant hormone signal transduction” are hypothesized to play vital roles during heat response in D. involucrata, and their potential interactions during heat stress are also discussed. In addition, 32 genes encoding putative heat shock transcription factors (Hsfs) were found to be associated with the response of D. involucrata to heat stress. Finally, the expression patterns of eight heat-responsive genes derived from qRT-PCR were in agreement with their transcript level alterations, as determined by a transcriptome analysis. Taken together, our transcriptomic data provide the first comprehensive transcriptional profile affected by heat stress in D. involucrata, which will facilitate further studies on the improvement of heat tolerance in this rare and endangered species.
1. Introduction
Davidia involucrata Baill., the sole species placed in the genus Davidia in the family Davidiaceae, is a well-known ornamental plant called the “dove tree.” It is an endangered and rare relict tree species endemic to China with prominent scientific, horticultural, ecological, and medicinal values [1,2,3,4]. Owing to the highly strict ecotope demand, the geographic distribution of D. involucrata populations is rare and scattered at high altitudes in southwestern and south-central China. D. involucrata is best adapted to cool climates, and high temperature is generally detrimental to its growth and development. Notably, it has been estimated that this species will be greatly vulnerable under future rapid climate warming [5]. For Davidia resources conservation, research has been undertaken on its introduction and ex situ cultivation since 1979 in China [6]. However, such research is not going smoothly, since high-temperature stress in summer is the major limiting factor for the efficient propagation of D. involucrata outside its natural habitats [7]. Previous studies of D. involucrata in relation to heat stress have mainly focused on physiological or biochemical alterations [8,9]. Presently, little information is available concerning the molecular regulatory mechanisms of D. involucrata when responding to heat stress.
High temperature can cause plant growth retardation and even death, which could become a critical issue in the near future due to global warming. Climate predictions suggest that, toward the end of the 21st century, the global average temperature is projected to be elevated by 1–4 °C [10]. Hence, a fundamental understanding of heat response mechanisms in plants is of vital importance for the effective and efficient conservation of rare and endangered species. In general, there are four main heat stress regimes: Direct applied heat stress (DAHS), which is used to examine basal thermotolerance; pre-induction heat stress (PIHS), which is used for acquired thermotolerance; gradually increasing heat stress (GIHS), which is used for determining acquired thermotolerance by imitating the natural condition; and mild chronic heat stress (MCHS), which is used to determine mild heat thermotolerance [11]. Plants respond to high-temperature stress through modulating the expression levels of numerous genes, thus changing physiological, cellular, biochemical, and metabolic processes. For instance, high temperatures typically induce the gene expression of heat shock proteins (Hsps), which act as molecular chaperones primarily engaged in the maintenance and/or restoration of protein homeostasis [12,13,14]. Based on their molecular weight, plant Hsps are classified into six major families: Hsp100 (Clp), Hsp90, Hsp70 (DnaK), Hsp60 (chaperonin and GroEL), Hsp40 (DnaJ) and small HSP (sHsp) [15]. Their functions have been thoroughly documented in heat response, during which Hsps are mainly transcriptionally controlled by heat shock transcription factors (Hsfs) [16,17]. Hsfs generally serve as the terminal components of a stress-triggered signal transduction chain, regulating the expression of downstream targets including HSP genes [18,19,20]. Following the initial characterization of three tomato Hsf genes [21,22,23], a lot of knowledge regarding plant Hsf function has been gained and comes predominantly from analyses of Hsfs in tomato (Solanum lycopersicum) and Arabidopsis thaliana [19,24]. Approximately 27 and 21 different Hsf members were identified in tomato and Arabidopsis, respectively [17,25]. These Hsfs have been differentiated into three classes (A, B and C), which are mainly based on the structural features of their oligomerization domains [16,17,18,19]. Each Hsf class is further divided into various sub-classes. Notably, the platform known as HEATSTER provides different webtools and datasets for the identification and classification of Hsfs in plants [17,26]. Apart from Hsps and Hsfs, many other regulators have been implicated in heat stress response, such as abscisic acid (ABA), auxin, salicylic acid (SA), ethylene, multiprotein bridging factor 1c (MBF1c), and FK506-binding proteins (FKBPs) [19,27,28,29]. There have been a lot of breakthroughs in the study of plant heat stress response; however, many functional genes involved in thermotolerance remain to be discovered and identified.
Transcriptomic studies have been widely adopted to dissect the molecular mechanisms by which plants adapt to adverse environmental conditions. In recent years, a broad spectrum of heat-responsive genes and/or pathways have been identified from various plant species by transcriptomic approaches. For instance, the de novo transcriptome sequencing and gene expression profiling of spinach (Spinacia oleracea) leaves under heat stress indicated that calcium signaling molecules and certain transcription factors are required for early heat response [30]. The comprehensive transcriptomic analysis of Korean fir (Abies koreana) revealed many Hsps and transcription factors as being differentially expressed upon heat treatment [31]. In addition to the previously described heat stress regulators, the transcriptional profiling of perennial ryegrass (Lolium perenne) under heat stress also identified four genes primarily involved in C4 carbon fixation as well as one target of rapamycin (TOR) gene [32]. In maize (Zea mays), a transcriptome analysis highlights the crucial roles of the protein processing in the endoplasmic reticulum pathway in heat stress response [33]. It has been suggested that heat tolerances of reproductive and vegetative tissues are distinct and do not always correlate; it has also been suggested that reproductive tissues are highly sensitive to heat stress and are directly associated with fruit and seed production [11]. Therefore, transcriptomic studies have also been applied to unravel molecular responses to heat stress in reproductive tissues at specific developmental stages. For example, an RNA-Seq analysis in rice floral organs revealed that the expressions of five pectinase genes are highly responsive to heat stress at the booting stage [34]. Furthermore, the analysis of the transcriptome and proteome during development and heat stress response of tomato pollen demonstrated that, with the exception of Hsfs and Hsps, protein levels under heat stress are modulated by the interplay between protein synthesis and degradation than by changes of transcript levels [35]. In D. involucrata, the mining of genes associated with seed abortion was enabled through a transcriptome analysis [36]. However, the transcriptomic response of D. involucrata to heat stress has not yet been characterized.
In the present study, heat stress-induced transcriptome changes in D. involucrata were investigated, and potential responsive genes and pathways were identified by RNA-sequencing (RNA-Seq). To our knowledge, this work presents the first transcriptome profiling of D. involucrata responding to high-temperature stress. These findings improve our understanding of the adaptation mechanisms of D. involucrata to environmental stress and will serve as valuable genetic resources to inform further studies regarding the improvement of heat tolerance in D. involucrata.
2. Materials and Methods
2.1. Sample Preparation and Heat Treatment
After stratification in moist sand for more than one year, D. involucrata seeds were sown in the field of Deyang nursery, Deyang city, Sichuan Province. Subsequently, one-year-old D. involucrata seedlings obtained from Deyang nursery were cultivated for one additional year in pots in the greenhouse of China West Normal University. Two-year-old seedlings at uniform stages of development were placed in growth chambers for a one-week adaptation with the following settings: Relative humidity 75%–85%, 22 °C/16 °C (day/night), and a 14-h photoperiod (lights from 7:00 AM to 9:00 PM) with 400 μmol s−1 m−2 light intensity. One week later, healthy mature leaves of approximately the same size were collected at 0, 1, 6, and 12 h after the onset of heat treatment (42 °C, starting at 8:00 AM). Three biological replicates were set for each time point. All harvested samples were immediately frozen in liquid nitrogen and subsequently stored in a −80 °C freezer until RNA extraction.
2.2. RNA Extraction, cDNA Library Preparation and Transcriptome Sequencing
Total RNA was separately extracted from each sample using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). RNA quality was analyzed with 1.0% agarose gel electrophoresis and using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). The cDNA libraries were generated from these RNA samples for Illumina sequencing according to the standard Illumina protocol. The library preparations were then subjected to sequencing on an Illumina HiSeq 4000 platform, and 150-bp paired-end reads were yielded. The sequencing raw data are available in NCBI Short Read Archive (SRA) database (accession: SRP186742).
2.3. Transcriptome Assembly and Functional Annotation
Raw reads/data in the FASTQ format were firstly processed using in-house perl scripts. In this step, clean reads/data were generated by filtering reads with adapter, reads in which unknown bases represented more than 10% as well as reads containing more than 50% of bases that had a Q-value ≤20 from raw data. At the same time, the Q20, Q30, GC-content and sequence duplication level of the clean data were calculated. All the downstream analyses were based on clean data with high quality. Since the genome information of D. involucrata remains unavailable, the resulting clean reads/data were used to de novo assemble the transcriptome with Trinity software [37]. The longest transcript of each gene was taken accordingly as the unigene. The putative functions of D. involucrata unigenes were annotated by searching against different public databases, including the protein family (Pfam), NCBI non-redundant protein sequences (Nr), Swiss-Prot, NCBI nucleotide sequences (Nt), Kyoto Encyclopedia of Genes and Genomes ortholog (KO), EuKaryotic Orthologous Groups (KOG), and Gene Ontology (GO) database.
2.4. Differential Expression Analysis
The read counts of each unigene in a sample were obtained through mapping clean reads back onto the assembled D. involucrata transcriptome by RSEM v1.2.15 [38], with the bowtie2 parameter set at mismatch 0. Differential expression analyses among distinct conditions/groups was implemented with the DESeq2 based on the read counts from the normalized RNA-Seq data [39]. DESeq2 provides statistical routines for determining differential expression in digital gene expression data using a model based on a negative binomial distribution [39]. The resulting P values were adjusted using the Benjamini and Hochberg’s approach for controlling the false discovery rate. The D. involucrata genes with an adjusted p-value of less than 0.05 detected by DESeq2 were deemed to be differentially expressed genes (DEGs). A GO functional enrichment analysis of DEGs was carried out using the GOseq R package based on a Wallenius non-central hyper-geometric distribution [40]. For the KEGG pathways analysis, the KOBAS software was utilized to determine the statistical enrichment of the DEGs [41].
2.5. qRT-PCR Analysis
To verify the reliability of our transcriptome sequencing data, eight heat-responsive D. involucrata genes were selected for a quantitative real-time PCR (qRT-PCR) analysis. qRT-PCR was conducted with gene-specific primers (Table S1). DiActin was used as an internal control for normalization [36]. The reactions were carried out using StepOnePlus Real-Time PCR system (Applied Biosystems, Foster, CA, USA) with the SYBR Premix Ex Taq Kit (TaKaRa, Dalian, China). Each reaction was performed in triplicate. The melt curve was generated to validate the specificity of amplification. The relative gene expression was calculated based on the 2−ΔΔCt method [42].
3. Results
3.1. Illumina Sequencing and Transcriptome Assembly
To unravel the molecular mechanisms underlying D. involucrata responses to heat stress, we prepared twelve cDNA libraries from plant samples grown for 0 h (HS0), 1 h (HS1), 6 h (HS6) and 12 h (HS12) at 42 °C. A total of 621,415,654 raw reads were yielded from these libraries (Table S2). After quality trimming, 599,448,362 clean reads were generated. Subsequently, the resulting clean reads were assembled to generate 405,798 transcripts (Table 1). In addition, we further clustered these transcripts into 138,923 unigenes, ranging from 201 to 17,633 bp in length (Table 1). The mean length and N50 value of the unigenes were 941 bp and 1493 bp, respectively (Table 1).

Table 1.
Length distribution of unigenes and transcripts.
3.2. Functional Annotation and Classification of D. involucrata Unigenes
Next, all of the assembled unigenes were verified and annotated by searching against various public databases (Table S3). Among them, 57,030 unigenes (41.05%) had significant matches in Nr, 37,370 (26.89%) in Nt, and 37,384 (26.9%) in Pfam. Altogether, 69,743 unigenes (50.2%) were annotated to at least one database, and 6087 unigenes (4.38%) were annotated in all databases.
Of the 138,923 assembled unigenes, 37,384 unigenes (26.9%) could be aligned to the GO database using Blast2GO (e-value = 1 × 10−6) (Figure 1). “Cellular process” (20,699 unigenes) and “metabolic process” (18,948 unigenes) were the most highly represented terms within the biological process category, indicating relatively high levels of basic metabolic activity in the heat-treated D. involucrata leaves. In the cellular component category, “cell” (11,586 unigenes) and “cell part” (11,586 unigenes) were found to be the most enriched classes. With respect to the molecular function category, “binding” (19,041 unigenes) and “catalytic activity” (14,935 unigenes) were mostly assigned. Consistent with our results, the aforementioned most highly represented GO terms were also observed in transcriptome analyses of other plant species under heat stress [31,32,43]. Those groups of “nucleic acid binding transcription factor activity” (1097 unigenes), “signaling” (2374 unigenes), “transporter activity” (2468 unigenes), and “response to stimulus” (4699 unigenes) have been implicated in plant stress responses, but with lower numbers of assigned unigenes. In addition, 18,327 (13.19%) of the unigenes had significant matches in the KEGG database using the KEGG automatic annotation server (e-value = 1 × 10−10) and were referred to 129 pathways (Figure 2). The KEGG pathway analysis suggested that “translation” (1872, 10.21%) was the largest subgroup, followed by “folding, sorting, and degradation” (1766, 9.64%) and “carbohydrate metabolism” (1640, 8.95%).
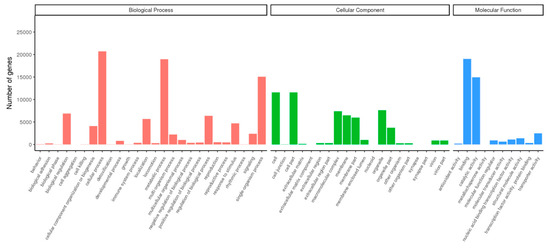
Figure 1.
Gene Ontology (GO) classification of D. involucrata unigenes.
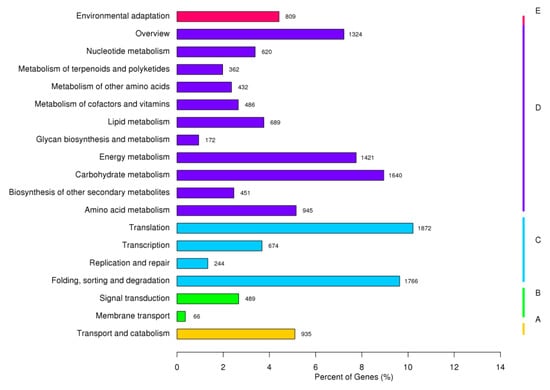
Figure 2.
KEGG classification of D. involucrata unigenes.
3.3. Identification of DEGs
Subsequently, the gene expression profiles were compared between different treatment groups to identify significant DEGs (Padj < 0.05) based on the criteria of twofold differential regulation [|log2fold change)| > 1] (Figure 3a). There were a total of 37,169 DEGs between the three heat-treated samples and the control samples. The comparisons between the HS1 and HS0, HS6 and HS0, and HS12 and HS0 resulted in 19,532, 20,497 and 27,716 DEGs, respectively (Figure 3b). Among the DEGs, 10,557, 11,607 and 14,138 were found to be up-regulated at HS1, HS6 and HS12, respectively, and 8975, 8890 and 13,578 were found to be down-regulated at HS1, HS6 and HS12, respectively (Figure 3b). Furthermore, 9537 D. involucrata genes displayed significantly altered expression levels under all three heat-treatment conditions (Figure 3c). A full list of assembled unigenes and their transcript abundance changes under heat stress have been provided in Table S4.
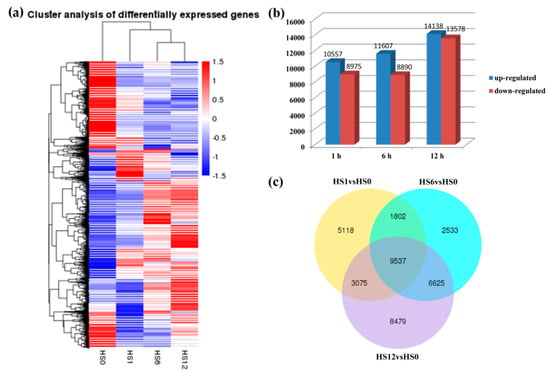
Figure 3.
Analysis of differentially expressed genes (DEGs) in response to heat stress in D. involucrata. (a) Hierarchical clustering of DEGs under heat stress based on gene expression profiles determined by RNA-Seq. The colors from red to blue in the heat map represent the expression levels toward low. (b) Numbers of DEGs under the three heat-treated conditions. The x-axis indicates the three time points, and the y-axis displays up- and down-regulated gene numbers. (c) Venn diagram analysis of DEGs in three comparisons.
3.4. GO and KEGG Enrichment Analyses of Heat-Responsive Genes
To obtain more insight into the biological functions of heat-responsive genes in D. involucrata, the DEGs were subjected to a GO term enrichment analysis. In general, all the GO-annotated DEGs—8471 (43.37%) at HS1, 8820 (43.03%) at HS6, and 12,426 (44.83%) at HS12—were enriched in 79 terms (Table S5). Moreover, “binding” and “catalytic activity” were found to be enriched by as many as 4658 (54.99%) at HS1 and 5524 (44.46%) at HS12, respectively (Table S5). Notably, the GO term “protein phosphorylation” was significantly enriched at all time points (Table S5), indicating the important role of protein phosphorylation in the D. involucrata response to heat stress.
To determine whether the heat-responsive genes were engaged in specific pathways, a KEGG pathway enrichment analysis was performed. The top 20 enriched pathways among DEGs from “HS1vsHS0,” “HS6vsHS0,” and “HS12vsHS0” are shown in Figure 4 and Table S6. The top four enriched pathways in “HS1vsHS0” were “Protein processing in endoplasmic reticulum,” “Plant-pathogen interaction,” “Endocytosis,” “Plant hormone signal transduction.” The top three enriched pathways in “HS6vsHS0” were “Protein processing in endoplasmic reticulum,” “Endocytosis,” “Plant hormone signal transduction.” The top two enriched pathways in “HS12vsHS0” were “Plant hormone signal transduction” and “Protein processing in endoplasmic reticulum.” These results unequivocally revealed that both “protein processing in endoplasmic reticulum” (ko04141) and “plant hormone signal transduction” (ko04075) were enriched in all three comparisons (Figure 4 and Table S6). In “protein processing in endoplasmic reticulum,” 555, 390, and 424 DEGs were identified at HS1, HS6, and HS12, respectively (Figure 5, Figure S1, and Table S6). There were 138, 123, and 175 DEGs engaged in “plant hormone signal transduction” at HS1, HS6, and HS12, respectively (Figure S2, Table S6, and Table S7).
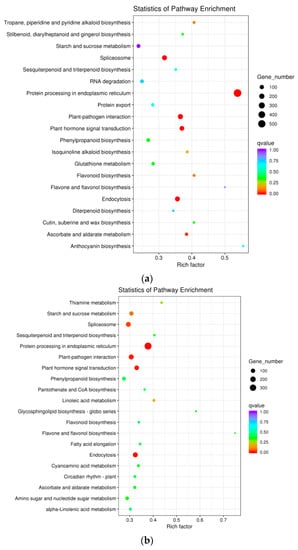
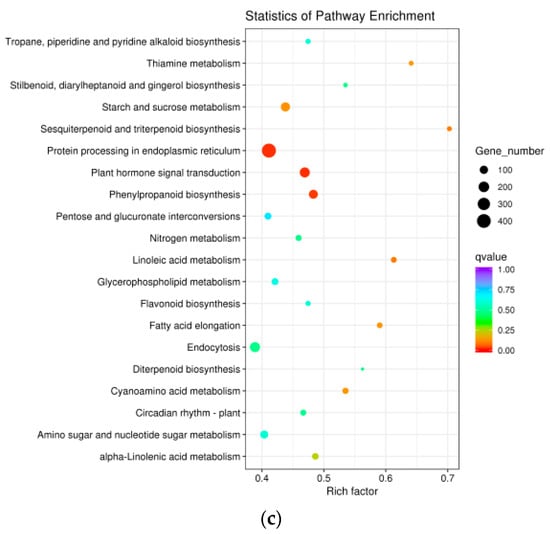
Figure 4.
KEGG analysis of DEGs from the comparisons of “HS1vsHS0” (a), “HS6vsHS0” (b), and “HS12vsHS0” (c). The size of the plots indicates the numbers of DEGs implicated in the corresponding pathway. A high/low q value is indicated by blue/red, respectively.
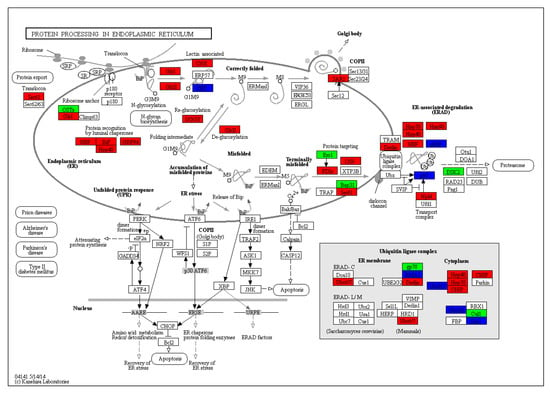
Figure 5.
Analysis of DEGs associated with protein processing in endoplasmic reticulum in the comparison of HS1vsHS0. Red and green reflect up- and down-regulation, respectively, while blue represents the mixed expression pattern.
3.5. Heat Shock Transcription Factors Responding to Heat Stress
The transcriptional responses of Hsf genes to high-temperature stress were then examined in D. involucrata. Hsfs with an altered expression in at least one of heat-treated conditions have been classified and listed in Table S8, and their cDNA sequences can be found in File S1. Totally, the expression levels of 32 Hsf members were remarkably changed upon heat treatment (Table S8). At HS1, 23 Hsf members were up-regulated, including classes A1, A2 A3, A4, A6, A7, B1, B2, B4, and C1, whereas six Hsf members were down-regulated, including classes A1, A4, B2, B3, and B4. At HS6, 15 Hsf genes (A1, A2 A3, A6, A7, B2, C1) were induced, and one member (HsfB4) was suppressed. At HS12, 22 differentially expressed Hsf genes were detected, with 18 members (A1, A2 A3, A6, A7, B2, C1) up-regulated and four members (A1, B1, B2, B4) down-regulated.
3.6. Verification of D. involucrata DEGs by qRT-PCR
To prove the reliability of our RNA-Seq results, eight candidate genes associated with heat stress response were selected for qRT-PCR. These genes included HSFA2b (Cluster-7138.49594), HSFB2b (Cluster-7138.27054), HSFC1 (Cluster-7138.47419), HSP90 (Cluster-7138.32229), HSP70 (Cluster-7138.44833), sHSP (Cluster-7138.10919), MBF1c (Cluster-7138.35285), and FKBP62 (Cluster-7138.54315) (Figure 6). Their cDNA sequences are shown in File S2. The expression patterns of all these DEGs derived from qRT-PCR were consistent with their transcript level alterations as detected by RNA-Seq, and a high correlation (R2 = 0.8237) was observed between the two experimental methods (Figure 6 and Figure S3), hence validating the accuracy of the transcriptome sequencing data.
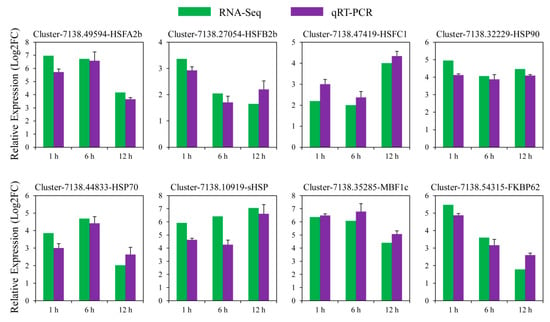
Figure 6.
qRT-PCR verification of D. involucrata DEGs. The qRT-PCR outcomes and RNA-Seq results were displayed by the blue and green bars, respectively. Error bars indicate standard deviations. Log2FC indicates the log2-transformed fold changes in transcript abundance between heat-treated and control samples.
4. Discussion
The augmented prevalence of high temperature worldwide is of global concern, as it dramatically affects plant growth, productivity, and distribution [44,45,46]. Though recent years have seen tremendous progress in unraveling the complex mechanisms underlying heat stress response in many different plants, more investigation is still required, particularly for uncharacterized species. The rare and endangered plant D. involucrata favors cold and humid climatic conditions in the mountains of southwestern and south-central China [5]. Therefore, high temperature is the crucial environmental factor that restrained its natural population distribution, ex situ conservation, as well as landscape application. At present, information on the large-scale regulation of gene expression profiles in response to heat stress is still lacking for D. involucrata. In this study, RNA-Seq, an invaluable approach allowing for the comprehensive characterization of transcriptomic events occurring in certain conditions, was utilized to uncover the heat response mechanism in D. involucrata. After de novo assembly, we generated 138,923 unigenes, of which 69,743 unigenes (50.2%) were annotated in public databases. Nevertheless, more complete transcriptome data of D. involucrata could be further obtained by integrating full-length transcriptome analyses in the future. Moreover, 19,532, 20,497 and 27,716 DEGs were identified after 1 h, 6 h, and 12 h of heat treatment in comparison to 0 h, respectively, and a functional enrichment analysis was subsequently performed for these DEGs. Taken together, our study gives the first insights into a global transcriptome picture of D. involucrata under heat stress, thus opening the door for adaptation research at the molecular level in this rare and endangered plant.
4.1. Heat Stress Treatment Used for Transcriptome Analysis in D. involucrata
Heat stress may result in either eustress or distress, both of which are determined by the thermic threshold of the organism, its state of acclimation and adaptation, as well as stress severity and duration [11,47]. Depending on the duration and strength of heat stress, plants can activate transient or long-term responses for protection and recovery which engage massive transcriptional and translational alterations. By contrast, extremely high temperature or long-term heat stress often lead to an irreversible disturbance of homeostasis, which can result in cell death [48]. Apart from basal thermotolerance, plants also employ conditionally expressed acquired thermotolerance at the cellular and molecular levels to withstand acute heat stress [11,49]. Acquired thermotolerance can be experimentally achieved through subjecting plants to a moderate level of heat stress (known as “priming”). Alternatively, priming can also be induced by a slow and continuous increase in temperature [11,49]. Heat stress is initially perceived by the leaf, which subsequently brings about physiological and biochemical alterations at the whole plant level [50,51]. Moreover, researchers have expended a lot of effort on studying heat response mechanisms in leaf tissues, including analyses of heat-induced leaf transcriptome changes [20,30,32,33,43]. Therefore, healthy mature leaves of approximately the same size were used for the transcriptome analysis of heat stress response in D. involucrata. Since photosystems in the leaves of D. involucrata seedlings were affected during a heat treatment of around 42 °C for several hours and for a longer period [9], a time-course experiment was performed at 42 °C in this study. After a one-week adaptation in growth chambers, D. involucrata seedlings were subjected to the 42 °C heat treatment, and leaf tissues were collected at time points 0, 1, 6, and 12 h after the start of heat treatment for transcriptome sequencing. Similarly, time-course gene expression profiling was conducted in carnation (Dianthus caryophyllus L.) leaves in response to the 42 °C heat treatment within 12 h [43].
4.2. Heat Stress Affected Expression of Genes Associated with Protein Processing in the ER
In plants, sensitive cellular processes including protein folding in the endoplasmic reticulum (ER) can be disrupted by environmental stresses. The limited capacity of the ER to process proteins leads to massive accumulation of unfolded or misfolded proteins, which is the greatest risk for plant development and survival during heat stress [52,53]. Hence, protein processing in the ER is a very important pathway under heat stress. In this study, a KEGG pathway analysis revealed that “protein processing in endoplasmic reticulum” was significantly enriched under all three heat-treatment conditions (Figure 4 and Table S6). There were 555 DEGs at HS1, with 14 of them down-regulated; there were 390 DEGs at HS6, with 21 of them down-regulated; and there were 424 DEGs at HS12, with 46 of them down-regulated (Figure 5, Figure S1, and Table S6). These results suggest that most of the genes engaged in protein processing were induced during heat stress to promote the efficiency of protein processing for stress response.
In higher plants, heat stress is known to redirect protein synthesis via a reduction in the synthesis of normal proteins concomitantly with a remarkable enhancement in the transcription and translation of Hsps [54]. Mounting evidence suggests that transcript abundance of Hsps along with chaperones is implicated in heat stress adaptation [30,31,32,33,43]. Hsps serve as molecular chaperones that are responsible for the folding, assembly, translocation and degradation of protein molecules, and they play paramount roles in combating stresses through re-establishing normal protein conformation and, thereby, cellular homeostasis [13,44]. In protein processing in the ER pathway, we observed that a lot of DEGs encoding Hsps (i.e., HSP90, HSP70, HSP40 and sHSP) were substantially up-regulated upon heat treatment. These results imply that inductions of Hsps facilitate protein processing in the ER and are important for acclimating to high-temperature stress in D. involucrata.
To eliminate misfolded proteins in the ER and maintain homeostasis, an ER-associated degradation (ERAD) process is usually activated [55]. Through ERAD, misfolded proteins are directed for degradation via the ubiquitin-proteasome system. DERLIN is the core component of the retrotranslocation machinery of the plant ERAD pathway, and it was reported to be induced responding to ER stress [56,57]. Consistently, we observed an up-regulation of DERLIN at HS1 and HS6 (Figure 5 and Figure S1). Ubc6 in yeast is a ubiquitin-conjugating enzyme that localizes to the ER membrane and is part of the Doa10 complex in the ERAD pathway [58]. In this study, the plant homolog of Ubc6, UBC32, was induced at all three heat treatment time points (Figure 5 and Figure S1). Likewise, the transcriptional levels of UBC32 in Arabidopsis were found to be elevated by various abiotic stresses and ER stress [55].
CHIP in Arabidopsis acts as the chaperone-associated E3 ubiquitin ligase and has been implicated in protein degradation and stress responses [59]. Importantly, CHIP is induced under stress conditions including high temperature [60]. In accord with this, we found CHIP was up-regulated in D. involucrata at the treatment time points HS1 and HS6 (Figure 5 and Figure S1). It has been demonstrated that disruption in CHIP resulted in elevated sensitivity to abiotic stresses [59]. However, transgenic plants overexpressing CHIP are hypersensitive to temperature stress [60]. The exact functions of CHIP in D. involucrata during heat stress need to be dissected in the future.
4.3. Influence of Heat Stress on Plant Hormone Signal Transduction
Phytohormones are regarded as master regulators of plant growth, development, and stress responses [61,62,63]. Our KEGG pathway analysis of DEGs revealed that “plant hormone signal transduction” was significantly altered under heat stress (Figure 4 and Table S6). More specifically, all hormone regulatory pathways were affected at three heat treatment time points, except for the salicylic acid pathway at HS6 (Figure S2). Though abscisic acid (ABA) is the best-characterized stress hormone, the roles of other hormones such as auxin, cytokinin (CK), ethylene (ET), jasmonic acid (JA) and brassinosteroid (BR) in plant stress responses are beginning to be better understood [64,65]. Furthermore, emerging evidence indicates that crosstalk between different hormonal signaling pathways leads to synergetic or antagonistic interactions, which modulate stress adaptations [66,67,68]. Based on our transcriptome data, it is tempting to assume that multiple phytohormones and their signaling crosstalk are engaged in the heat stress response in D. involucrata.
Many studies have revealed that phytohormone signaling and protein processing in the ER interact coordinately and cooperatively in stress responses. For instance, Arabidopsis UBC32 was demonstrated to be engaged in salt stress responses through the regulation of BR signaling by the ERAD pathway [55]. In maize, the crosstalk between the ER quality control (ERQC) and ABA signaling engages the transcription factor ZmbZIP17 [69]. Fascinatingly, ER stress was shown to negatively regulate auxin signaling in Arabidopsis [70]. Furthermore, the ER-based auxin homoeostasis is crucial for unfolded protein response (UPR) activation [70]. In our study, auxin signaling might have been the primary hormone regulatory pathway in the heat stress response in D. involucrata based on the maximum amount of DEGs (i.e., 82 DEGs) assigned to the auxin pathway (Table S7). Moreover, majority of the DEGs (86.3% at HS1, 91.5% at HS6, 90.1% at HS12) in the auxin pathway were suppressed during heat stress (Table S7). It has been previously demonstrated that hormone synthesis, transport, and response for most part of the phytohormone pathways are conserved among different plant species [71]. In particular, the regulatory regime of auxin signaling can be transferred to all phototrophic eukaryotes containing co-orthologues except for the unicellular green algae Chlamydomonas reinhardtii [71]. Therefore, it is likely that the UPR in D. involucrata leads to changes in the auxin signaling pathway to cope with heat-induced ER stress.
4.4. Hsfs May Play a Pivotal Role During Heat Stress
Accumulating data indicate the existence of a heat stress-activated transcriptional network that modulates the expression of heat-responsive genes [53]. In such a network, Hsfs were documented to play a central and highly conserved role in eukaryotes [72]. In tomatoes, HsfA1a possesses a unique role as a master regulator for acquired thermotolerance [20,73]. The knockdown of HsfA1a expression in tomato practically eliminates heat stress-triggered synthesis of HsfA2, HsfB1, and molecular chaperones, suggesting no substitution of function by other Hsfs [73]. In Arabidopsis, the HsfA1 group comprises Hsf A1a, A1b, A1d and A1e. It has been demonstrated that HsfA1s in Arabidopsis act as major regulators in heat-responsive gene expression [74,75]. Transcriptomic studies using HsfA1s quadruple knockout mutants revealed that more than 65% of up-regulated genes during heat stress were HsfA1-dependent [74]. These induced genes that are under the control of HsfA1s include several Hsfs, such as HsfA2, HsfA7a, HsfB1, HsfB2a, and HsfB2b [74]. Consistent with these studies, we observed that most of the DEGs belonging to class A1a, A2, A7a, B1, B2a, and B2b were up-regulated upon heat treatment (Table S8), implying the possibility that HsfA1a-dependent transcriptional regulation networks also exist in D. involucrata. Other DEGs encoding Hsfs belong to class A3, A4, A6, B3, B4, and C1 (Table S8). Future studies may dissect the molecular functions of these Hsfs in D. involucrata during heat stress.
5. Conclusions
The endangered and rare tertiary relict tree D. involucrata is highly sensitive to high temperatures. To dissect the dynamic and complicated gene regulatory network in D. involucrata seedlings under heat stress, we carried out time-course gene expression profiling using Illumina HiSeq 4000 sequencing. Compared to the control (0 h), we identified 19,532, 20,497 and 27,716 DEGs after 1 h, 6 h, and 12 h of heat treatment, respectively. Furthermore, a KEGG pathway analysis of DEGs revealed that protein processing in the ER and plant hormone signaling may play crucial roles in D. involucrata responses to high-temperature stress. Additionally, the expression levels of 32 genes encoding putative Hsfs were significantly altered during heat stress. Our transcriptomic data will facilitate future studies on such heat-sensitive and endangered species and will provide the theoretical basis for conservation and utilization of these valuable plant resources.
Supplementary Materials
The following are available online at https://www.mdpi.com/1999-4907/10/8/656/s1, Table S1: Primers used for the qRT-PCR; Table S2: Summary of RNA-Seq data from all samples; Table S3: Annotation of unigenes in different databases; Table S4: A full list of assembled unigenes and their annotation and expression levels during heat stress; Table S5: DEGs enriched in GO terms; Table S6: KEGG pathway analysis of DEGs; Table S7: Classification of DEGs associated with plant hormone signal transduction; Table S8: DEGs encoding heat shock transcription factors (Hsfs); Figure S1: Analysis of DEGs associated with protein processing in endoplasmic reticulum in two comparisons [HS6vsHS0 (a) and HS12vsHS0 (b)]; Figure S2: Analysis of DEGs associated with plant hormone signal transduction in three comparisons [HS1vsHS0 (a), HS6vsHS0 (b) and HS12vsHS0 (c)]; Figure S3: Correlation analysis between data of RNA-Seq (x-axis) and qRT-PCR (y-axis); File S1: cDNA sequences of 32 Hsfs that displayed differential expressions under heat stress; File S2: cDNA sequences of eight heat-responsive genes used for qRT-PCR.
Author Contributions
Q.L. and X.X. conceived and designed the research. Q.L. performed the experiments. Q.L., R.R.V., W.X. and X.X. analyzed the data. Q.L. wrote the manuscript with input from R.R.V. and X.X. All authors read and approved the manuscript.
Funding
This work was supported by Sichuan Science and Technology Program (2019YFH0130) and the Fundamental Research Funds of China West Normal University (17B008, 17E069).
Acknowledgments
We thank Novogene Corporation (Beijing, China) for assistance in transcriptome sequencing. We are grateful to three anonymous reviewers whose constructive comments enabled us to improve the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Li, Y.X.; Chen, L.; Juan, L.; Li, Y.; Chen, F. Suppression subtractive hybridization cloning of cDNAs of differentially expressed genes in dovetree (Davidia involucrata) bracts. Plant Mol. Biol. Rep. 2002, 20, 231–238. [Google Scholar] [CrossRef]
- He, Z.C.; Li, J.Q.; Wang, H.C. Karyomorphology of Davidia involucrata and Camptotheca acuminata, with special reference to their systematic positions. Bot. J. Linn. Soc. 2004, 144, 193–198. [Google Scholar] [CrossRef]
- Sun, J.F.; Gong, Y.B.; Renner, S.S.; Huang, S.Q. Multifunctional bracts in the dove tree Davidia involucrata (Nyssaceae: Cornales): Rain protection and pollinator attraction. Am. Nat. 2008, 171, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Shimozu, Y.; Kimura, Y.; Esumi, A.; Aoyama, H.; Kuroda, T.; Sakagami, H.; Hatano, T. Ellagitannins of Davidia involucrata. I. structure of davicratinic acid A and effects of davidia tannins on drug-resistant bacteria and human oral squamous cell carcinomas. Molecules 2017, 22, 470. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.Q.; Dong, Y.F.; Herrando-Moraira, S.; Matsui, T.; Ohashi, H.; He, L.Y.; Nakao, K.; Tanaka, N.; Tomita, M.; Li, X.S.; et al. Potential effects of climate change on geographic distribution of the Tertiary relict tree species Davidia involucrata in China. Sci. Rep. 2017, 7, 43822. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, J.; Zhou, B.; Lian, X. Natural distribution of Davidia involucrata and introduction analysis. J. Beijing For. Univ. 1995, 17, 25–30. [Google Scholar]
- Chen, Y.; Su, Z.X. Research on the protection of Davidia involucrata populations, a rare and endangered plant endemic to China. Acta Ecol. Sin. 2011, 31, 5466–5474. [Google Scholar]
- Li, Y.Q.; Lei, N.F.; Xu, Y.; Chen, F. Effects of high temperature stress on physiological and biochemical indicators of Davidia involucrata leaves. J. Sichuan Univ. 2009, 46, 809–813. [Google Scholar]
- Zhang, C.C.; Zhou, Q.; Ren, S.H.; Zou, J.; Li, Y.Y. Study on high temperature stress test of Davidia involucrata Baill. Hortic. Seed 2017, 5, 45–47. [Google Scholar]
- Driedonks, N.; Rieu, I.; Vriezen, W.H. Breeding for plant heat tolerance at vegetative and reproductive stages. Plant Reprod. 2016, 29, 67–79. [Google Scholar] [CrossRef]
- Mesihovic, A.; Iannacone, R.; Firon, N.; Fragkostefanakis, S. Heat stress regimes for the investigation of pollen thermotolerance in crop plants. Plant Reprod. 2016, 29, 93–105. [Google Scholar] [CrossRef]
- Vierling, E. The roles of heat shock proteins in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1991, 42, 579–620. [Google Scholar] [CrossRef]
- Wang, W.; Vinocur, B.; Shoseyov, O.; Altman, A. Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 2004, 9, 244–252. [Google Scholar] [CrossRef]
- Fragkostefanakis, S.; Röth, S.; Schleiff, E.; Scharf, K.D. Prospects of engineering thermotolerance in crops through modulation of heat stress transcription factor and heat shock protein networks. Plant Cell Environ. 2015, 38, 1881–1895. [Google Scholar] [CrossRef]
- Fragkostefanakis, S.; Simm, S.; Paul, P.; Bublak, D.; Scharf, K.D.; Schleiff, E. Chaperone network composition in Solanum lycopersicum explored by transcriptome profiling and microarray meta-analysis. Plant Cell Environ. 2015, 38, 693–709. [Google Scholar] [CrossRef]
- Nover, L.; Bharti, K.; Döring, P.; Mishra, S.K.; Ganguli, A.; Scharf, K.D. Arabidopsis and the heat stress transcription factor world: How many heat stress transcription factors do we need? Cell Stress Chaperones 2001, 6, 177–189. [Google Scholar] [CrossRef]
- Scharf, K.D.; Berberich, T.; Ebersberger, I.; Nover, L. The plant heat stress transcription factor (Hsf) family: Structure, function and evolution. Biochim. Biophys. Acta 2012, 1819, 104–119. [Google Scholar] [CrossRef]
- Nover, L.; Scharf, K.D.; Gagliardi, D.; Vergne, P.; Czarnecka-Verner, E.; Gurley, W.B. The Hsf world: Classification and properties of plant heat stress transcription factors. Cell Stress Chaperones 1996, 1, 215–223. [Google Scholar] [CrossRef]
- Kotak, S.; Larkindale, J.; Lee, U.; von Koskull-Döring, P.; Vierling, E.; Scharf, K.D. Complexity of the heat stress response in plants. Curr. Opin. Plant Biol. 2007, 10, 310–316. [Google Scholar] [CrossRef]
- von Koskull-Döring, P.; Scharf, K.D.; Nover, L. The diversity of plant heat stress transcription factors. Trends Plant Sci. 2007, 12, 452–457. [Google Scholar] [CrossRef]
- Scharf, K.D.; Rose, S.; Zott, W.; Schöffl, F.; Nover, L. Three tomato genes code for heat stress transcription factors with a region of remarkable homology to the DNA-binding domain of the yeast HSF. EMBO J. 1990, 9, 4495–4501. [Google Scholar] [CrossRef]
- Scharf, K.D.; Rose, S.; Thierfelder, J.; Nover, L. Two cDNAs for tomato heat stress transcription factors. Plant Physiol. 1993, 102, 1355–1356. [Google Scholar] [CrossRef][Green Version]
- Treuter, E.; Nover, L.; Ohme, K.; Scharf, K.D. Promoter specificity and deletion analysis of three heat stress transcription factors of tomato. Mol. Gen. Genet. 1993, 240, 113–125. [Google Scholar] [CrossRef]
- Fragkostefanakis, S.; Simm, S.; El-Shershaby, A.; Hu, Y.; Bublak, D.; Mesihovic, A.; Darm, K.; Mishra, S.K.; Tschiersch, B.; Theres, K.; et al. The repressor and co-activator HsfB1 regulates the major heat stress transcription factors in tomato. Plant Cell Environ. 2019, 42, 874–890. [Google Scholar] [CrossRef]
- Fragkostefanakis, S.; Mesihovic, A.; Simm, S.; Paupière, M.J.; Hu, Y.; Paul, P.; Mishra, S.K.; Tschiersch, B.; Theres, K.; Bovy, A.; et al. HsfA2 controls the activity of developmentally and stress-regulated heat stress protection mechanisms in tomato male reproductive tissues. Plant Physiol. 2016, 170, 2461–2477. [Google Scholar] [CrossRef]
- Berz, J.; Simm, S.; Schuster, S.; Scharf, K.D.; Schleiff, E.; Ebersberger, I. HEATSTER: A database and web server for identification and classification of heat stress transcription factors in plants. Bioinform. Biol. Insights 2019, 13, 1177932218821365. [Google Scholar] [CrossRef]
- Suzuki, N.; Bajad, S.; Shuman, J.; Shulaev, V.; Mittler, R. The transcriptional co-activator MBF1c is a key regulator of thermotolerance in Arabidopsis thaliana. J. Biol. Chem. 2008, 283, 9269–9275. [Google Scholar] [CrossRef]
- Meiri, D.; Breiman, A. Arabidopsis ROF1 (FKBP62) modulates thermotolerance by interacting with HSP90.1 and affecting the accumulation of HsfA2-regulated sHSPs. Plant J. 2009, 59, 387–399. [Google Scholar] [CrossRef]
- Zhang, C.; Li, G.; Chen, T.; Feng, B.; Fu, W.; Yan, J.; Islam, M.R.; Jin, Q.; Tao, L.; Fu, G. Heat stress induces spikelet sterility in rice at anthesis through inhibition of pollen tube elongation interfering with auxin homeostasis in pollinated pistils. Rice 2018, 11, 14. [Google Scholar] [CrossRef]
- Yan, J.; Yu, L.; Xuan, J.; Lu, Y.; Lu, S.; Zhu, W. De novo transcriptome sequencing and gene expression profiling of spinach (Spinacia oleracea L.) leaves under heat stress. Sci. Rep. 2016, 6, 19473. [Google Scholar] [CrossRef]
- Hwang, J.E.; Kim, Y.J.; Shin, M.H.; Hyun, H.J.; Bohnert, H.J.; Park, H.C. A comprehensive analysis of the Korean fir (Abies koreana) genes expressed under heat stress using transcriptome analysis. Sci. Rep. 2018, 8, 10233. [Google Scholar] [CrossRef]
- Wang, K.; Liu, Y.; Tian, J.; Huang, K.; Shi, T.; Dai, X.; Zhang, W. Transcriptional profiling and identification of heat-responsive genes in perennial ryegrass by RNA-sequencing. Front. Plant Sci. 2017, 8, 1032. [Google Scholar] [CrossRef]
- Qian, Y.; Ren, Q.; Zhang, J.; Chen, L. Transcriptomic analysis of the maize (Zea mays L.) inbred line B73 response to heat stress at the seedling stage. Gene 2019, 692, 68–78. [Google Scholar] [CrossRef]
- Wu, L.; Taohua, Z.; Gui, W.; Xu, L.; Li, J.; Ding, Y. Five pectinase gene expressions highly responding to heat stress in rice floral organs revealed by RNA-seq analysis. Biochem. Biophys. Res. Commun. 2015, 463, 407–413. [Google Scholar] [CrossRef]
- Keller, M.; Consortium, S.; Simm, S. The coupling of transcriptome and proteome adaptation during development and heat stress response of tomato pollen. BMC Genom. 2018, 19, 447. [Google Scholar] [CrossRef]
- Li, M.; Dong, X.; Peng, J.; Xu, W.; Ren, R.; Liu, J.; Cao, F.; Liu, Z. De novo transcriptome sequencing and gene expression analysis reveal potential mechanisms of seed abortion in dove tree (Davidia involucrata Baill.). BMC Plant Biol. 2016, 16, 82. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆CT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Wan, X.L.; Zhou, Q.; Wang, Y.Y.; Wang, W.E.; Bao, M.Z.; Zhang, J.W. Identification of heat-responsive genes in carnation (Dianthus caryophyllus L.) by RNA-seq. Front. Plant Sci. 2015, 6, 519. [Google Scholar] [CrossRef]
- Wahid, A.; Gelani, S.; Ashraf, M.; Foolad, M.R. Heat tolerance in plants: An overview. Environ. Exp. Bot. 2007, 61, 199–223. [Google Scholar] [CrossRef]
- Xu, X.; Peng, G.; Wu, C.; Han, Q. Global warming induces female cuttings of Populus cathayana to allocate more biomass, C and N to aboveground organs than do male cuttings. Aust. J. Bot. 2010, 58, 519–526. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, J.; Liu, Q.; He, H.; Xu, X.; Dong, T. Sexual differences in growth and defence of Populus yunnanensis under drought stress. Can. J. Forest Res. 2019, 49, 491–499. [Google Scholar] [CrossRef]
- Kranner, I.; Minibayeva, F.V.; Beckett, R.P.; Seal, C.E. What is stress? Concepts, definitions and applications in seed science. New Phytol. 2010, 188, 655–673. [Google Scholar] [CrossRef]
- Richter, K.; Haslbeck, M.; Buchner, J. The heat shock response: Life on the verge of death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef]
- Bokszczanin, K.L.; Fragkostefanakis, S. Perspectives on deciphering mechanisms underlying plant heat stress response and thermotolerance. Front. Plant Sci. 2013, 4, 315. [Google Scholar] [CrossRef]
- Zhang, Y.; Mian, M.A.; Chekhovskiy, K.; So, S.; Kupfer, D.; Lai, H.; Roe, B.A. Differential gene expression in Festuca under heat stress conditions. J. Exp. Bot. 2005, 56, 897–907. [Google Scholar] [CrossRef]
- Vidya, S.M.; Kumar, H.S.V.; Bhatt, R.M.; Laxman, R.H.; Ravishankar, K.V. Transcriptional profiling and genes involved in acquired thermotolerance in Banana: A non-model crop. Sci. Rep. 2018, 8, 10683. [Google Scholar] [CrossRef]
- Fragkostefanakis, S.; Mesihovic, A.; Hu, Y.; Schleiff, E. Unfolded protein response in pollen development and heat stress tolerance. Plant Reprod. 2016, 29, 81–91. [Google Scholar] [CrossRef]
- Li, B.; Gao, K.; Ren, H.; Tang, W. Molecular mechanisms governing plant responses to high temperatures. J. Integr. Plant Biol. 2018, 60, 757–779. [Google Scholar] [CrossRef]
- Zhang, Y.; Du, H. Differential accumulation of proteins in leaves and roots associated with heat tolerance in two Kentucky bluegrass genotypes differing in heat tolerance. Acta Physiol. Plant. 2016, 38, 213. [Google Scholar] [CrossRef]
- Cui, F.; Liu, L.; Zhao, Q.; Zhang, Z.; Li, Q.; Lin, B.; Wu, Y.; Tang, S.; Xie, Q. Arabidopsis ubiquitin conjugase UBC32 is an ERAD component that functions in brassinosteroid-mediated salt stress tolerance. Plant Cell 2012, 24, 233–244. [Google Scholar] [CrossRef]
- Kirst, M.E.; Meyer, D.J.; Gibbon, B.C.; Jung, R.; Boston, R.S. Identification and characterization of endoplasmic reticulum-associated degradation proteins differentially affected by endoplasmic reticulum stress. Plant Physiol. 2005, 138, 218–231. [Google Scholar] [CrossRef]
- Qian, D.; Chen, G.; Tian, L.; Qu, L.Q. OsDER1 is an ER-associated protein degradation factor that responds to ER Stress. Plant Physiol. 2018, 178, 402–412. [Google Scholar] [CrossRef]
- Kostova, Z.; Tsai, Y.C.; Weissman, A.M. Ubiquitin ligases, critical mediators of endoplasmic reticulum-associated degradation. Semin. Cell Dev. Biol. 2007, 18, 770–779. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, Y.; Qi, J.; Chi, Y.; Fan, B.; Yu, J.Q.; Chen, Z. E3 ubiquitin ligase CHIP and NBR1-mediated selective autophagy protect additively against proteotoxicity in plant stress responses. PLoS Genet. 2014, 10, e1004116. [Google Scholar] [CrossRef]
- Yan, J.; Wang, J.; Li, Q.; Hwang, J.R.; Patterson, C.; Zhang, H. AtCHIP, a U-box-containing E3 ubiquitin ligase, plays a critical role in temperature stress tolerance in Arabidopsis. Plant Physiol. 2003, 132, 861–869. [Google Scholar] [CrossRef]
- Jaillais, Y.; Chory, J. Unraveling the paradoxes of plant hormone signaling integration. Nat. Struct. Mol. Biol. 2010, 17, 642–645. [Google Scholar] [CrossRef]
- Liu, Q.; Vain, T.; Viotti, C.; Doyle, S.M.; Tarkowská, D.; Novák, O.; Zipfel, C.; Sitbon, F.; Robert, S.; Hofius, D. Vacuole integrity maintained by DUF300 proteins is required for brassinosteroid signaling regulation. Mol. Plant 2018, 11, 553–567. [Google Scholar] [CrossRef]
- Ku, Y.S.; Sintaha, M.; Cheung, M.Y.; Lam, H.M. Plant hormone signaling crosstalks between biotic and abiotic stress responses. Int. J. Mol. Sci. 2018, 19, 3206. [Google Scholar] [CrossRef]
- Wani, S.H.; Kumar, V.; Shriram, V.; Sah, S.K. Phytohormones and their metabolic engineering for abiotic stress tolerance in crop plants. Crop J. 2016, 4, 162–176. [Google Scholar] [CrossRef]
- Nie, S.; Huang, S.; Wang, S.; Mao, Y.; Liu, J.; Ma, R.; Wang, X. Enhanced brassinosteroid signaling intensity via SlBRI1 overexpression negatively regulates drought resistance in a manner opposite of that via exogenous BR application in tomato. Plant Physiol. Biochem. 2019, 138, 36–47. [Google Scholar] [CrossRef]
- Peleg, Z.; Blumwald, E. Hormone balance and abiotic stress tolerance in crop plants. Curr. Opin. Plant Biol. 2011, 14, 290–295. [Google Scholar] [CrossRef]
- Jiroutova, P.; Oklestkova, J.; Strnad, M. Crosstalk between brassinosteroids and ethylene during plant growth and under abiotic stress conditions. Int. J. Mol. Sci. 2018, 19, 3283. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, P.; Li, C.; Xia, G. The moss jasmonate ZIM-domain protein PnJAZ1 confers salinity tolerance via crosstalk with the abscisic acid signalling pathway. Plant Sci. 2019, 280, 1–11. [Google Scholar] [CrossRef]
- Yang, Y.G.; Lv, W.T.; Li, M.J.; Wang, B.; Sun, D.M.; Deng, X. Maize membrane-bound transcription factor Zmbzip17 is a key regulator in the cross-talk of ER quality control and ABA signaling. Plant Cell Physiol. 2013, 54, 2020–2033. [Google Scholar] [CrossRef]
- Chen, Y.; Aung, K.; Rolčík, J.; Walicki, K.; Friml, J.; Brandizzi, F. Inter-regulation of the unfolded protein response and auxin signaling. Plant J. 2014, 77, 97–107. [Google Scholar] [CrossRef]
- Simm, S.; Scharf, K.D.; Jegadeesan, S.; Chiusano, M.L.; Firon, N.; Schleiff, E. Survey of genes involved in biosynthesis, transport, and signaling of phytohormones with focus on Solanum lycopersicum. Bioinform. Biol. Insights 2016, 10, 185–207. [Google Scholar] [CrossRef]
- Liu, H.C.; Charng, Y.Y. Common and distinct functions of Arabidopsis class A1 and A2 heat shock factors in diverse abiotic stress responses and development. Plant Physiol. 2013, 163, 276–290. [Google Scholar] [CrossRef]
- Mishra, S.K.; Tripp, J.; Winkelhaus, S.; Tschiersch, B.; Theres, K.; Nover, L.; Scharf, K.D. In the complex family of heat stress transcription factors, HsfA1 has a unique role as master regulator of thermotolerance in tomato. Genes Dev. 2002, 16, 1555–1567. [Google Scholar] [CrossRef]
- Liu, H.C.; Liao, H.T.; Charng, Y.Y. The role of class A1 heat shock factors (HSFA1s) in response to heat and other stresses in Arabidopsis. Plant Cell Environ. 2011, 34, 738–751. [Google Scholar] [CrossRef]
- Yoshida, T.; Ohama, N.; Nakajima, J.; Kidokoro, S.; Mizoi, J.; Nakashima, K.; Maruyama, K.; Kim, J.M.; Seki, M.; Todaka, D.; et al. Arabidopsis HsfA1 transcription factors function as the main positive regulators in heat shock-responsive gene expression. Mol. Genet. Genom. 2011, 286, 321–332. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).