
Cationic Site-Preference in the Yb14-xCaxAlSb11 (4.81 ≤ x ≤ 10.57) Series: Theoretical and Experimental Studies

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
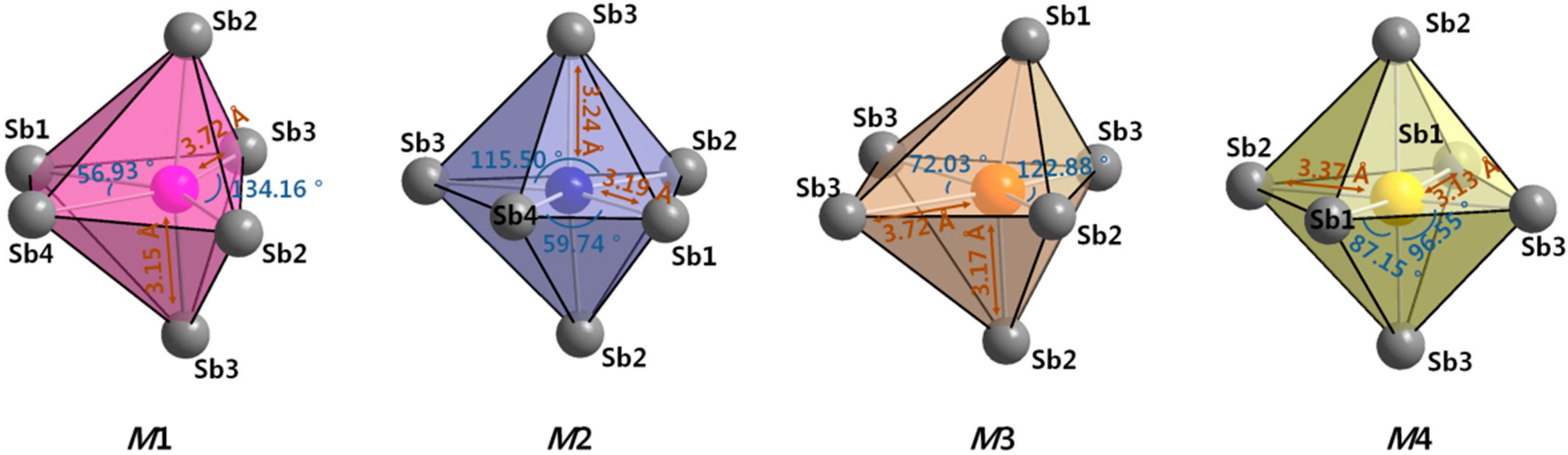
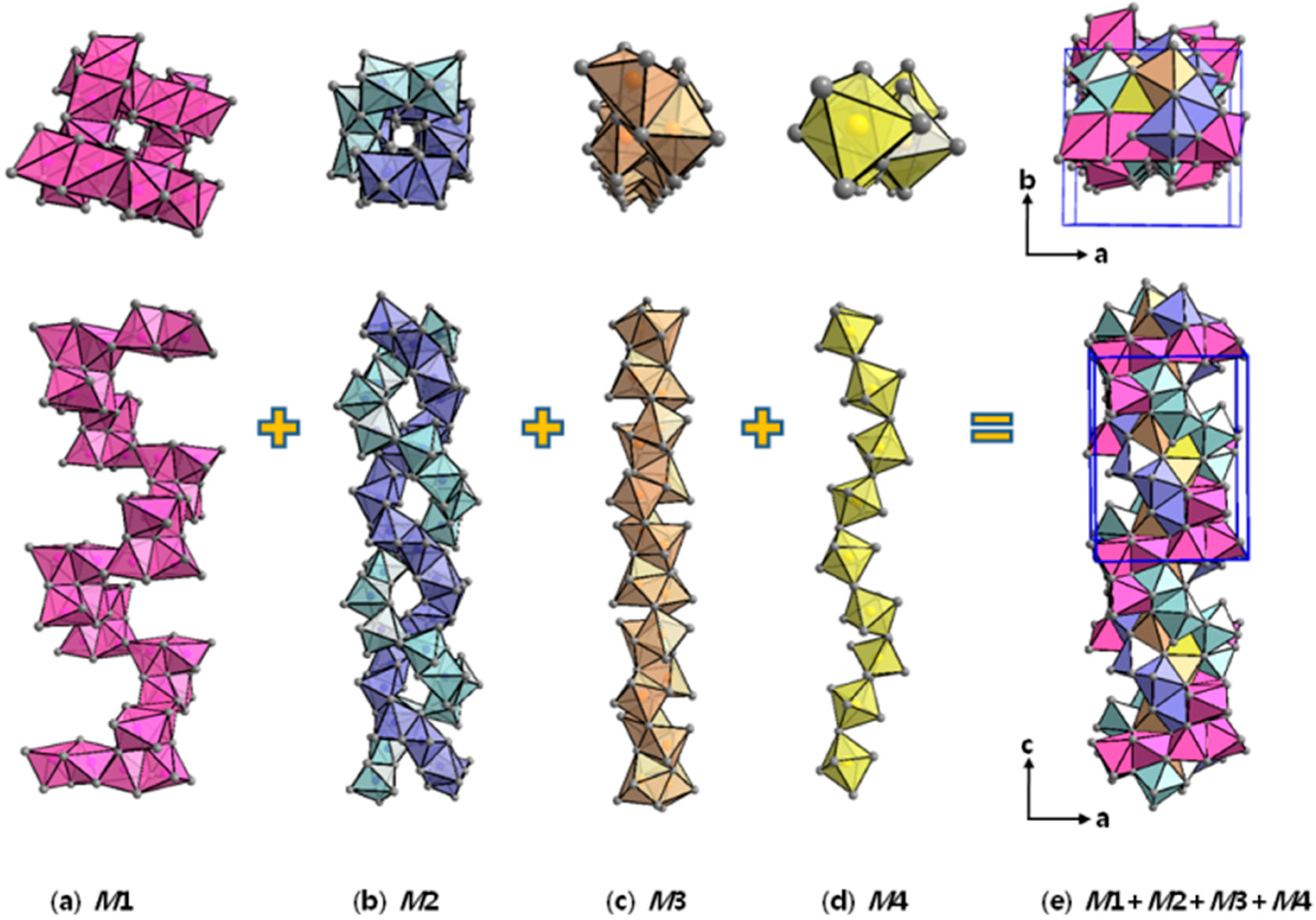
2.1. Crystal Structure Analysis
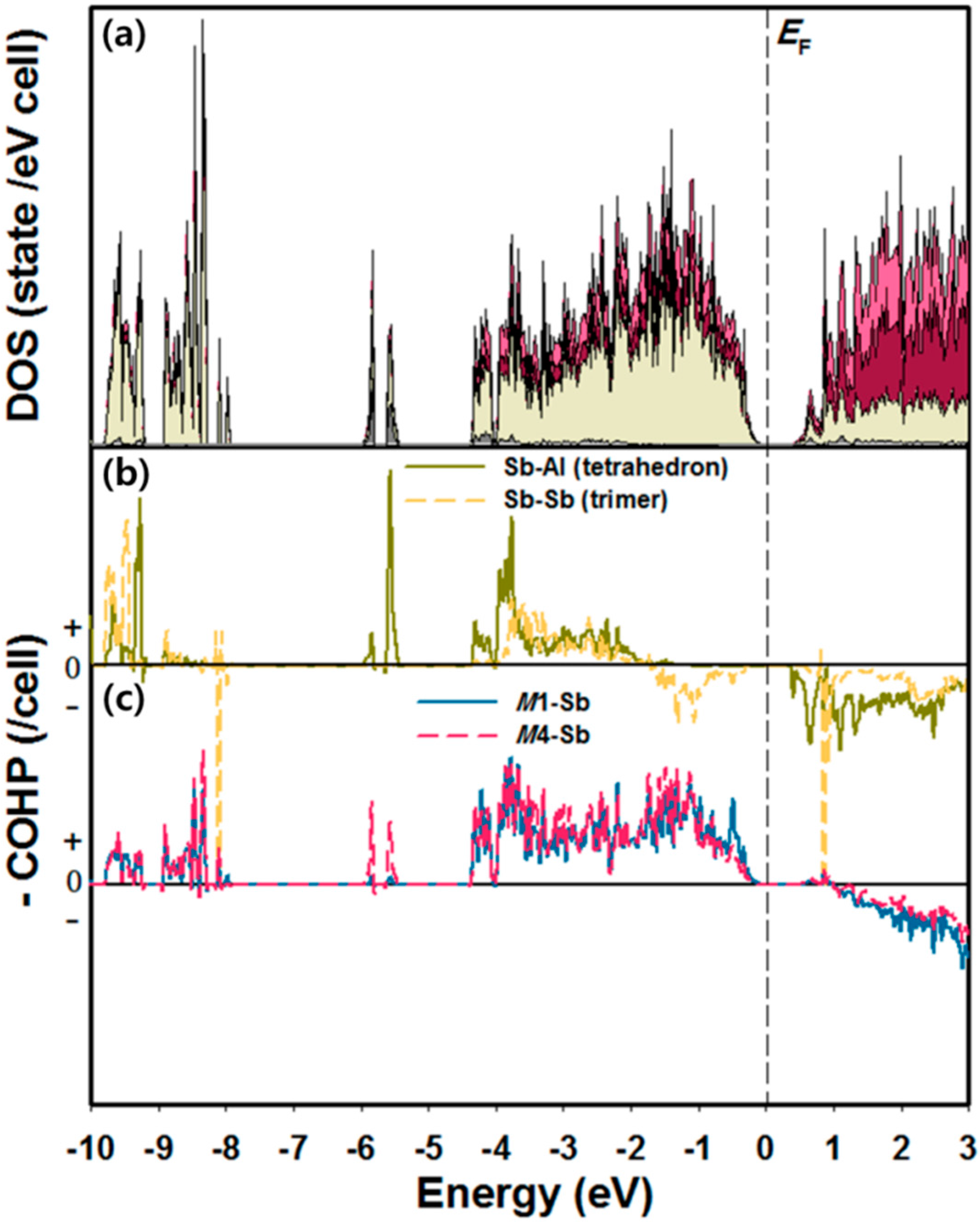
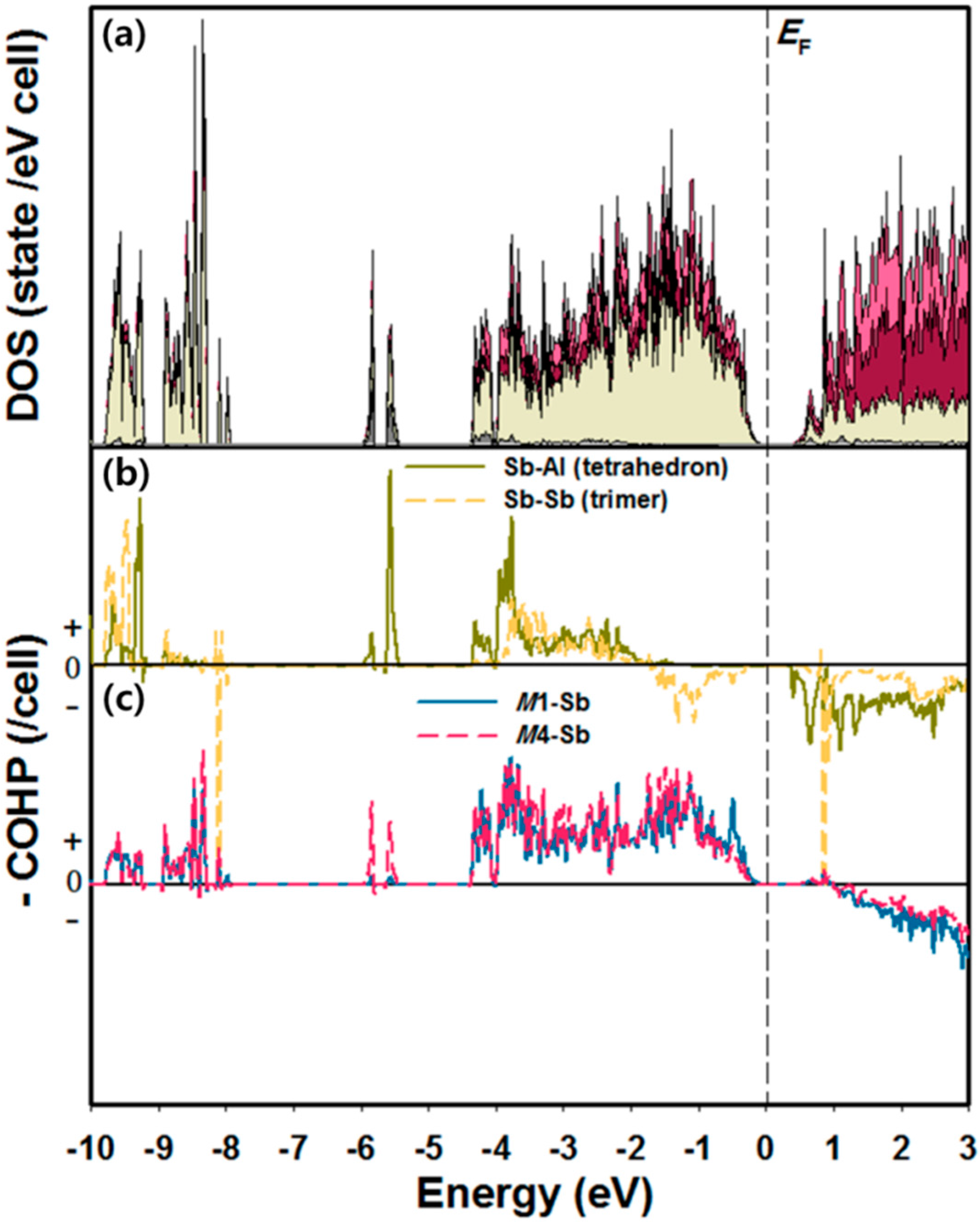
2.2. Site Preference and Electronic Structure
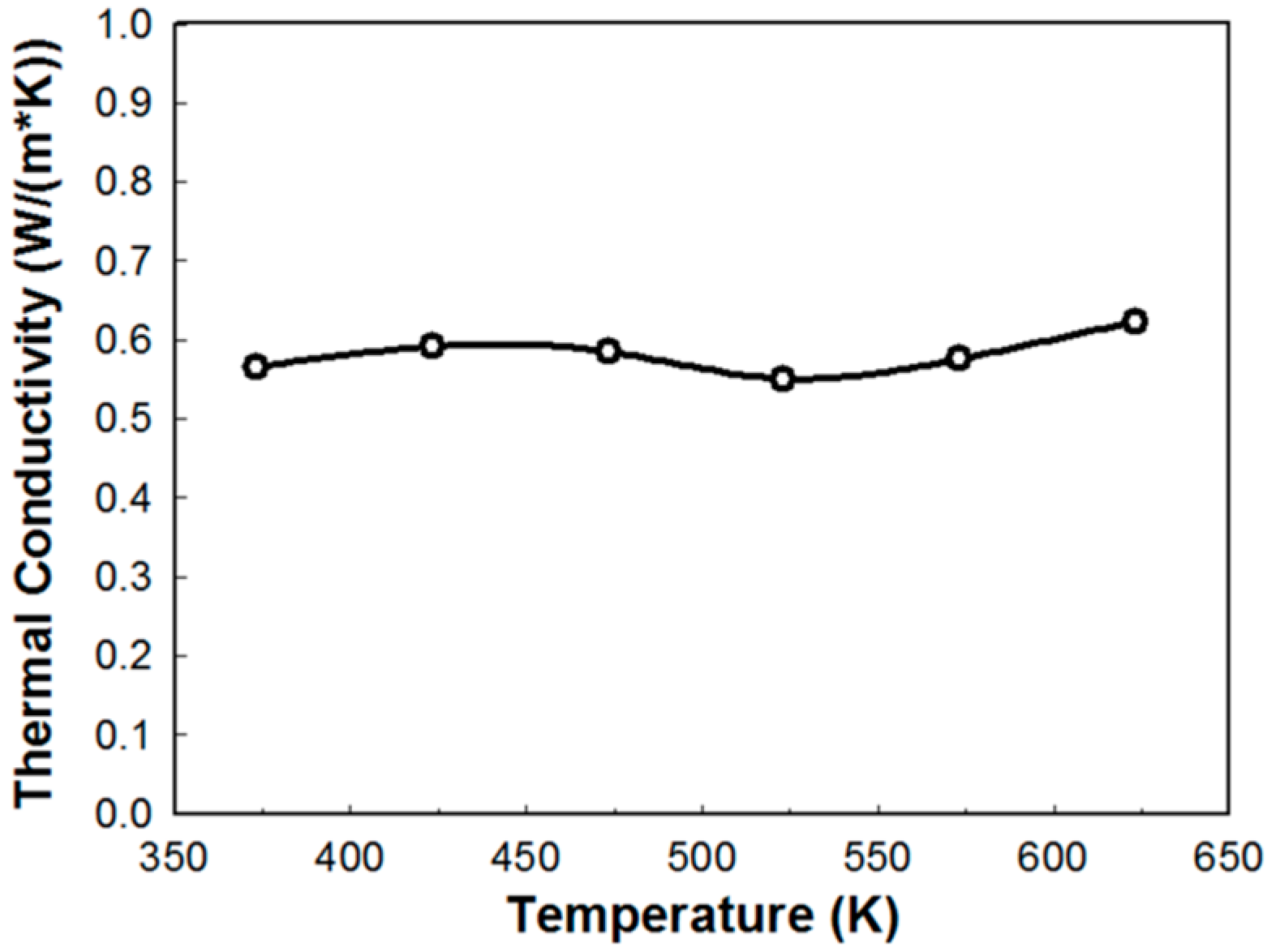
2.3. Thermal Transport Property
3. Materials and Methods
3.1. Synthesis
3.2. Powder and Single-Crystal X-ray Diffraction Experiments
3.3. Electronic Structure Calculations
3.4. Thermal Conductivity Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Snyder, G.J.; Toberer, E.S. Complex thermoelectric materials. Nat. Mater. 2008, 7, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Yang, J.; Salvador, J.R.; Chi, M.; Cho, J.Y.; Wang, H.; Bai, S.; Yang, J.; Zhang, W.; Chen, L. Multiple-Filled Skutterudites: High Thermoelectric Figure of Merit through Separately Optimizing Electrical and Thermal Transports. J. Am. Chem. Soc. 2011, 133, 7837–7846. [Google Scholar] [CrossRef] [PubMed]
- Yanzhong, P.; Xiaoya, S.; LaLonde, A.; Heng, W.; Lidong, C.; Snyder, G.J. Convergence of electronic bands for high performance bulk thermoelectrics. Nature 2011, 473, 66–69. [Google Scholar]
- Biswas, K.; He, J.; Blum, I.D.; Wu, C.I.; Hogan, T.P.; Seidman, D.N.; Dravid, V.P.; Kanatzidis, M.G. High-performance bulk thermoelectrics with all-scale hierarchical architectures. Nature 2012, 489, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-D.; Lo, S.-H.; Zhang, Y.; Sun, H.; Tan, G.; Uher, C.; Wolverton, C.; Dravid, V.P.; Kanatzidis, M.G. Ultralow thermal conductivity and high thermoelectric figure of merit in SnSe crystals. Nature 2014, 508, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Pope, A.L.; Littleton, R.T., IV; Terry, M.T. Effect of Sb doping on the thermoelectric properties of Ti-based half-Heusler compounds, TiNiSn1-xSbx. Appl. Phys. Lett. 2000, 77, 2476–2478. [Google Scholar] [CrossRef]
- Chung, D.Y.; Hogan, T.P.; Rocci-Lane, M.; Brazis, P.; Ireland, J.R.; Kannewurf, C.R.; Bastea, M.; Uher, C.; Kanatzidis, M.G. A New Thermoelectric Material: CsBi4Te6. J. Am. Chem. Soc. 2004, 126, 6414–6428. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Sootsman, J.R.; Girard, S.N.; Zheng, J.-C.; Wen, J.; Zhu, Y.; Kanatzidis, M.G.; Dravid, V.P. On the Origin of Increased Phonon Scattering in Nanostructured PbTe Based Thermoelectric Materials. J. Am. Chem. Soc. 2010, 132, 8669–8675. [Google Scholar] [CrossRef] [PubMed]
- Soni, A.; Yanyuan, Z.; Ligen, Y.; Aik, M.K.K.; Dresselhaus, M.S.; Xiong, Q. Enhanced Thermoelectric Properties of Solution Grown Bi2Te3–xSex Nanoplatelet Composites. Nano Lett. 2012, 12, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.R.; Kauzlarich, S.M.; Gascoin, F.; Snyder, G.J. Yb14MnSb11: New High Efficiency Thermoelectric Material for Power Generation. Chem. Mater. 2006, 18, 1873–1877. [Google Scholar] [CrossRef]
- Toberer, E.S.; Catherine, A.C.; Brown, S.R.; Ikeda, T.; May, A.F.; Kauzlarich, S.M.; Snyder, G.J. Traversing the Metal-Insulator Transition in a Zintl Phase: Rational Enhancement of Thermoelectric Efficiency in Yb14Mn1−xAlxSb11. Adv. Funct. Mater. 2008, 18, 2795–2800. [Google Scholar] [CrossRef]
- Brown, S.R.; Toberer, E.S.; Ikeda, T.; Cox, C.A.; Gascoin, F.; Kauzlarich, S.M.; Snyder, G.J. Improved Thermoelectric Performance in Yb14Mn1−xZnxSb11 by the Reduction of Spin-Disorder Scattering. Chem. Mater. 2008, 20, 3412–3419. [Google Scholar] [CrossRef]
- Yi, T.; Abdusalyamova, M.N.; Makhmudov, F.; Kauzlarich, S.M. Magnetic and transport properties of Te doped Yb14MnSb11. J. Mater. Chem. 2012, 22, 14378–14384. [Google Scholar] [CrossRef]
- Uvarov, C.A.; Ortega-Alvarez, F.; Kauzlarich, S.M. Enhanced High-Temperature Thermoelectric Performance of Yb14−xCaxMnSb11. Inorg. Chem. 2012, 51, 7617–7624. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Chen, Y.; Xie, H.; Snyder, G.J.; Fu, C.; Xu, J.; Zhao, X.; Zhu, T. Improved Thermoelectric Properties in Lu-doped Yb14MnSb11 Zintl Compounds. Appl. Phys. Express 2012, 5, 031801. [Google Scholar] [CrossRef]
- Woo, H.; Nam, G.; Jang, E.; Kim, J.; Lee, Y.; Ahn, K.; You, T.-S. Single-Crystal Growth and Size Control of Three Novel Polar Intermetallics: Eu2.94(2)Ca6.06In8Ge8, Eu3.13(2)Ca5.87In8Ge8 and Sr3.23(3)Ca5.77In8Ge8 with Crystal Structure, Chemical Bonding and Magnetism Studies. Inorg. Chem. 2014, 53, 4669–4677. [Google Scholar] [CrossRef] [PubMed]
- Jeon, B.-Y.; Jeon, J.; Lee, J.; Kim, J.; You, T.-S. Experimental and Theoretical Investigations for Site-Preference and Anisotropic Size-Change of RE11Ge4In6-xMx (RE = La, Ce; M = Li or Ge/In-mixed site; x = 1, 1.96). J. Alloys Compd. 2015, 620, 269–276. [Google Scholar] [CrossRef]
- Jang, E.; Nam, G.; Woo, H.; Lee, J.; Ham, M.-K.; Kim, S.-J.; You, T.-S. Li-filled Double-deck Layered Structure of the RELixCu2-yP2 (RE = La, Pr, Nd, Gd, Er; 0.82 < x <1; 1.19 < y <1.54) Series: Experimental and Theoretical Studies. Eur. J. Inorg. Chem. 2015, 17, 2786–2793. [Google Scholar]
- Jeon, B.-Y.; Nam, G.; Lee, D.; Ok, K.; You, T.-S. Ce11Ge3.73(2)In6.27: Solid-state synthesis, crystal structure and site-preference. J. Sol. State Chem. 2016, 236, 195–202. [Google Scholar] [CrossRef]
- Cordier, G.; Schäfer, H.; Stelter, M. Preparation and structure of calcium aluminum antimonide (Ca14AlSb11). Z. Anorg. Allg. Chem. 1984, 519, 183–188. [Google Scholar] [CrossRef]
- Emsley, J. The Elements; Clarendon Press: Oxford, UK, 1989; pp. 40–208. [Google Scholar]
- Schellenberg, I.; Eul, M.; Hermes, W.; Pöttgen, R. A 121Sb and 151Eu Mössbauer Spectroscopic Investigation of EuMn2Sb2, EuZn2Sb2, YbMn2Sb2, and YbZn2Sb2. Z. Anorg. Allg. Chem. 2010, 636, 85–93. [Google Scholar] [CrossRef]
- Bobev, S.; Fritsch, V.; Thompson, J.D.; Sarrao, J.L.; Eck, B.; Dronskowski, R.; Kauzlarich, S.M. Synthesis, structure and properties of the new rare-earth Zintl phase Yb11GaSb9. J. Solid State Chem. 2005, 178, 1071–1079. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. 1976, A32, 751–767. [Google Scholar] [CrossRef]
- Andersen, O.K. Linear methods in band theory. Phys. Rev. B 1975, 12, 3060–3083. [Google Scholar] [CrossRef]
- Andersen, O.K.; Jepsen, O. Explicit, first-principles tight-binding theory. Phys. Rev. Lett. 1984, 53, 2571–2574. [Google Scholar] [CrossRef]
- Lambrecht, W.R.L.; Andersen, O.K. Minimal basis sets in the linear muffin-tin orbital method: Application to the diamond-structure crystals C, Si, and Ge. Phys. Rev. B 1986, 34, 2439–2449. [Google Scholar] [CrossRef]
- Jepsen, O.; Burkhardt, A.; Andersen, O.K. The TB-LMTO-ASA Program, version 4.7; Max-Plank-Institut fur Festkorperforschung: Shuttgart, Germany, 1999. [Google Scholar]
- Andersen, O.K.; Jepsen, O.; Glötzel, D. Canonical Description of the Band Structures of Metals. In Highlights of Condensed Matter Theory; Bassani, F., Fumi, F., Tosi, M., Eds.; Elsevier: North Holland, NY, USA, 1985. [Google Scholar]
- Misra, S.; Miller, G.J. Gd5−xYxTt4 (Tt = Si or Ge): Effect of Metal Substitution on Structure, Bonding, and Magnetism. J. Am. Chem. Soc. 2008, 130, 13900–13911. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Klavins, P.; Kauzlarich, S.M. Structure, Magnetism, and Magnetoresistance of the Rare-Earth Transition Metal Compounds Eu13AMnSb11 (A = Ca, Sr, Ba, and Yb). Chem. Mater. 2002, 14, 2308–2316. [Google Scholar] [CrossRef]
- You, T.-S.; Nam, G.; Kim, Y.; Darone, G.; Bobev, S. Structural Characterization of the Intermetallic Phase EuZnxIn4-x (x = 1.1–1.2). Zn and In site-preference in the BaAl4 structure type from cmputational analysis. Bull. Korean Chem. Soc. 2013, 34, 1656–1662. [Google Scholar] [CrossRef]
- Nam, G.; Jeon, J.; Kim, Y.; Kang, S.; Ahn, K.; You, T.-S. Combined effect of chemical pressure and valence electron concentration through the electron-deficient Li substitution on the RE4LiGe4 (RE=La, Ce, Pr, and Sm) system. J. Sol. State Chem. 2013, 205, 10–20. [Google Scholar] [CrossRef]
- Nam, G.; Jang, E.; You, T.-S. Site-Preference among Three Anions in the Quaternary BaAl4-Type Structure: Experimental and Computational Investigations for BaLi1.09(1)In0.91Ge2. Bull. Korean Chem. Soc. 2013, 34, 3847–3850. [Google Scholar] [CrossRef]
- Wang, H.; Misra, S.; Wang, F.; Miller, G.J. Structural and Magnetic Characteristics of Gd5GaxSi4−x. Inorg. Chem. 2010, 49, 4586–4593. [Google Scholar] [CrossRef] [PubMed]
- You, T.-S.; Miller, G.J. Phase Width and Site Preference in the EuMgxGa4-x Series. Z. Anorg. Allg. Chem. 2008, 634, 2845–2852. [Google Scholar] [CrossRef]
- Hunter, B.A. International Union of Crystallography, Commission for Powder Diffraction; Newsletter: Chester, UK, 1998; Volume 20, p. 21. [Google Scholar]
- Bruker, APEX2; Bruker AXS Inc.: Madison, WI, USA, 2007.
- Bruker, SAINT; Bruker AXS Inc.: Madison, WI, USA, 2002.
- Sheldrick, G.M. SADABS; University of Göttingen: Göttingen, Germany, 2003. [Google Scholar]
- Gelato, L.M.; Parthé, E. STRUCTURE TIDY—A computer program to standardize crystal structure data. J. Appl. Crystallogr. 1987, 20, 139–143. [Google Scholar] [CrossRef]
- Jepsen, O.; Andersen, O.K. Calculated electronic structure of the sandwich d1 metals LaI2 and CeI2: Application of new LMTO techniques. Z. Phys. B 1995, 97, 35–47. [Google Scholar] [CrossRef]
- Dronskowski, R.; Blöchl, P.E. Crystal orbital Hamilton populations (COHP): Energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 1993, 97, 8617–8624. [Google Scholar] [CrossRef]
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223–16233. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Compositions | Yb9.19(3)Ca4.81AlSb11 | Yb8.42(4)Ca5.58AlSb11 | Yb5.12(2)Ca8.98AlSb11 | Yb3.43(2)Ca10.57AlSb11 | |
|---|---|---|---|---|---|
| Formula weight (g/mol) | 3149.25 | 3046.87 | 2640.45 | 2383.40 | |
| space group; Z | I41/acd; 8 | ||||
| Lattice parameters Å | A | 16.5704(5) | 16.6366(8) | 16.6374(6) | 16.6488(2) |
| C | 22.1967(7) | 22.2244(12) | 22.3365(8) | 22.3684(3) | |
| Volume (Å3) | 6094.7(4) | 6151.2(7) | 6182.8(5) | 6200.1(2) | |
| dcalcd (g/cm3) | 6.864 | 6.580 | 5.609 | 5.107 | |
| θ range for data collection | 2.90°–27.86° | 2.89°–26.37° | 2.45°–29.13° | 2.45°–28.27° | |
| Reflections collected | 73,570 | 28,005 | 22,936 | 22,847 | |
| Data/restraints/parameters | 1826/0/65 | 1579/0/65 | 2086/0/65 | 1929/0/65 | |
| GOF on F2 | 1.075 | 1.044 | 1.177 | 1.227 | |
| R 1 indices (all data) | R1 | 0.0364 | 0.0498 | 0.0319 | 0.0307 |
| wR2 | 0.0427 | 0.0590 | 0.0565 | 0.0600 | |
| Largest differences of peak/hole (e/Å3) | 1.196/−1.283 | 1.356/−1.335 | 1.183/−2.946 | 1.020/−3.155 | |
| Hypothetical Structures | Model 1 | Model 2 |
|---|---|---|
| ETotal | 0 | 8.15 |
| EBand | 0 | 6.57 |
| Site energies | ||
| Ca1 | 105.41 | 106.88 |
| M2 a | 162.28 | 137.92 |
| M3 b | 101.00 | 124.25 |
| Ca3 | 56.00 | 56.00 |
| Al | −52.21 | −52.32 |
| Sb1 | −1089.05 | −1086.61 |
| Sb2 | −1014.10 | −1013.22 |
| Sb3 | −517.63 | −517.46 |
| Sb4 | −244.61 | −242.43 |
| Total | −2492.90 | −2486.98 |
| Bond energies | ||
| Ca1-Sb | −4.62 | −4.64 |
| M2 a-Sb | −4.81 | −4.59 |
| M3 b-Sb | −4.51 | −4.77 |
| Ca3-Sb | −5.08 | −5.09 |
| Al-Sb1 (tetrahedron) | −11.27 | −11.29 |
| Sb3-Sb4 (trimer) | −1.85 | −1.88 |
| Total | −32.16 | −32.27 |
| Site energies + bond energies | −2525.06 | −2519.25 |
| Relative total | 0 | 6.81 |
| Atom | Ca1 | Ca2 | Ca3 | Ca4 | Al | Sb1 | Sb2 | Sb3 | Sb4 |
|---|---|---|---|---|---|---|---|---|---|
| Wyckoff site | 32g | 32g | 32g | 16e | 8a | 32g | 32g | 16f | 8b |
| QVAL | 1.746 | 1.821 | 1.659 | 1.759 | 2.843 | 5.323 | 4.597 | 5.400 | 5.322 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nam, G.; Jang, E.; Jo, H.; Han, M.-K.; Kim, S.-J.; Ok, K.M.; You, T.-S. Cationic Site-Preference in the Yb14-xCaxAlSb11 (4.81 ≤ x ≤ 10.57) Series: Theoretical and Experimental Studies. Materials 2016, 9, 553. https://doi.org/10.3390/ma9070553
Nam G, Jang E, Jo H, Han M-K, Kim S-J, Ok KM, You T-S. Cationic Site-Preference in the Yb14-xCaxAlSb11 (4.81 ≤ x ≤ 10.57) Series: Theoretical and Experimental Studies. Materials. 2016; 9(7):553. https://doi.org/10.3390/ma9070553
Chicago/Turabian StyleNam, Gnu, Eunyoung Jang, Hongil Jo, Mi-Kyung Han, Sung-Jin Kim, Kang Min Ok, and Tae-Soo You. 2016. "Cationic Site-Preference in the Yb14-xCaxAlSb11 (4.81 ≤ x ≤ 10.57) Series: Theoretical and Experimental Studies" Materials 9, no. 7: 553. https://doi.org/10.3390/ma9070553
APA StyleNam, G., Jang, E., Jo, H., Han, M.-K., Kim, S.-J., Ok, K. M., & You, T.-S. (2016). Cationic Site-Preference in the Yb14-xCaxAlSb11 (4.81 ≤ x ≤ 10.57) Series: Theoretical and Experimental Studies. Materials, 9(7), 553. https://doi.org/10.3390/ma9070553