Comparison of Genetic Diversity in Naturally Regenerated Norway Spruce Stands and Seed Orchard Progeny Trials




Abstract
:1. Introduction
2. Materials and Methods
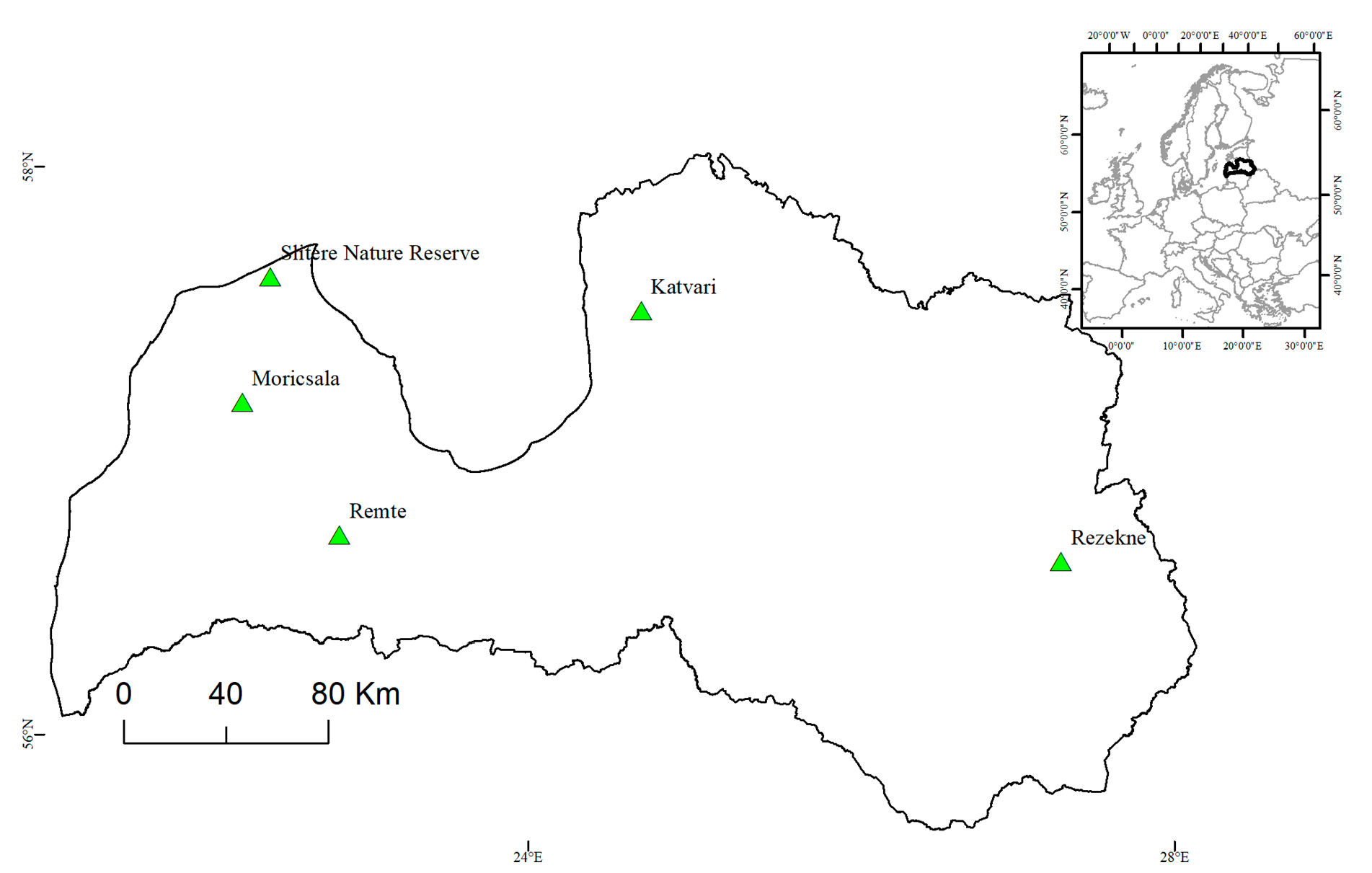
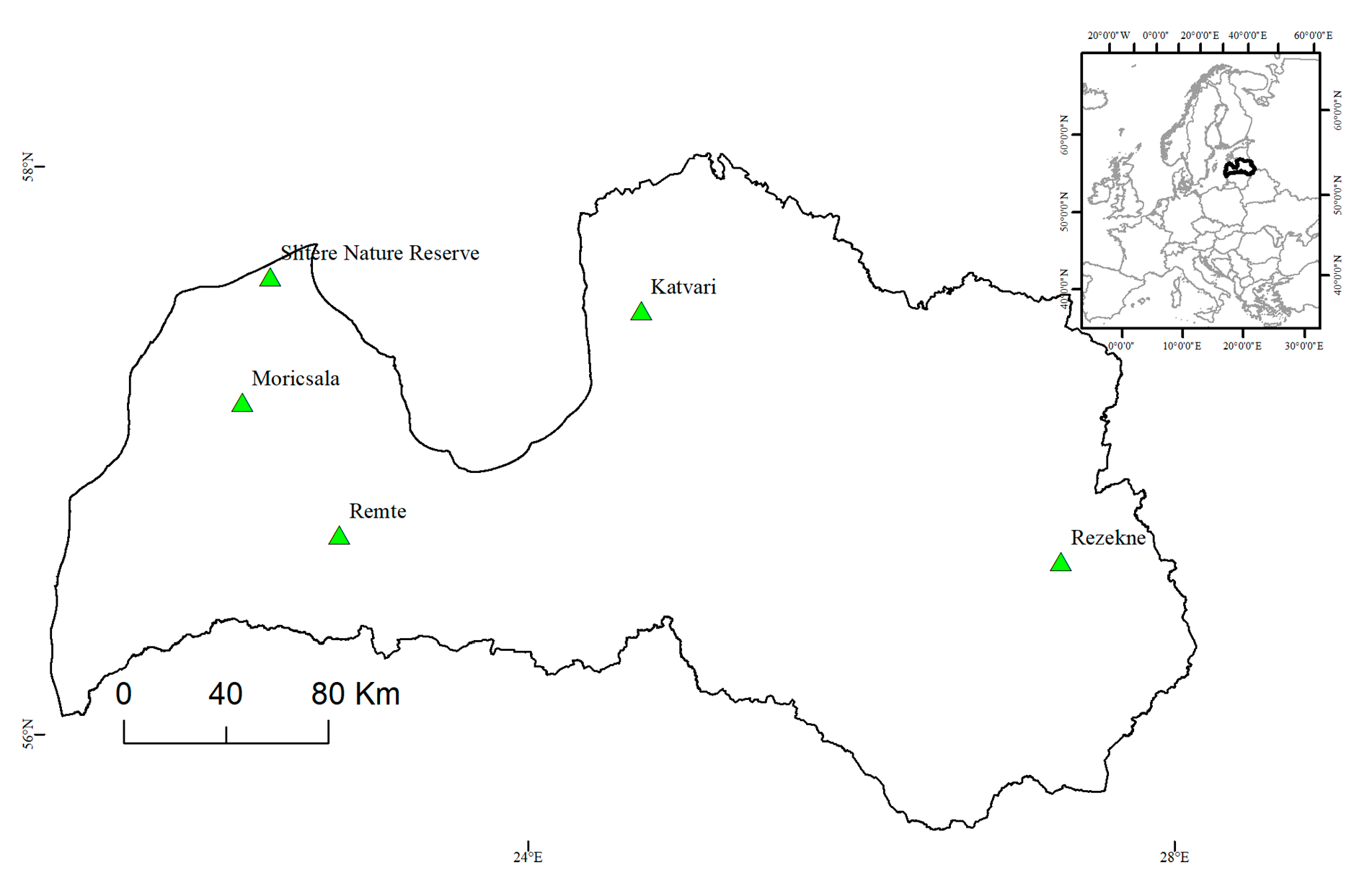
2.1. Analyzed Populations
2.2. Genetic Analysis
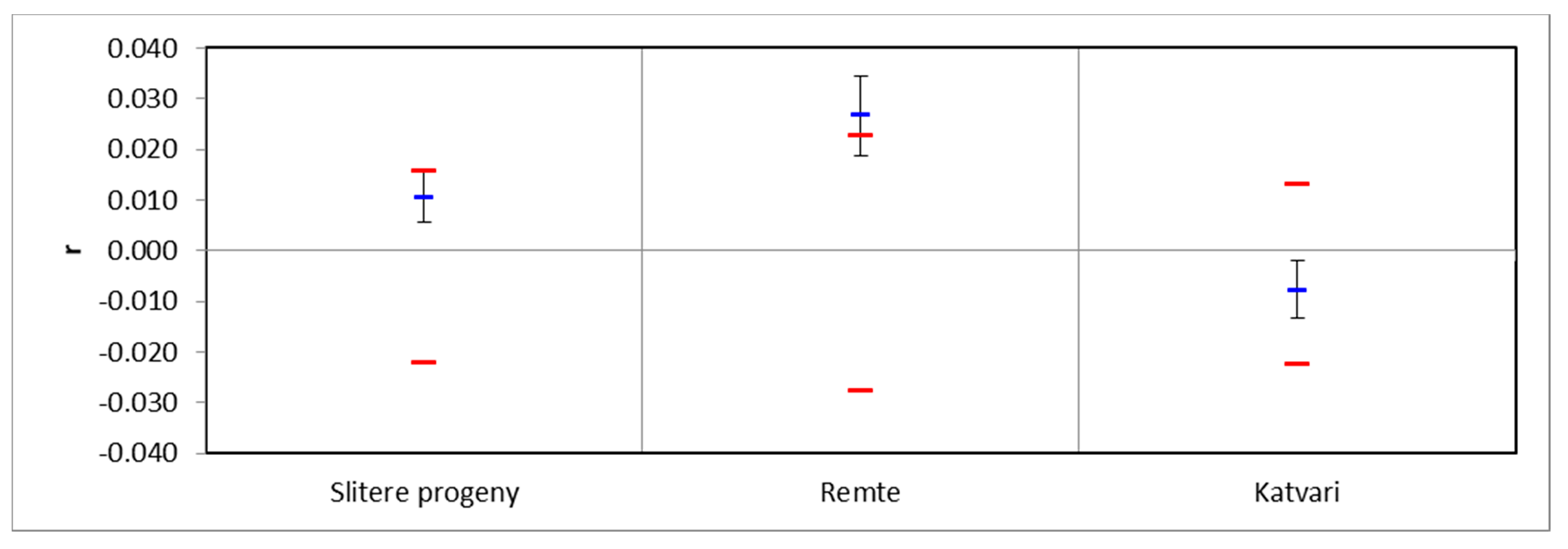
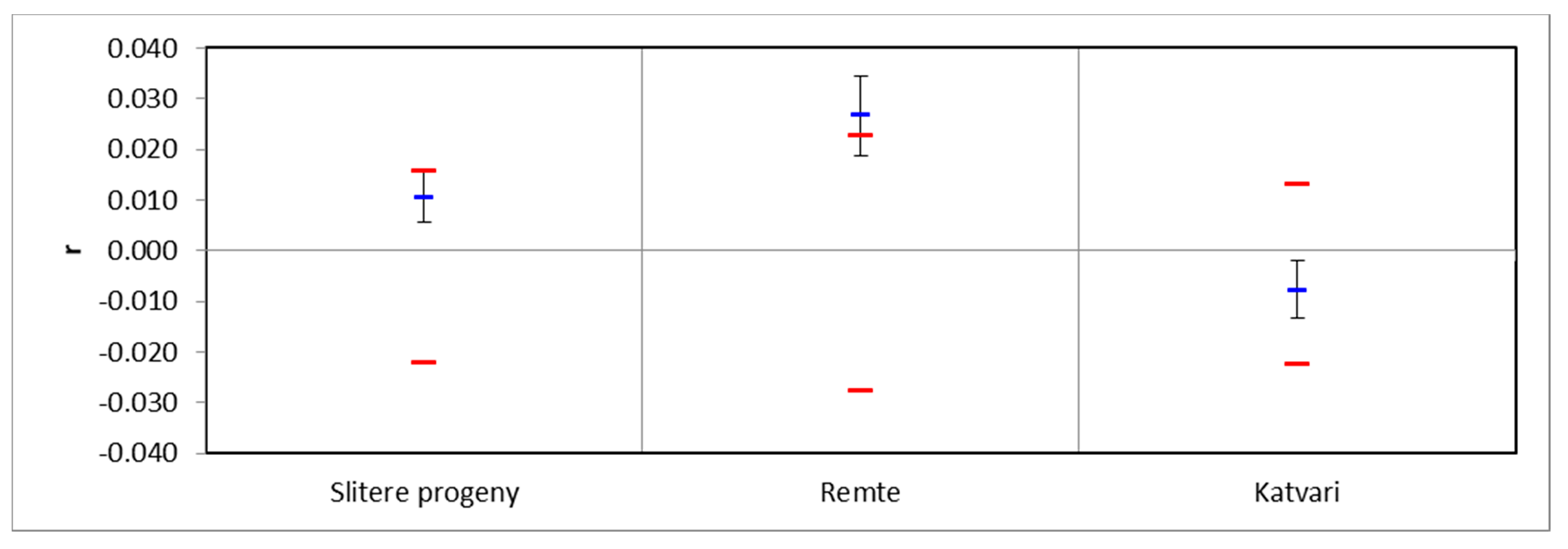
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lindner, M.; Fitzgerald, J.B.; Zimmermann, N.E.; Reyer, C.; Delzon, S.; van der Maaten, E.; Schelhaas, M.-J.; Lasch, P.; Eggers, J.; van der Maaten-Theunissen, M.; et al. Climate change and European forests: What do we know, what are the uncertainties, and what are the implications for forest management? J. Environ. Manag. 2014, 146, 69–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Intergovernmental Panel on Climate Change (IPCC). Managing the Risks of Extreme Events and Disasters to Advance Climate Change Adaptation; A Special Report of Working Groups I and II of the Intergovernmental Panel on Climate Change; Field, C.B., Barros, V., Stocker, T.F., Qin, D., Dokken, D.J., Ebi, K.L., Mastrandrea, M.D., Mach, K.J., Plattner, G.-K., Allen, S.K., Eds.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2012. [Google Scholar]
- Hickler, T.; Vohland, K.; Feehan, J.; Miller, P.A.; Smith, B.; Costa, L.; Giesecke, T.; Fronzek, S.; Carter, T.R.; Cramer, W.; et al. Projecting the future distribution of European potential natural vegetation zones with a generalized, tree species-based dynamic vegetation model. Glob. Ecol. Biogeogr. 2012, 21, 50–63. [Google Scholar] [CrossRef]
- Mäkinen, H.; Nöjd, P.; Mielikäinen, K. Climatic signal in annual growth variation in damaged and healthy stands of Norway spruce [Picea abies (L.) Karst.] in Southern Finland. Trees 2001, 15, 177–185. [Google Scholar] [CrossRef]
- Reich, P.B.; Oleksyn, J. Climate warming will reduce growth and survival of Scots pine except in the far north. Ecol. Lett. 2008, 11, 588–597. [Google Scholar] [CrossRef]
- Seidl, R.; Schelhaas, M.-J.; Rammer, W.; Verkerk, P.J. Increasing forest disturbances in Europe and their impact on carbon storage. Nat. Clim. Chang. 2014, 4, 806–810. [Google Scholar] [CrossRef] [Green Version]
- Banks, S.C.; Cary, G.J.; Smith, A.L.; Davies, I.D.; Driscoll, D.A.; Gill, A.M.; Lindenmayer, D.B.; Peakall, R. How does ecological disturbance influence genetic diversity? Trends Ecol. Evol. 2013, 28, 670–679. [Google Scholar] [CrossRef]
- Namkoong, G. Biodiversity issues in genetics, forestry and ethics. For. Chron. 1992, 68, 438–443. [Google Scholar] [CrossRef]
- Thom, D.; Rammer, W.; Seidl, R. Disturbances catalyze the adaptation of forest ecosystems to changing climate conditions. Glob. Chang. Biol. 2017, 23, 269–282. [Google Scholar] [CrossRef]
- Alfaro, R.I.; Fady, B.; Vendramin, G.G.; Dawson, I.K.; Fleming, R.A.; Sáenz-Romero, C.; Lindig-Cisneros, R.A.; Murdock, T.; Vinceti, B.; Navarro, C.M.; et al. The role of forest genetic resources in responding to biotic and abiotic factors in the context of anthropogenic climate change. Ecol. Manag. 2014, 333, 76–87. [Google Scholar] [CrossRef]
- Alberto, F.J.; Aitken, S.N.; Alía, R.; González-Martínez, S.C.; Hänninen, H.; Kremer, A.; Lefèvre, F.; Lenormand, T.; Yeaman, S.; Whetten, R. Potential for evolutionary responses to climate change-evidence from tree populations. Glob. Chang. Biol. 2013, 19, 1645–1661. [Google Scholar] [CrossRef]
- Leimu, R.; Mutikainen, P.; Koricheva, J.; Fisher, M. How general are positive relationships between plant population size, fitness and genetic variation? J. Ecol. 2006, 94, 942–952. [Google Scholar] [CrossRef]
- Hamrick, J.L. Response of forest trees to global environmental changes. For. Ecol. Manag. 2004, 197, 323–335. [Google Scholar] [CrossRef]
- González-Díaz, P.; Jump, A.S.; Perry, A.; Wachowiak, W.; Lapshina, E.; Cavers, S. Ecology and management history drive spatial genetic structure in Scots pine. For. Ecol. Manag. 2017, 400, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Rungis, D.; Bérubé, Y.; Zhang, J.; Ralph, S.; Ritland, C.E.; Ellis, B.E.; Douglas, C.; Bohlmann, J.; Ritland, K. Robust simple sequence repeat markers for spruce (Picea spp.) from expressed sequence tags. Appl. Genet. 2004, 109, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Keller, L.F.; Waller, D.M. Inbreeding effects in wild populations. Trends Ecol. Evol. 2002, 17, 230–241. [Google Scholar] [CrossRef]
- Namkoong, G.; Kang, H.C.; Brouard, J.S. Tree Breeding: Principles and Strategies. Monographs on Theoretical and Applied Genetics 11; Springer: New York, NY, USA, 1988. [Google Scholar]
- Androsiuk, P.; Shimono, A.; Westin, J.; Lindgren, D.; Fries, A.; Wang, X.-R. Genetic status of Norway spruce (Picea abies) breeding populations for northern Sweden. Silvae Genet. 2013, 62, 127–136. [Google Scholar] [CrossRef]
- Godt, M.J.W.; Hamrik, J.L.; Edwards-Burke, M.A.; Williams, J.H. Comparisons of genetic diversity in white spruce (Picea glauca) and jack pine (Pinus banksiana) seed orchards with natural populations. Can. J. For. Res. 2001, 31, 943–949. [Google Scholar] [CrossRef]
- Eriksson, G.; Ekberg, I.; Clapham, D. Genetics Applied to Forestry: An Introduction, 3rd ed.; Elanders Sverige AB: Uppsala, Sweden, 2013; p. 206. [Google Scholar]
- Ahti, T.; Hämet-Ahti, L.; Jalas, J. Vegetation zones and their sections on northwestern Europe. Ann. Bot. Fenn. 1968, 5, 169–211. [Google Scholar]
- Bāders, E.; Puriņa, L.; Lībiete, Z.; Nartišs, M.; Jansons, Ā. Fragmentācijas ilgtermiņa dinamika meža ainavā bez cilvēka saimnieciskās darbības ietekmes. [Long-term fragmentation dynamics in semi-natural forest landscape]. Mežzinātne 2014, 28, 91–107. (In Latvian) [Google Scholar]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Pfeiffer, A.; Olivieri, A.M.; Morgante, M. Identification and characterization of microsatellites in Norway spruce (Picea abies K.). Genome 1997, 40, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Hodgetts, R.B.; Aleksiuk, M.A.; Brown, A.; Clarke, C.; Macdonald, E.; Nadeem, S.; Khasa, D.; Macdonald, E. Development of microsatellite markers for white spruce (Picea glauca) and related species. Appl. Genet. 2001, 102, 1252–1258. [Google Scholar] [CrossRef]
- Besnard, G.; Acheré, V.; Rampant, P.F.; Favre, J.M.; Jeandroz, S. A set of cross-species amplifying microsatellite markers developed from DNA sequence databanks in Picea (Pinaceae). Mol. Ecol. Resour. 2003, 3, 380–383. [Google Scholar] [CrossRef]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–553. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenALEx 6.5: Genetic analysis in Excel—Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Queller, D.C.; Goodnight, K.F. Estimating relatedness using genetic markers. Evolution 1989, 258–275. [Google Scholar] [CrossRef]
- Goudet, J. FSTAT (Version 1.2): A computer program to calculate F-statistics. J. Hered. 1995, 86, 485–486. [Google Scholar] [CrossRef]
- Waples, R.S.; Do, C. Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: A largely untapped resource for applied conservation and evolution. Evol. Appl. 2010, 3, 244–262. [Google Scholar] [CrossRef]
- Funda, T.; El-Kassaby, Y.A. Seed orchard genetics. Plant Sci. Rev. 2012, 21–43. [Google Scholar] [CrossRef]
- Chaisurisri, K.; El-Kassaby, Y.A. Genetic diversity in a seed production population vs. natural populations of Sitka spruce. Biodivers. Conserv. 1994, 3, 512–523. [Google Scholar] [CrossRef]
- Sved, J.A.; Cameron, E.C.; Gilchrist, A.S. Estimating effective population size from linkage disequilibrium between unlinked loci: Theory and application to fruit fly outbreak populations. PLoS ONE 2013, 8, e69078. [Google Scholar] [CrossRef] [PubMed]
- Sønstebø, J.H.; Tollefsrud, M.M.; Myking, T.; Steffenrem, A.; Nilsen, A.E.; Edvardsen, Ø.M.; Johnskås, O.R.; El-Kassaby, Y.A. Genetic diversity of Norway spruce (Picea abies (L.) Karst.) seed orchard crops: Effects of number of parents, seed year, and pollen contamination. For. Ecol. Manag. 2018, 411, 132–141. [Google Scholar] [CrossRef]
- Wang, J.; Santiago, E.; Caballero, A. Prediction and estimation of effective population size. Heredity 2016, 117, 193–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burczyk, J.; Lewandowski, A.; Chalupka, W. Local pollen dispersal and distant gene flow in Norway spruce (Picea abies [L.] Karst.). For. Ecol. Manag. 2004, 197, 39–48. [Google Scholar] [CrossRef]
- Verbylaitė, R.; Pliūra, A.; Lygis, V.; Suchockas, V.; Jankauskienė, J.; Labokas, J. Genetic Diversity and Its Spatial Distribution in Self-Regenerating Norway Spruce and Scots Pine Stands. Forests 2017, 8, 470. [Google Scholar] [CrossRef]
- Kantorowicz, W. Half a century of seed years in major tree species of Poland. Silvae Genet. 2000, 49, 245–249. [Google Scholar]

{kind=link}
{kind=link}
| Population | Type | Establishment Year | Age (Years) | Number of Clones | Nearest Con-Specific Stand | Location (N; E) |
|---|---|---|---|---|---|---|
| Slitere progeny | Autochthonous—naturally regenerated | >1969 | <50 | - | - | 57.63; 22.28 |
| Slitere parents | Autochthonous | <1969 | >50 | - | - | 57.63; 22.28 |
| Rezekne | Autochthonous | ~1912 | ≥100 | - | 100 m | 56.60; 27.41 |
| Moricsala | Autochthonous | 1910-1930 | ~90 | - | 2400 m | 57.20; 22.15 |
| Remte | Seed orchard progeny trial | 1965 | 54 | 50 | 1000 m | 56.74; 22.80 |
| Katvari | Seed orchard progeny trial | 1975 | 44 | 20 | 500 m | 57.53; 24.75 |
| Locus | Primer Sequences (5′-3′) | Label (F Primer) | Repeat Motif | Fixation Index (Over All Progeny Populations) (SE) | Estimated null Allele Frequencies |
|---|---|---|---|---|---|
| SpAGC1 1 | F:TTCACCTTAGCCGAGAACC | 6-FAM | (TC)5TT(TC)10 | 0.091 (0.032) | 0.022 |
| R:CACTGGAGATCTTCGTTCTGA | |||||
| SpAGC2 1 | F:TACCATTCAACGCAAGGG | HEX | (TA)11(GA)20 | 0.563 (0.054) | 0.301 |
| R:GTGTATGGTTTTCTTTTCGCA | |||||
| SpAGG3 1 | F:CTCCAACATTCCCATGTAGC | TMR | (GA)23 | 0.026 (0.012) | 0.018 |
| R:AGCATGTTGTCCCATATAGACC | |||||
| UAPgTG25 2 | F:TCAAGCTCTCCAACCCAGAT | HEX | (TG)27 | 0.629 (0.040) | 0.280 |
| R:TGTCGAGTTTGACTTGTACCAA | |||||
| UAPgAG150 2 | F:ACCAATGCTTTTACCAAACG | TMR | (AG)19 | 0.172 (0.082)/0.260 (0.038) | 0.043/0.106 |
| R:TTGATTGCAAGTGATGGTTG | |||||
| WS0033.A18 3 | F:GGCTGCTCTCTTATCCGTTTT | 6-FAM | (TA)26 | 0.689 (0.031) | 0.300 |
| R:TGGCTCTCATCCAGAAAAGAA | |||||
| WS0022.B15 3 | F:TTTGTAGGTGCTGCAGAGATG | HEX | (AG)12 | 0.086 (0.049) | 0.087 |
| R:TGGCTTTTTATTCCAGCAAGA | |||||
| WS0073.H08 3 | F:TGCTCTCTTATTCGGGCTTC | TMR | (AT)14 | 0.066 (0.018) | 0.042 |
| R:AAGAACAAGGCTTCCCAATG | |||||
| WS0073cG10 5 | F:AGCATGGAGGTTTGCACTTT | 6-FAM | (GGC)8 | 0.194 (0.028) | 0.096 |
| R:CGCTGAGAAAGAAATTCAGG | |||||
| paGB3 4 | F:AGTGATTAAACTCCTGACCAC | HEX | (AT)11 | 0.062 (0.018) | 0.014 |
| R:CACTGAATACACCCATTATCC |
| Slitere Progeny | Slitere Parents | Rezekne | Moricsala | Remte | Katvari | |
|---|---|---|---|---|---|---|
| N | 95 | 58 | 96 | 48 | 72 | 90 |
| Total Na | 134 | 112 | 122 | 110 | 106 | 123 |
| Average Na | 12.18 (1.75) | 10.18 (1.48) | 11.09 (1.86) | 10.00 (1.61) | 9.64 (1.28) | 11.18 (1.57) |
| Total Na Frequency ≤ 5% | 85 | 56 | 69 | 55 | 62 | 70 |
| Average Na Frequency ≥ 5% | 4.45 (0.59) | 5.09 (0.89) | 4.82 (0.64) | 5.00 (0.65) | 4.00 (0.40) | 4.82 (0.70) |
| Neff | 5.14 (1.26) | 5.18 (1.31) | 5.00 (1.10) | 4.87 (0.96) | 4.02 (0.59) | 5.03 (1.03) |
| I | 1.67 (0.19) | 1.65 (0.18) | 1.66 (0.18) | 1.67 (0.16) | 1.57 (0.14) | 1.70 (0.17) |
| Ho | 0.55 (0.07) | 0.52 (0.18) | 0.55 (0.07) | 0.52 (0.08) | 0.52 (0.07) | 0.53 (0.07) |
| He | 0.71 (0.05) | 0.71 (0.04) | 0.71 (0.05) | 0.72 (0.05) | 0.69 (0.05) | 0.72 (0.05) |
| F | 0.22 (0.08) | 0.28 (0.08) | 0.24 (0.07) | 0.27 (0.09) | 0.27 (0.08) | 0.27 (0.08) |
| No. of unique alleles | 15 | 8 | 10 | 9 | 3 | 8 |
| Average gene diversity | 0.71 (0.05) | 0.72 (0.04) | 0.72 (0.05) | 0.73 (0.05) | 0.70 (0.05) | 0.73 (0.05) |
| Average Allelic richness | 10.22 (1.52) | 9.65 (1.43) | 9.79 (1.49) | 9.87 (1.57) | 8.99 (1.12) | 10.01 (1.41) |
| Population | Ne | Lower CI | Upper CI |
|---|---|---|---|
| Slitere progeny | 167.3 | 103.3 | 361.7 |
| Slitere parents | 251.4 | 114.2 | Infinite |
| Rezekne | 685.5 | 220.7 | Infinite |
| Moricsala | 714.6 | 141.3 | Infinite |
| Remte | 68.2 | 47.5 | 108.4 |
| Katvari | 48.5 | 38.5 | 62.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruņģis, D.; Luguza, S.; Bāders, E.; Šķipars, V.; Jansons, Ā. Comparison of Genetic Diversity in Naturally Regenerated Norway Spruce Stands and Seed Orchard Progeny Trials. Forests 2019, 10, 926. https://doi.org/10.3390/f10100926
Ruņģis D, Luguza S, Bāders E, Šķipars V, Jansons Ā. Comparison of Genetic Diversity in Naturally Regenerated Norway Spruce Stands and Seed Orchard Progeny Trials. Forests. 2019; 10(10):926. https://doi.org/10.3390/f10100926
Chicago/Turabian StyleRuņģis, Dainis, Solveiga Luguza, Endijs Bāders, Vilnis Šķipars, and Āris Jansons. 2019. "Comparison of Genetic Diversity in Naturally Regenerated Norway Spruce Stands and Seed Orchard Progeny Trials" Forests 10, no. 10: 926. https://doi.org/10.3390/f10100926
APA StyleRuņģis, D., Luguza, S., Bāders, E., Šķipars, V., & Jansons, Ā. (2019). Comparison of Genetic Diversity in Naturally Regenerated Norway Spruce Stands and Seed Orchard Progeny Trials. Forests, 10(10), 926. https://doi.org/10.3390/f10100926