To assess the accuracy and impact of our new DTLx reconciliation framework, we implemented our ODTLx-Sampling algorithm and applied it to both simulated and real datasets. We describe the results of this experimental evaluation below.
4.1. Results on Simulated Datasets
We used simulated datasets to systematically evaluate the accuracy of DTLx reconciliations and to compare its accuracy and functionality against ecceTERA, the only other parsimony-based reconciliation model that handles
and
events. Our implementation is built upon the open-source RANGER-DTL software package [
17] and we refer to the new software implementation as RANGER-DTLx.
We used the recently developed phylogenetic tree simulation software ZOMBI [
29] to evolve simulated gene trees with known ground-truth evolutionary histories containing explicit transfers from extinct lineages. ZOMBI tracks species lineages that eventually go extinct and allows for such lineages to participate in horizontal gene transfer events before going extinct. In our analysis, we simulated two datasets, each consisting of 500 gene tree/species tree pairs. Note that each of these 500 pairs consist of a unique (i.e., different) species tree and gene tree, where each of the species trees has exactly 50 taxa. The specific parameters used to generate the datasets are shown in
Table 1. Notably, Dataset-2 has a higher rate of transfers and more
events in the gene trees due to a higher extinction rate. For increased realism, the transfer rate was split evenly between additive and replacing transfers.
The final gene trees, obtained after pruning out all gene lineages with no surviving gene descendants, had, on average, 152.5 leaves, 11.6 duplications, 18.1 transfers, 3.0 events, and 121.7 speciations for Dataset-1, and 180.3 leaves, 10.5 duplications, 26.7 transfers, 5.7 events, and 142 speciations for Dataset-2. For reference, the unpruned gene trees showed on average, for Dataset-1, 16.1 losses and 42 extinctions, and for Dataset-2, 24.2 losses and 67.4 extinctions. Thus, Datasets 1 and 2 correspond to low and moderate rates, respectively, of the relevant evolutionary events.
We used these two simulated datasets to address the following three questions: (i) Does using DTLx reconciliation, i.e., RANGER-DTLx, improve upon the overall accuracy of DTL reconciliation in the presence of events? (ii) How well do RANGER-DTLx and ecceTERA detect events? Furthermore, (iii) How do RANGER-DTLx and ecceTERA compare in their ability to detect the phylogenetic location of the extinct donor of a event?
Comparing accuracies of DTL and DTLx reconciliation. Since both RANGER-DTLx and ecceTERA can account for additional evolutionary scenarios that cannot be correctly handled in traditional DTL reconciliation, it is reasonable to expect that both RANGER-DTLx and ecceTERA should result in greater overall reconciliation accuracy than traditional DTL reconciliation. We therefore applied RANGER-DTLx, ecceTERA, and RANGER-DTL (which implements the traditional DTL reconciliation framework) to both simulated datasets and evaluated the overall accuracies of the resulting reconciliations. Note that, for a fair comparison, we only compared reconciliation accuracy across the nodes present on the input gene trees, without inclusion of any hidden nodes inferred by RANGER-DTLx.
For an inferred reconciliation
, let
,
,
,
, and
denote the sets of speciation nodes, duplication nodes, standard transfer nodes, mappings for internal nodes of
G, and transfer recipients for standard transfers, respectively. The sets
,
,
,
, and
are defined analogously for the ground truth reconciliation
. Overall accuracy is quantified through the following three metrics:
To account for multiple optimal reconciliations, we used the standard approach of assigning the most well supported mapping and event type for each of the three methods/software. Specifically, 100 random optimal reconciliations were sampled for each gene tree/species tree pair, for both RANGER-DTLx and RANGER-DTL, and each gene tree node was assigned the most frequently observed mapping and event type among the 100 samples [
11]. A similar procedure was followed for ecceTERA, but based on the reconciliation graph computed by ecceTERA [
12], rather than on uniform random sampling.
Recall that, unlike ecceTERA, our DTLx reconciliation framework allows for independent assignment of and event costs. We therefore tried two different costs for events in all our experiments: 3 and 4. We fixed the cost of a event at 4. The costs for other events were fixed as follows for all three methods: Loss cost = 1, duplication cost = 2, and transfer cost = 3, which correspond to the defaults used in RANGER-DTL and ecceTERA. Based on these costs, ecceTERA automatically assigns events a cost of 4 (equivalent to one transfer plus one loss), and events a cost of 3 (equivalent to one transfer event).
The results of our analysis appear in
Table 2. As the table shows, RANGER-DTL, RANGER-DTLx with
cost 4, and ecceTERA have nearly indistinguishable reconciliation accuracies across both datasets, while RANGER-DTLx with
cost 3 shows slightly worse event and mapping accuracies but slightly better recipient accuracy. These results suggest that models that account for
and
events may not result in improved overall reconciliation accuracy compared to traditional DTL reconciliation, at least for low to moderate rates of
events as in our datasets. Importantly, these results also show that allowing for
and
events does not worsen reconciliation accuracy, even when the rate of
events is low (Dataset-1).
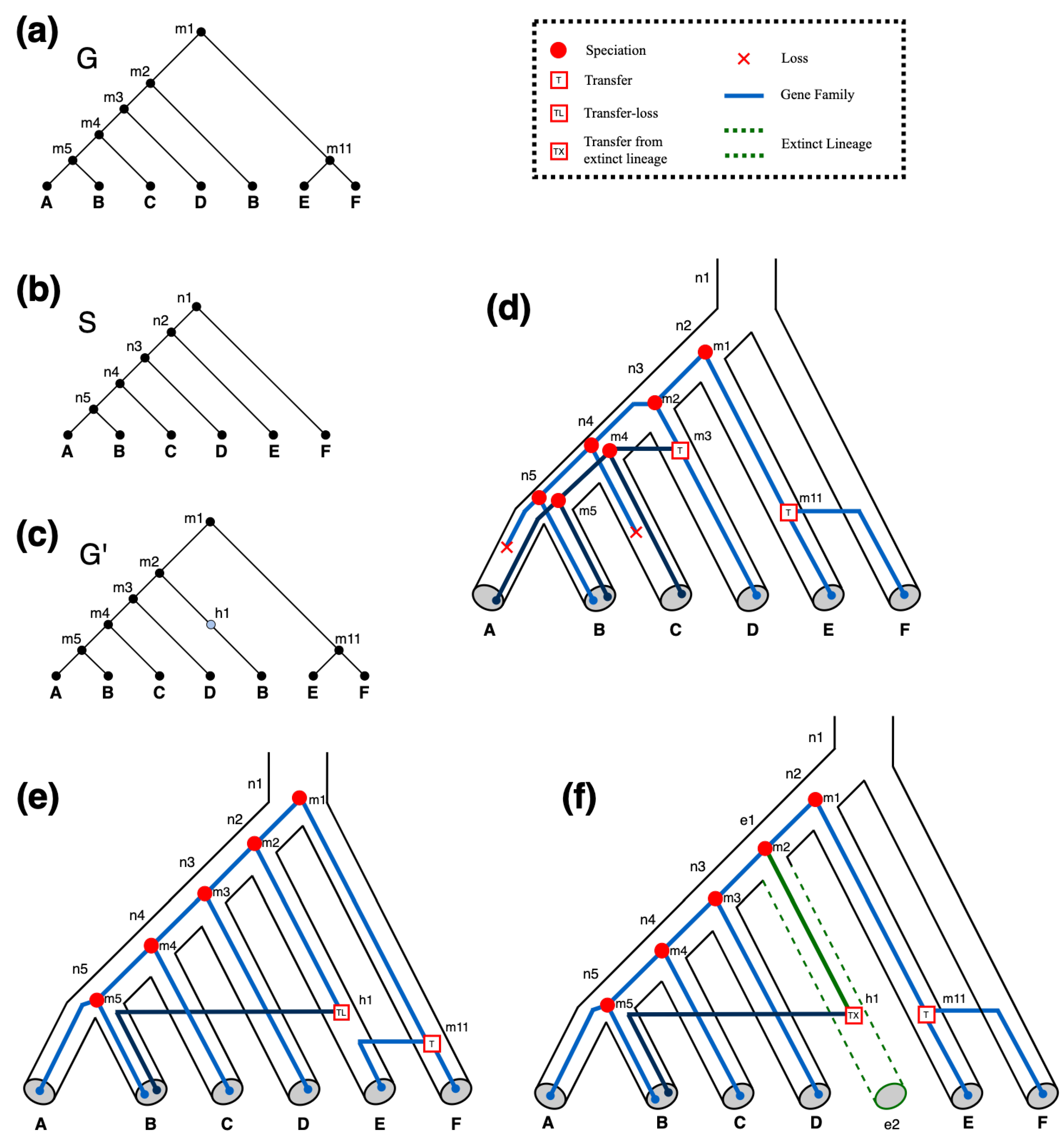
Accuracy of event detection. Next, we assessed how well RANGER-DTLx and ecceTERA can infer ground-truth events. We labelled transfers from unsampled lineages as ground truth events if, in the event log generated by ZOMBI, the gene survives in an extant lineage but the original copy goes extinct. While the original name of the internal gene tree node representing the extinct gene is absent from the input gene tree, we can verify if either method correctly detects this transfer by looking for a event with the same recipient as the ground truth transfer.
We first assessed how well
events were detected, in terms of precision and recall, in a single random optimal reconciliation computed by each method.
Table 3 shows the results of this analysis, which reveals several interesting insights. First, and perhaps most strikingly, we find that none of the methods shows high precision in detecting
events, with ecceTERA and RANGER-DTLx with
cost 3 both showing 10% precision in the two datasets, and RANGER-DTLx with
cost 4 showing much higher, but still relatively low, precision of 17.24% in Dataset-1 and 29.41% in Dataset-2. Second, we find that RANGER-DTLx with
cost 3 and ecceTERA, despite using effectively the same event costs, differ dramatically in the number of
events they infer. For example, for Dataset-1, ecceTERA infers only 413
events across all 500 gene trees with recall and precision of 2.61% and 9.44%, respectively, and RANGER-DTLx with
cost 3 infers 4515
events with recall and precision of 30.12% and 9.97%. Furthermore, third, these results show that there is a clear tradeoff between sensitivity and precision of
event inference and suggest that RANGER-DTLx with
cost 4 should be the method of choice for
event inference if precision is more important than recall, while RANGER-DTLx with
cost 3 should be used if recall is more important than precision. This experiment also highlights the utility of allowing for a user-specifiable cost for
events as permitted under our model but not under ecceTERA.
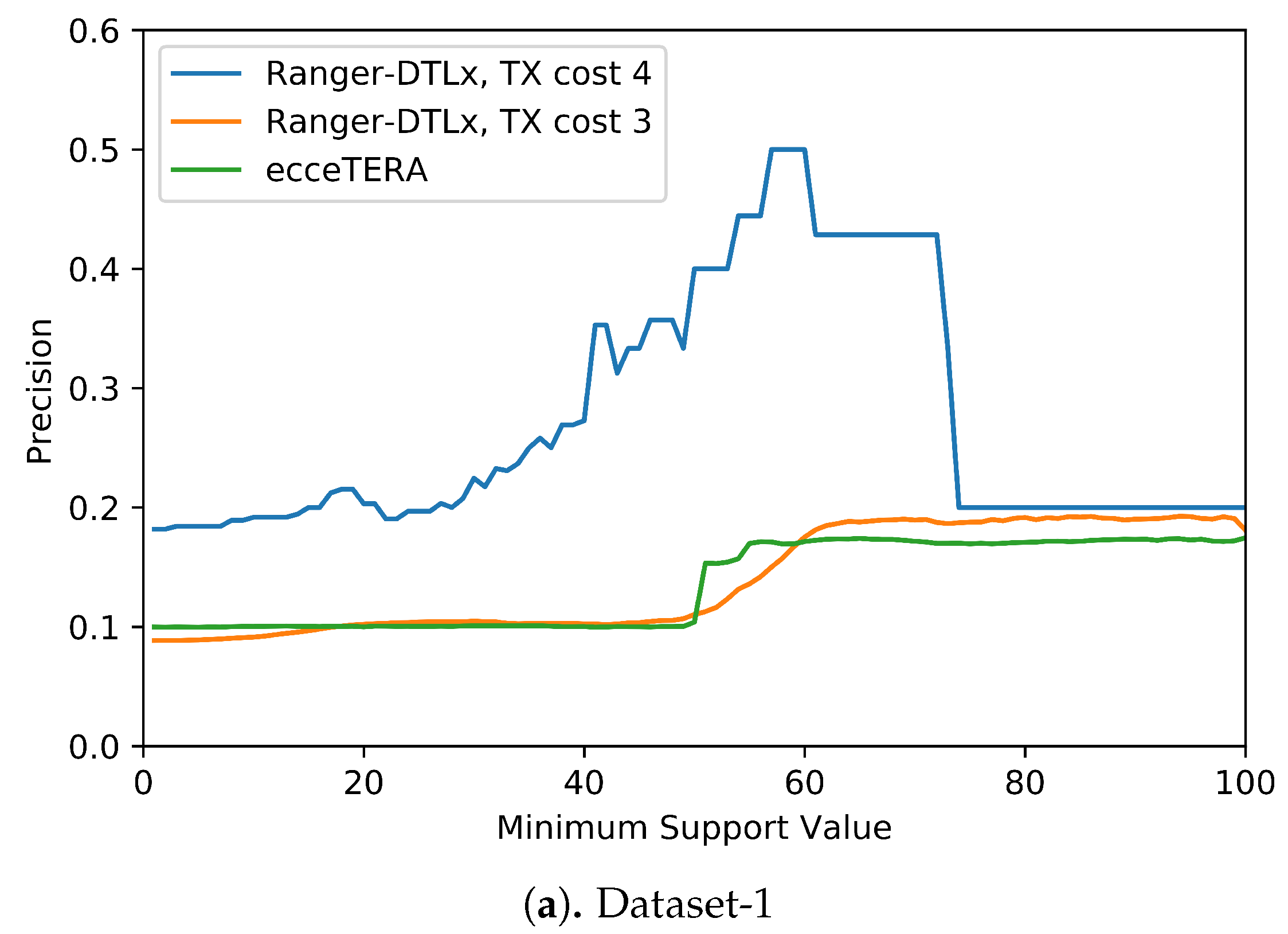
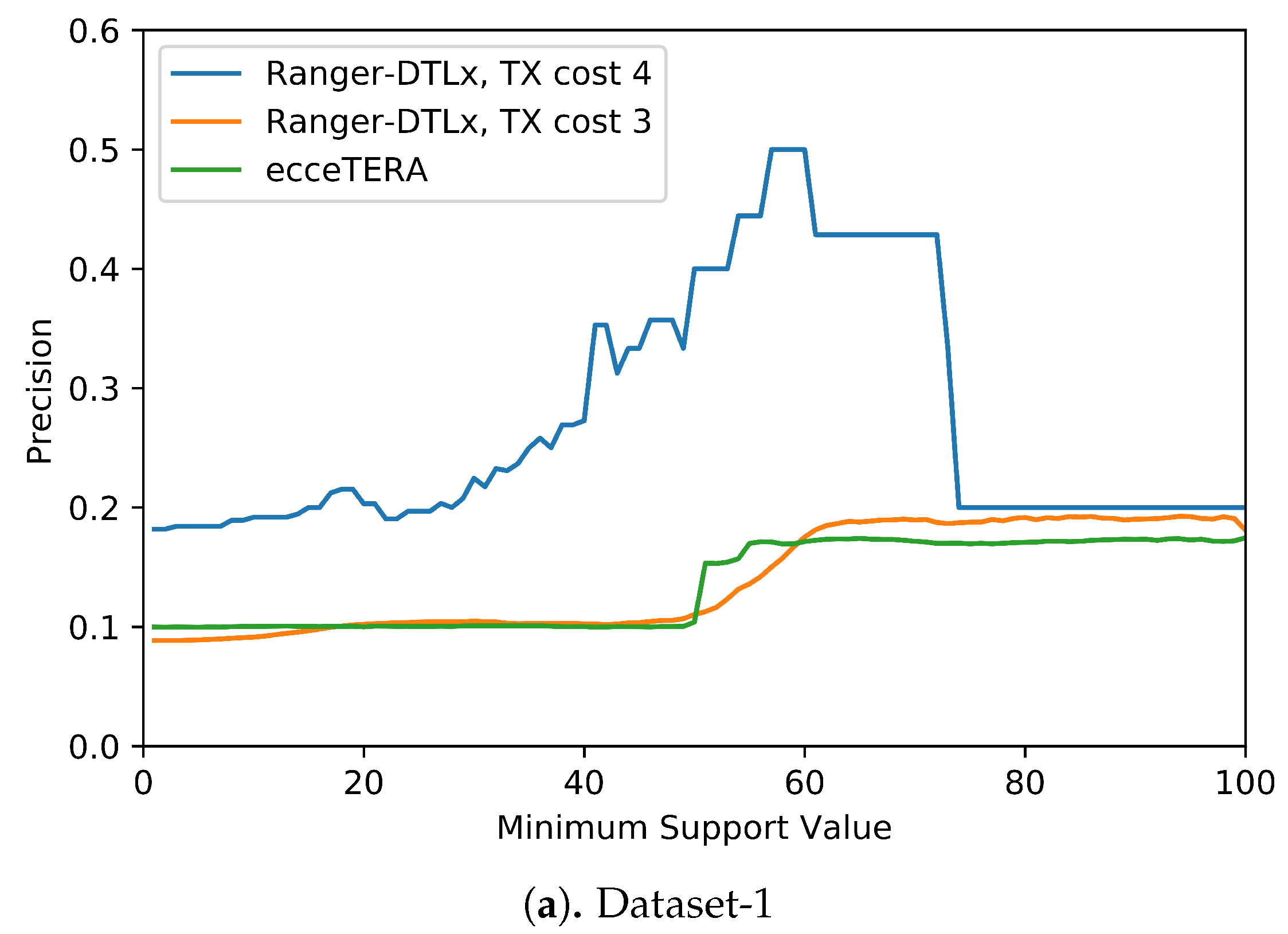
We also assessed if inferred
events that have higher support (i.e., are inferred in at least some fraction of all optimal reconciliations for that gene tree) are more likely to be correct (i.e., show higher precision).
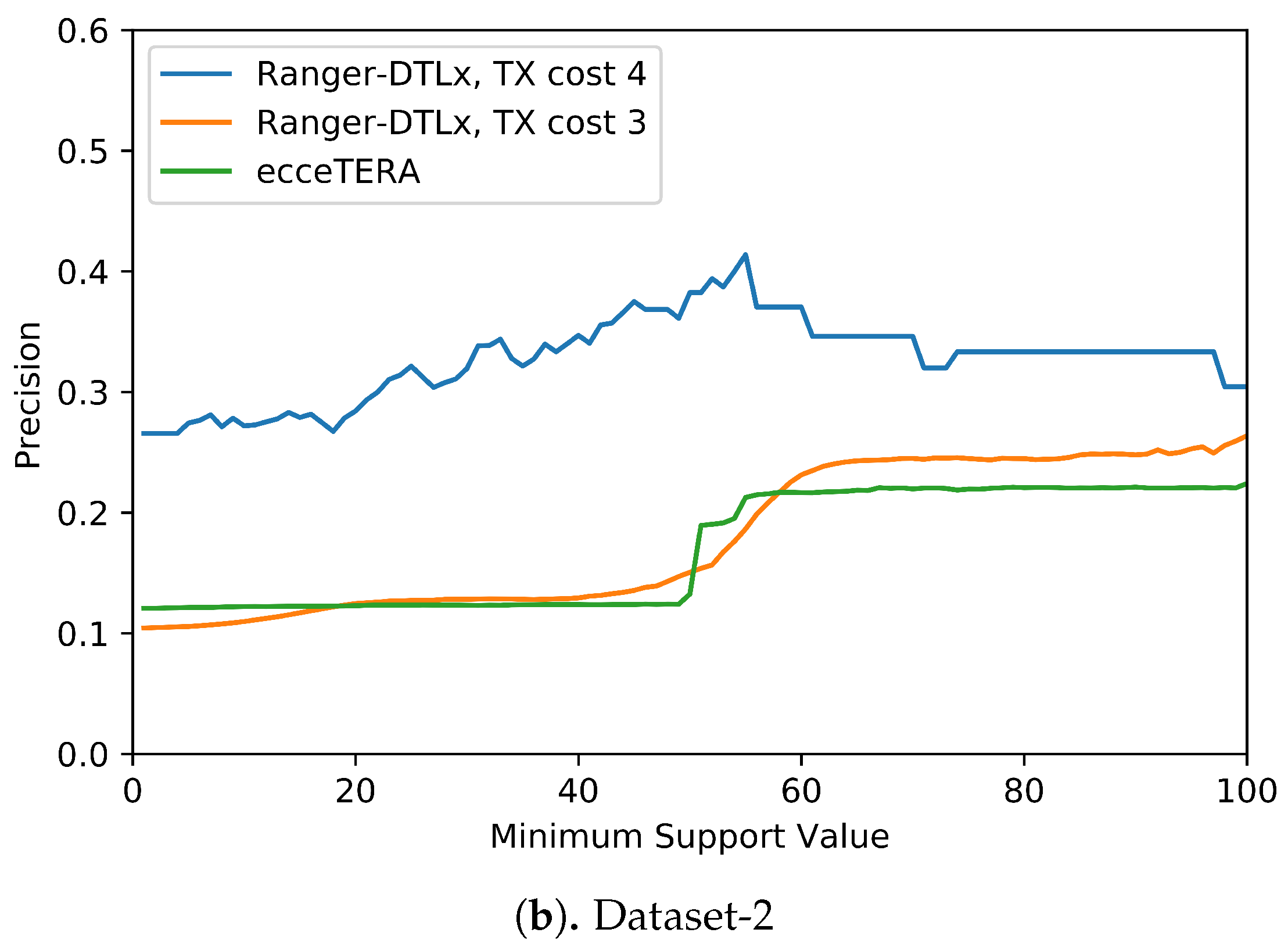
Figure 5 shows the results of this analysis. These results suggest that higher support values do tend to result in greater precision. More precisely, we find that RANGER-DTLx with
cost 3 and ecceTERA both shown similar trends in precision, with precision on Dataset-1 increasing from a low of about 0.1 for all inferred
events irrespective of support to a high of about 0.18 if only considering
events with 100% support, and precision on Dataset-2 increasing from about 0.1 for all inferred
events irrespective of support to a high of approximately 0.25 if only considering
events with 100% support. However, as expected, the increase in precision comes at the expense of recall (
Table 4). For example, on Dataset-2, RANGER-DTLx with
cost 3 infers a total of 14,134 distinct
events with support
among 100 optimal reconciliation samples but only 940 of these have 100% support, and the corresponding numbers for ecceTERA are 12,884 and 2088, respectively. Remarkably, as
Figure 5 shows, RANGER-DTLx with
cost 4 achieves a precision of almost 50% on Dataset-1 and over 40% on Dataset-2 when using a support threshold of about 55%. However, as
Table 4 shows, the number of
events inferred at that support level by RANGER-DTLx with
cost 4 is relatively small. The small number of these events likely explains the drop in precision for RANGER-DTLx with
cost 4 as the support threshold in increased past ∼60.
Phylogenetic placement of event donors. Identifying the locations on the species tree of unsampled lineages that served as donors for
events can offer important insights regarding major unsampled species lineages and can help to better understand the evolutionary histories of gene families. By design, RANGER-DTLx identifies a specific edge/lineage in the species tree where any given
event originates from. However, ecceTERA handles
events by introducing a single unsampled branch existing outside of the species tree, which represents all unsampled lineages on the species tree and serves as the donor of all
events. This makes it difficult to infer the location of unsampled donor lineages on the species tree using ecceTERA. To assess the accuracy of RANGER-DTLx in identifying/placing the unsampled species donor on the species tree, we used all
events (across all 100 samples, regardless of support) inferred by RANGER-DTLx that mapped to the correct recipient species and computed the distance on the species tree between the inferred location of the donor and the actual location of the donor (as given by Zombi). This distance is defined as the number of edges between the inferred donor lineage and the true lineage on the full species tree (including extinct lineages) as simulated by Zombi. As
Table 5 shows, RANGER-DTLx performs remarkably well at inferring the location of the unsampled species donor on the species tree, identifying the exact location for almost half the
events when the
event cost is 3. Results are worse for RANGER-DTLx with
cost 4, but this is likely due to small sample sizes. These results also show that even when the donor lineage is not identified exactly, the inferred placement is often close to the actual placement of the unsampled donor. For completeness, results are also shown for ecceTERA and, as expected, the performance of ecceTERA is much worse than that of RANGER-DTLx, both for exact matches and average distance.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}