
Network Inference from Gene Expression Data with Distance Correlation and Network Topology Centrality




Abstract
1. Introduction
2. Methods
2.1. Distance Correlation
2.2. Network Topology Centrality
2.3. GRNs Inference with DCNTC Algorithm
2.3.1. Initialization of Regulatory Relationships
2.3.2. Calculation of the Network Topology Centrality and Optimization of the Ranking
2.3.3. Inference of the Regulatory Network Structure
| Algorithm 1 DCNTC algorithm |
| Input: Microarray data , the threshold Output: A gene network
|
3. Results
3.1. Results for DREAM3 Challenge Network
3.2. Result for SOS Network in E. coil
4. Discussion
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bansal, M.; Belcastro, V.; Ambesi-Impiombato, A.; Bernardo, D.D. How to infer gene networks from expression profiles. Mol. Syst. Biol. 2007, 3, 78. [Google Scholar] [CrossRef]
- Markowetz, F.; Spang, R. Inferring cellular networks—A review. BMC Bioinform. 2007, 8, 1–17. [Google Scholar] [CrossRef]
- Basso, K.; Margolin, A.A.; Stolovitzky, G.; Klein, U.; Dalla-Favera, R.; Califano, A. Reverse Engineering of Regulatory Networks in Human B Cells. Nat. Genet. 2005, 37, 382–390. [Google Scholar] [CrossRef]
- Margolin, A.A.; Wang, K.; Lim, W.K.; Kustagi, M.; Nemenman, I.; Califano, A. Reverse engineering cellular networks. Nat. Protoc. 2006, 1, 662–671. [Google Scholar] [CrossRef]
- Marbach, D.; Prill, R.J.; Schaffter, T.; Mattiussi, C.; Floreano, D.; Stolovitzky, G. Revealing strengths and weaknesses of methods for gene network inference. Proc. Natl. Acad. Sci. USA 2010, 107, 6286–6291. [Google Scholar] [CrossRef]
- Liang, S. REVEAL, a general reverse engineering algorithm for inference of genetic network architectures. In Proceedings of the Pacific Symposium on Biocomputing Pacific Symposium on Biocomputing, Hawaii, HI, USA, 4–9 January 1998. [Google Scholar]
- Ruz, G.A.; Goles, E. Learning gene regulatory networks using the bees algorithm. Neural Comput. Appl. 2013, 22, 63–70. [Google Scholar] [CrossRef]
- Butte, A.J.; Kohane, I.S. Mutual information relevance networks: Functional genomic clustering using pairwise entropy measurements. In Proceedings of the Pacific Symposium on Biocomputing. Pacific Symposium on Biocomputing, Hawaii, HI, USA, 4–9 January 2000; Volume 5, pp. 418–429. [Google Scholar]
- Faith, J.J.; Hayete, B.; Thaden, J.T.; Mogno, I.; Wierzbowski, J.; Cottarel, G.; Kasif, S.; Collins, J.J.; Gardner, T.S. Large-Scale Mapping and Validation of Escherichia coli Transcriptional Regulation from a Compendium of Expression Profiles. PLoS Biol. 2007, 5, e8. [Google Scholar] [CrossRef]
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.H.; Stolovitzky, G.; Favera, R.D.; Califano, A. ARACNE: An Algorithm for the Reconstruction of Gene Regulatory Networks in a Mammalian Cellular Context. BMC Bioinform. 2006, 7, 1–15. [Google Scholar] [CrossRef]
- Meyer, P.E.; Kontos, K.; Lafitte, F.; Bontempi, G. Information-Theoretic Inference of Large Transcriptional Regulatory Networks. Eurasip J. Bioinform. Syst. Biol. 2007, 2007, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, X.; He, K.; Lu, L.; Cao, Y.; Liu, J.; Hao, J.; Liu, Z.; Chen, L. Inferring gene regulatory networks from gene expression data by path consistency algorithm based on conditional mutual information. Bioinformatics 2012, 28, 98–104. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, J.; Hao, J.; Zhao, X.; Chen, L. Conditional mutual inclusive information enables accurate quantification of associations in gene regulatory networks. Nucleic Acids Res. 2015, 43, e31. [Google Scholar] [CrossRef]
- Szekely, G.J.; Rizzo, M.L.; Bakirov, N.K. Measuring and testing dependence by correlation of distances. Ann. Stat. 2007, 35, 2769–2794. [Google Scholar] [CrossRef]
- Li, R.; Zhong, W.; Zhu, L. Feature Screening via Distance Correlation Learning. J. Am. Stat. Assoc. 2012, 107, 1129–1139. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, Y.; Hu, W.; Tan, H.; Wang, X. Inferring Nonlinear Gene Regulatory Networks from Gene Expression Data Based on Distance Correlation. PLoS ONE 2014, 9, e87446. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, D.; Chu, J.; Zhu, Y.; Cheng, X. A Sparse Bayesian Learning Method for Structural Equation Model-based Gene Regulatory Network Inference. IEEE Access 2020, 8, 40067–40080. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, M.; Zhang, D. Two-Stage Penalized Least Squares Method for Constructing Large Systems of Structural Equations. Statistics 2015, 19, 40–73. [Google Scholar]
- Kim, J.; Kim, I.; Han, S.K.; Bowie, J.U.; Kim, S. Network rewiring is an important mechanism of gene essentiality change. Sci. Rep. 2012, 2, 900. [Google Scholar] [CrossRef]
- Estrada, E.; Rodriguezvelazquez, J.A. Subgraph centrality in complex networks. Phys. Rev. E 2005, 71, 056103. [Google Scholar] [CrossRef]
- Ruyssinck, J.; Demeester, P.; Dhaene, T.; Saeys, Y. Netter: Re-ranking gene network inference predictions using structural network properties. BMC Bioinform. 2016, 17, 76. [Google Scholar] [CrossRef]
- Jeong, H.; Mason, S.P. Lethality andcentrality in protein networks. Nature 2001, 411, 41–42. [Google Scholar] [CrossRef]
- Liu, W. Research on the Structure Prediction Algorithm of Gene Regulation Network Based on Information Theory. Ph.D. Thesis, Hunan University, Hunan, China, 2017. (In Chinese). [Google Scholar]
- Kosorok, M.R. Discussion of: Brownian distance covariance. Ann. Appl. Stat. 2009, 3, 1270–1278. [Google Scholar] [CrossRef]
- Rizzo ML, S.G. Energy: E-Statistics (Energy Statistics). R Package Version 1.6.2. 2014. Available online: http://CRAN.R-project.org/package=energy (accessed on 1 May 2019).
- Batool, K.; Niazi, M.A. Correction: Towards a Methodology for Validation of Centrality Measures in Complex Networks. PLoS ONE 2014, 9, e98379. [Google Scholar] [CrossRef]
- Liu, W.; Zhu, W.; Liao, B.; Chen, H.; Ren, S.; Cai, L. Improving gene regulatory network structure using redundancy reduction in the MRNET algorithm. RSC Adv. 2017, 7, 23222–23233. [Google Scholar] [CrossRef]
- Meyer, P.E.; Lafitte, F.; Bontempi, G. minet: A R/Bioconductor Package for Inferring Large Transcriptional Networks Using Mutual Information. BMC Bioinform. 2008, 9, 1–10. [Google Scholar] [CrossRef]
- Ronen, M.; Rosenberg, R.; Shraiman, B.I.; Alon, U. Assigning numbers to the arrows: Parameterizing a gene regulation network by using accurate expression kinetics. Proc. Natl. Acad. Sci. USA 2002, 99, 10555–10560. [Google Scholar] [CrossRef]
- Shenorr, S.S.; Milo, R.; Mangan, S.; Alon, U. Network motifs in the transcriptional regulation network of Escherichia coli. Nat. Genet. 2002, 31, 64–68. [Google Scholar] [CrossRef]
- Matthews, B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Hache, H.; Wierling, C.; Lehrach, H.; Herwig, R. GeNGe: Systematic Generation of Gene Regulatory Networks. Bioinformatics 2009, 25, 1205–1207. [Google Scholar] [CrossRef] [PubMed]
- Gabrys, B. Propagation Phenomena in Real World Networks. Intell. Syst. Ref. Libr. 2015, 85, 1–24. [Google Scholar]
- Dablander, F.; Hinne, M. Node Centrality Measures are a Poor Substitute for Causal Inference. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Freeman, L.C. Centrality in social networks conceptual clarification. Soc. Netw. 1978, 1, 215–239. [Google Scholar] [CrossRef]



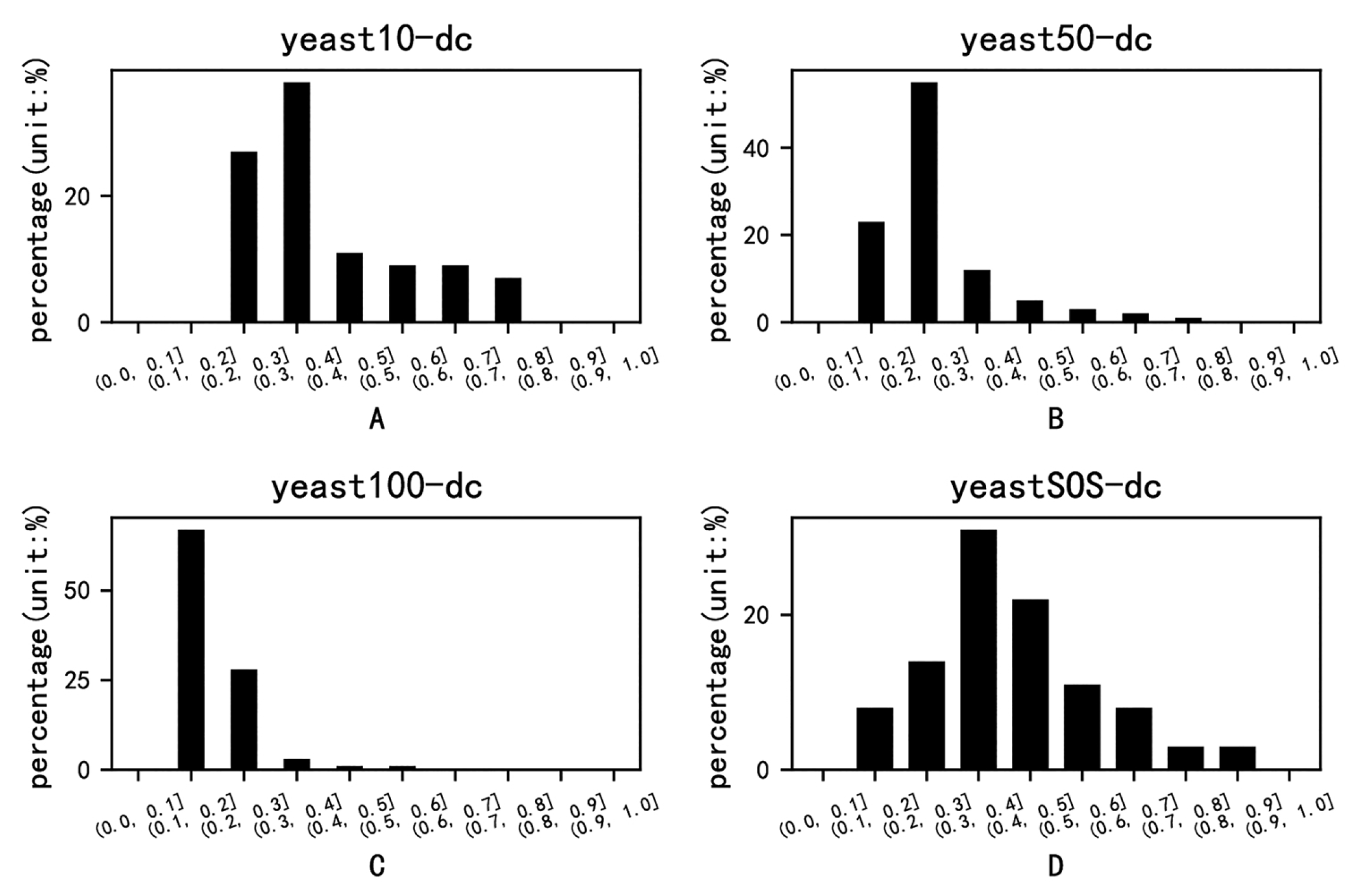{kind=link}
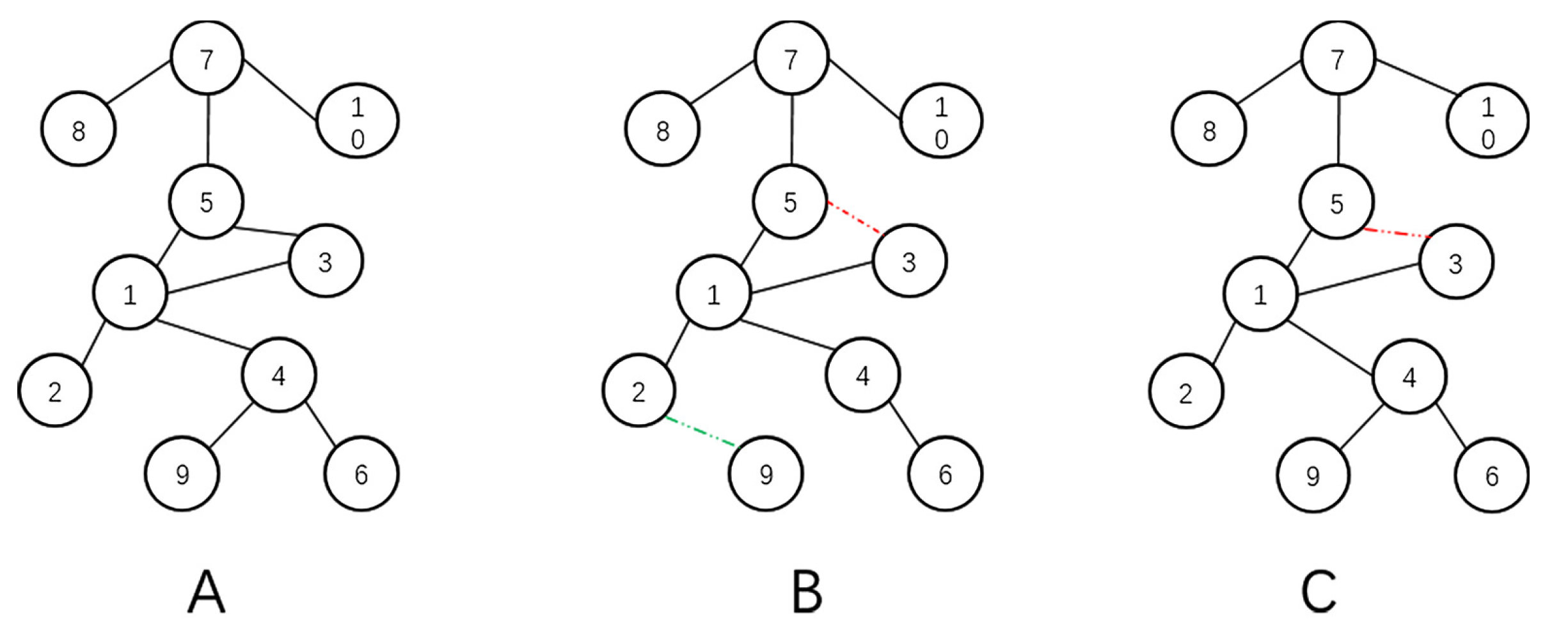{kind=link}
| Method | TP | FP | TN | FN | TPR | FPR | PPV | ACC | MCC |
|---|---|---|---|---|---|---|---|---|---|
| CLR | 6 | 10 | 25 | 4 | 0.600 | 0.286 | 0.375 | 0.689 | 0.273 |
| ARACNE | 6 | 6 | 29 | 4 | 0.600 | 0.171 | 0.500 | 0.778 | 0.403 |
| MRNET | 6 | 12 | 23 | 4 | 0.600 | 0.343 | 0.333 | 0.644 | 0.218 |
| REL-DC | 10 | 4 | 31 | 0 | 1 | 0.114 | 0.714 | 0.911 | 0.795 |
| MRNET-DC | 10 | 13 | 22 | 0 | 1 | 0.371 | 0.435 | 0.711 | 0.523 |
| LDCNET | 8 | 1 | 34 | 2 | 0.8 | 0.029 | 0.889 | 0.933 | 0.802 |
| DCNTC | 9 | 0 | 35 | 1 | 0.9 | 0 | 1 | 0.978 | 0.936 |
| Method | TP | FP | TN | FN | TPR | FPR | PPV | ACC | MCC |
|---|---|---|---|---|---|---|---|---|---|
| CLR | 19 | 165 | 983 | 58 | 0.247 | 0.144 | 0.103 | 0.818 | 0.070 |
| ARACNE | 13 | 125 | 1023 | 64 | 0.170 | 0.109 | 0.094 | 0.846 | 0.046 |
| MRNET | 21 | 215 | 933 | 56 | 0.273 | 0.187 | 0.089 | 0.779 | 0.053 |
| REL-DC | 34 | 49 | 1099 | 43 | 0.442 | 0.043 | 0.410 | 0.925 | 0.385 |
| MRNET-DC | 70 | 465 | 683 | 7 | 0.909 | 0.405 | 0.131 | 0.615 | 0.247 |
| LDCNET | 23 | 26 | 1122 | 54 | 0.299 | 0.023 | 0.469 | 0.935 | 0.342 |
| DCNTC | 29 | 20 | 1128 | 48 | 0.377 | 0.017 | 0.592 | 0.945 | 0.445 |
| Method | TP | FP | TN | FN | TPR | FPR | PPV | ACC | MCC |
|---|---|---|---|---|---|---|---|---|---|
| CLR | 39 | 713 | 4071 | 127 | 0.235 | 0.149 | 0.052 | 0.830 | 0.044 |
| ARACNE | 20 | 417 | 4367 | 146 | 0.121 | 0.087 | 0.046 | 0.886 | 0.403 |
| MRNET | 49 | 984 | 3800 | 117 | 0.295 | 0.206 | 0.047 | 0.778 | 0.040 |
| REL-DC | 121 | 386 | 4398 | 45 | 0.729 | 0.081 | 0.239 | 0.913 | 0.385 |
| MRNET-DC | 145 | 2011 | 2773 | 21 | 0.874 | 0.421 | 0.067 | 0.590 | 0.165 |
| LDCNET | 45 | 54 | 4730 | 121 | 0.271 | 0.011 | 0.455 | 0.965 | 0.334 |
| DCNTC | 55 | 42 | 4742 | 111 | 0.331 | 0.009 | 0.567 | 0.969 | 0.419 |
| Method | TP | FP | TN | FN | TPR | FPR | PPV | ACC | MCC |
|---|---|---|---|---|---|---|---|---|---|
| CLR | 12 | 5 | 7 | 12 | 0.500 | 0.417 | 0.706 | 0.528 | 0.079 |
| ARACNE | 7 | 3 | 9 | 17 | 0.292 | 0.250 | 0.700 | 0.444 | 0.044 |
| MRNET | 17 | 6 | 6 | 7 | 0.708 | 0.500 | 0.739 | 0.639 | 0.205 |
| REL-DC | 6 | 3 | 9 | 18 | 0.250 | 0.250 | 0.667 | 0.417 | 0 |
| MRNET-DC | 12 | 9 | 3 | 12 | 0.500 | 0.750 | 0.572 | 0.417 | −0.239 |
| LDCNET | 6 | 1 | 11 | 18 | 0.250 | 0.083 | 0.857 | 0.472 | 0.199 |
| DCNTC | 6 | 2 | 10 | 18 | 0.25 | 0.167 | 0.75 | 0.444 | 0.095 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, K.; Liu, H.; Sun, D.; Zhang, L. Network Inference from Gene Expression Data with Distance Correlation and Network Topology Centrality. Algorithms 2021, 14, 61. https://doi.org/10.3390/a14020061
Liu K, Liu H, Sun D, Zhang L. Network Inference from Gene Expression Data with Distance Correlation and Network Topology Centrality. Algorithms. 2021; 14(2):61. https://doi.org/10.3390/a14020061
Chicago/Turabian StyleLiu, Kuan, Haiyuan Liu, Dongyan Sun, and Lei Zhang. 2021. "Network Inference from Gene Expression Data with Distance Correlation and Network Topology Centrality" Algorithms 14, no. 2: 61. https://doi.org/10.3390/a14020061
APA StyleLiu, K., Liu, H., Sun, D., & Zhang, L. (2021). Network Inference from Gene Expression Data with Distance Correlation and Network Topology Centrality. Algorithms, 14(2), 61. https://doi.org/10.3390/a14020061