
Room Temperature Consolidation of a Porous Poly(lactic-co-glycolic acid) Matrix by the Addition of Maltose to the Water-in-Oil Emulsion



Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Samples Preparation
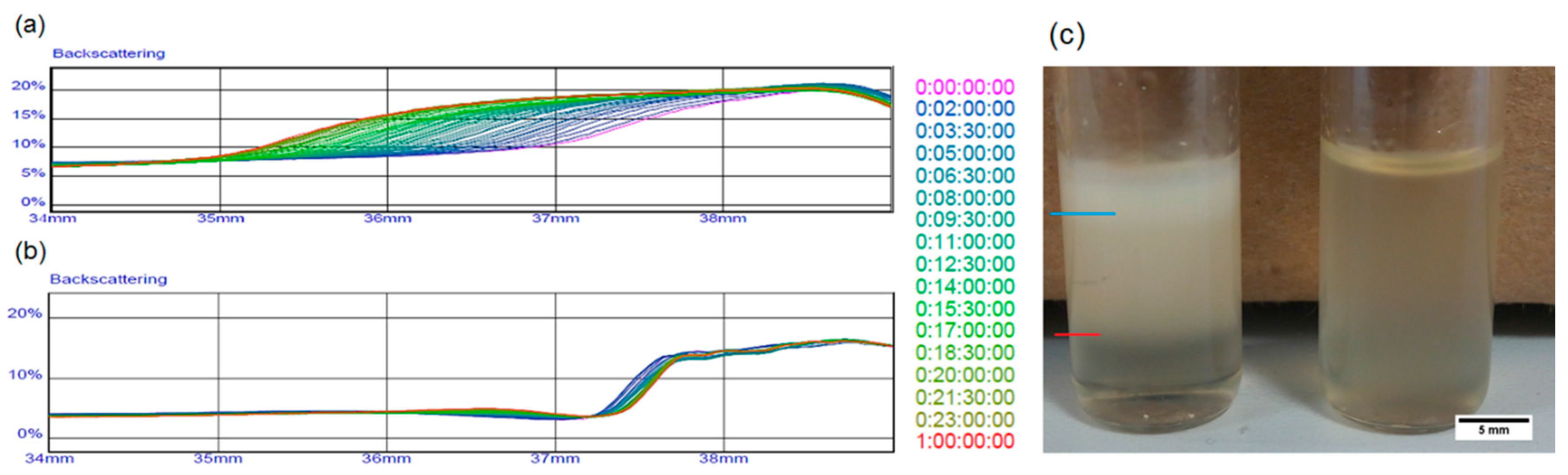
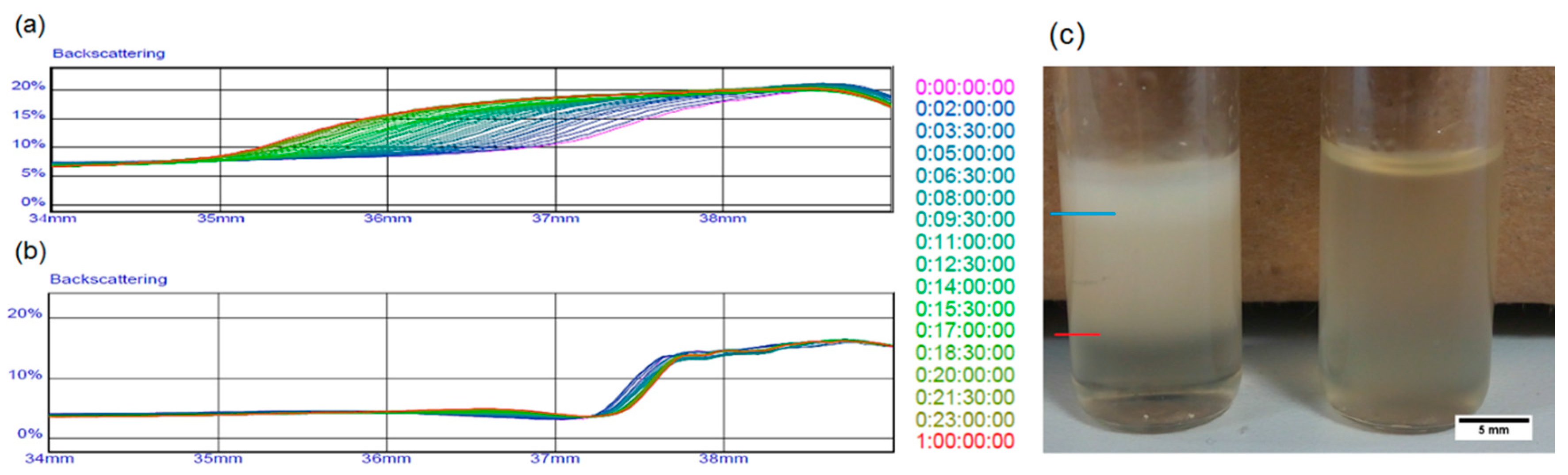
2.3. Emulsion Stability
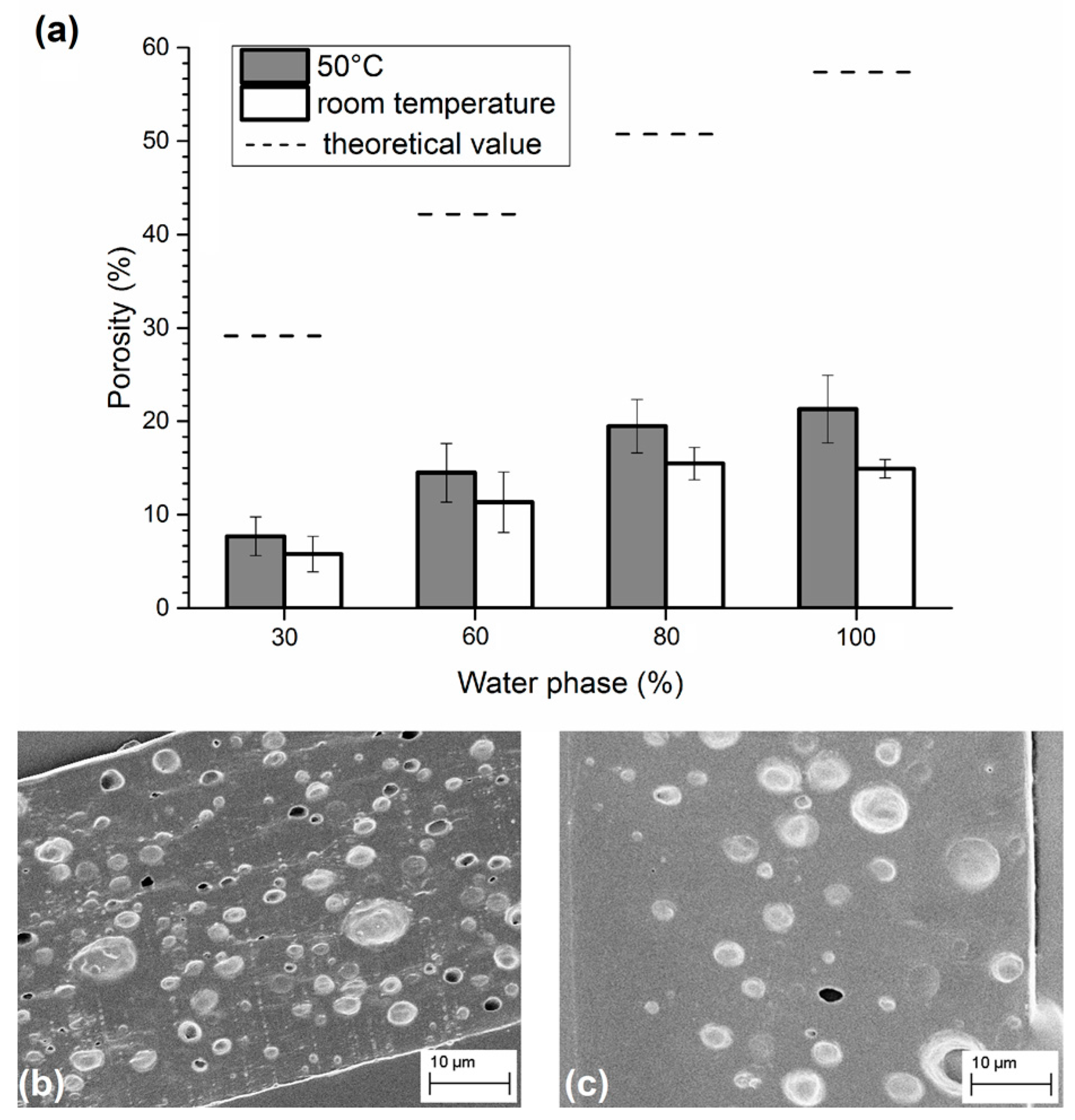
2.4. Morphological Analysis of Porosity
2.5. Interfacial Tension Measurements
2.6. Rheological Measurements
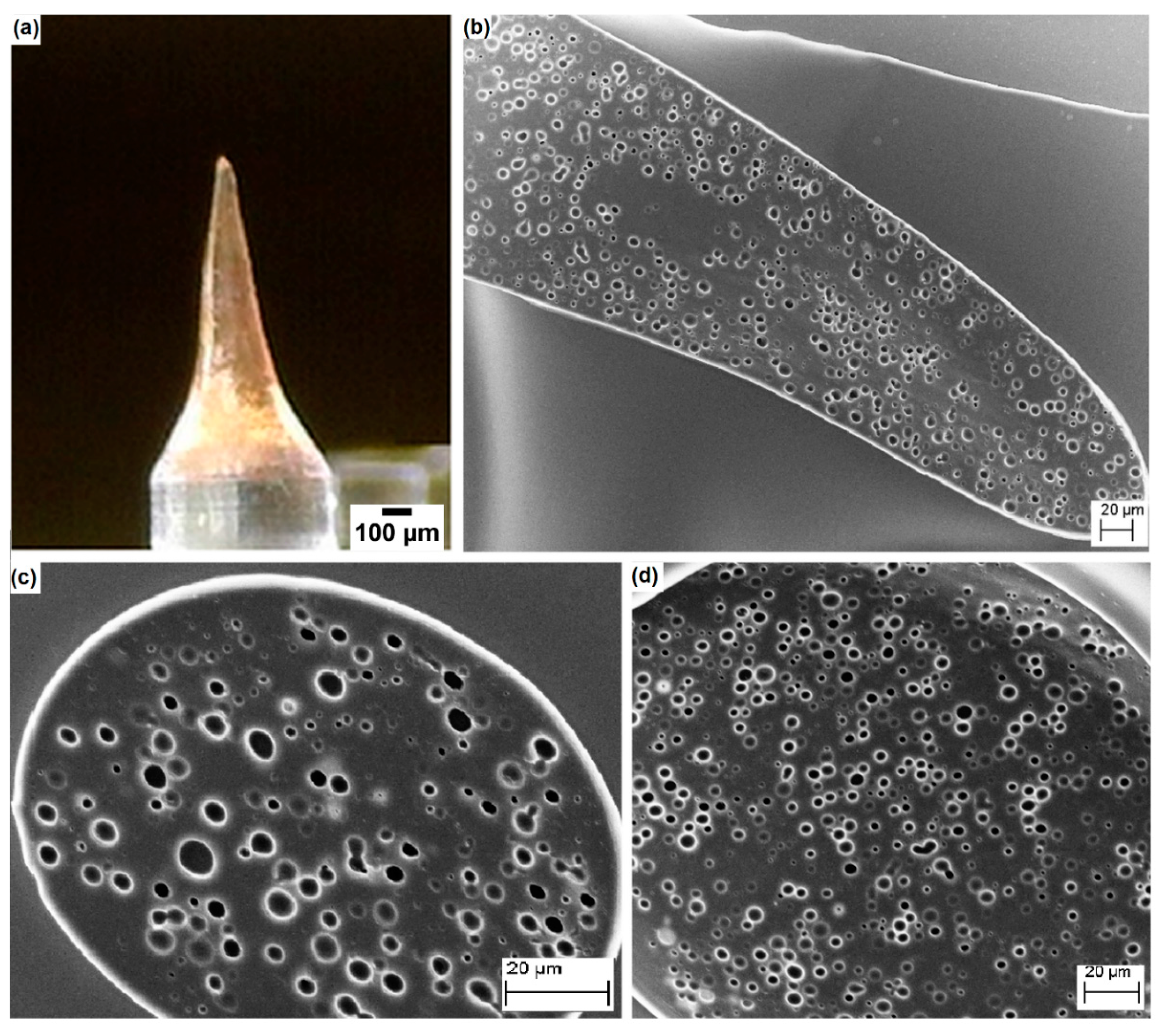
2.7. Microneedles Fabrication
2.8. Indentation Test
3. Results and Discussion
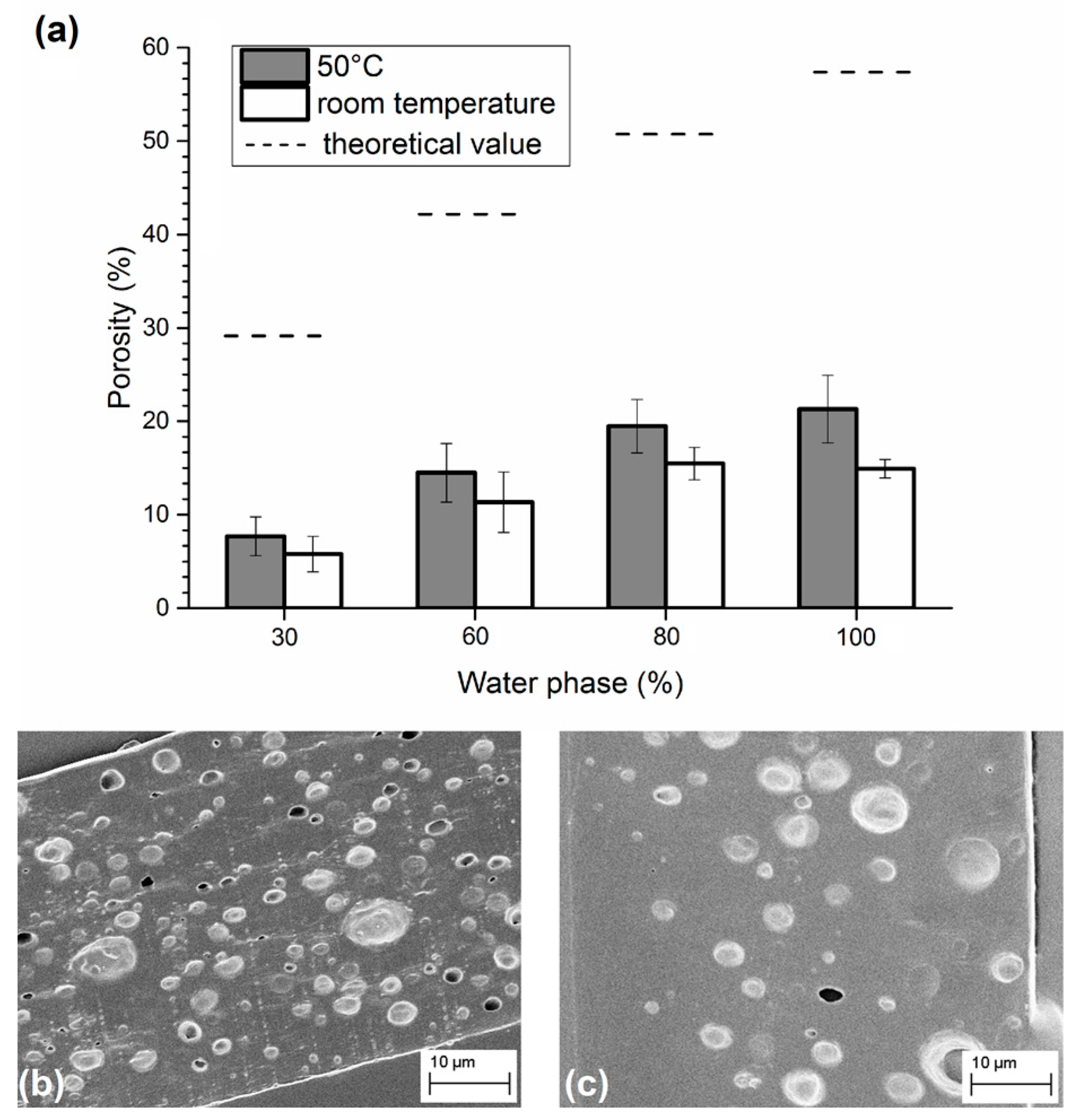
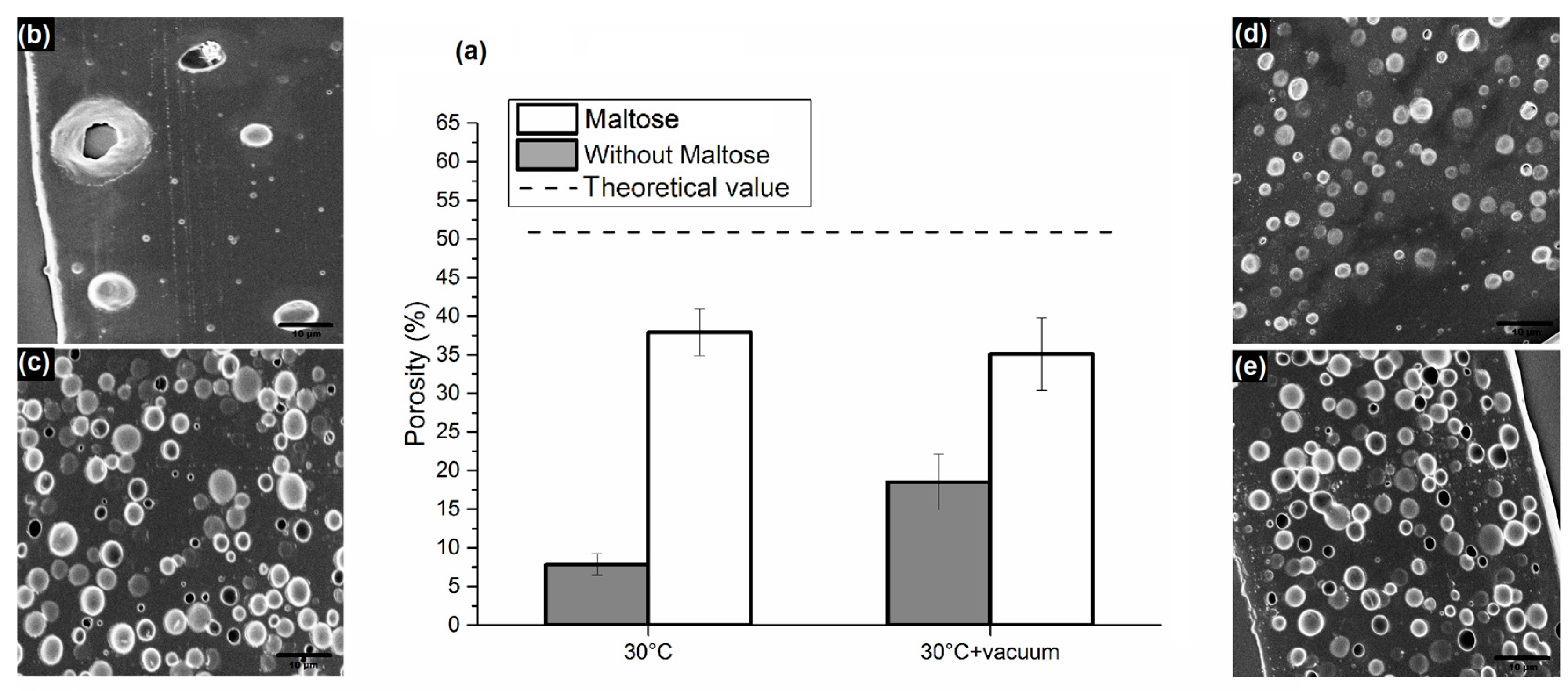
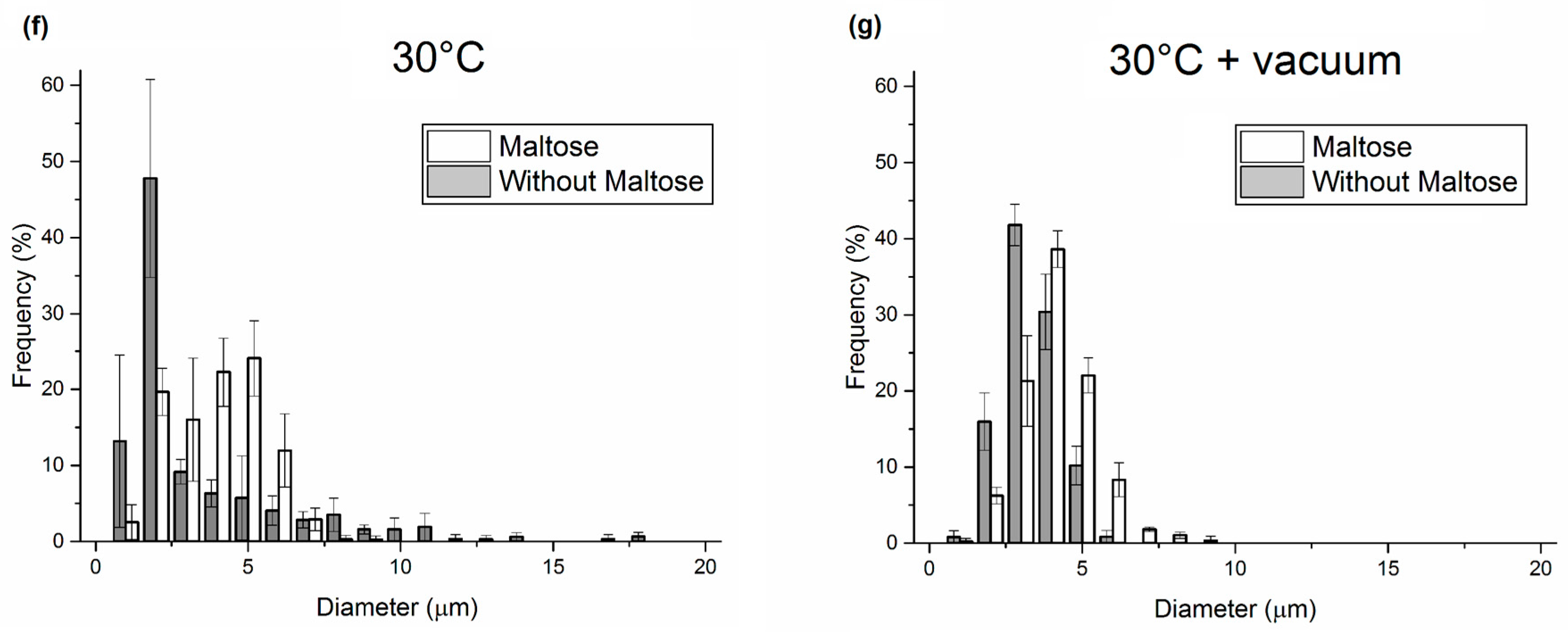
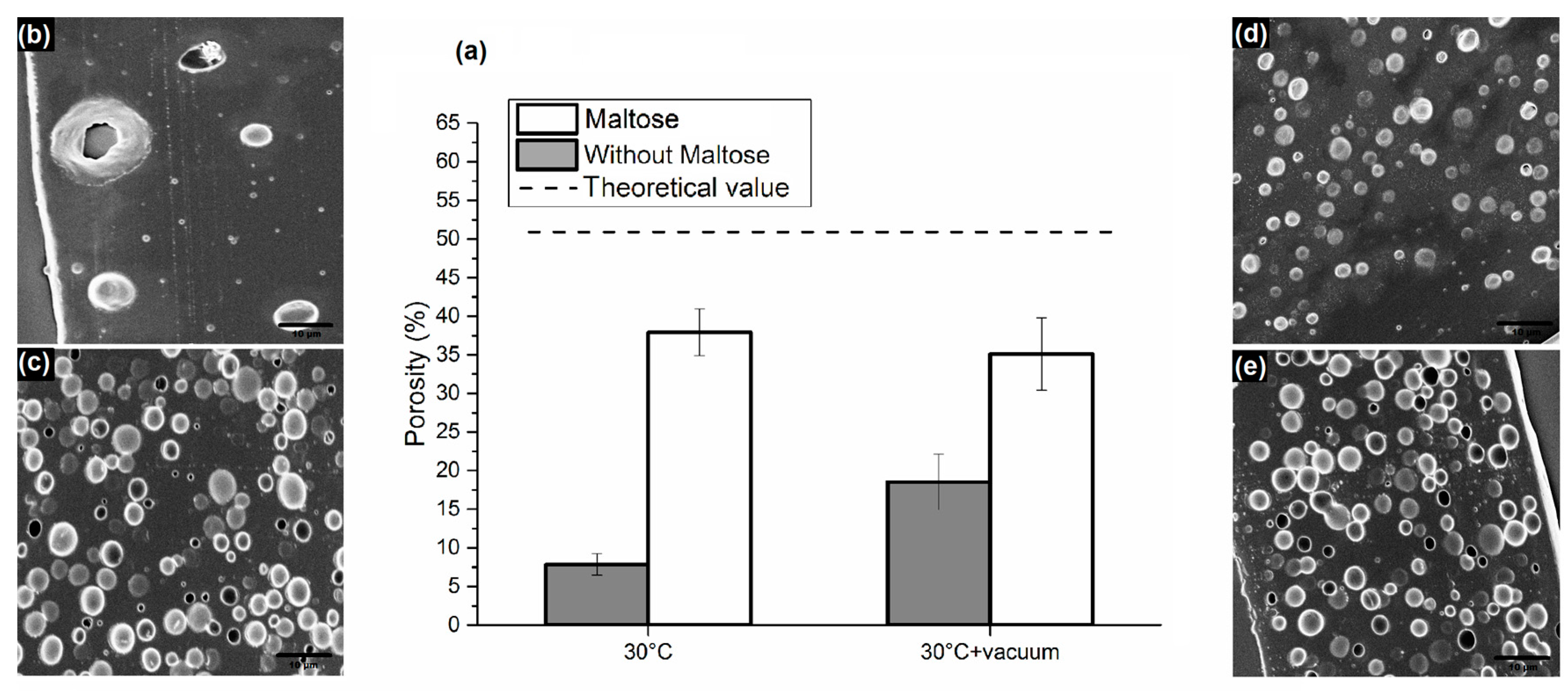
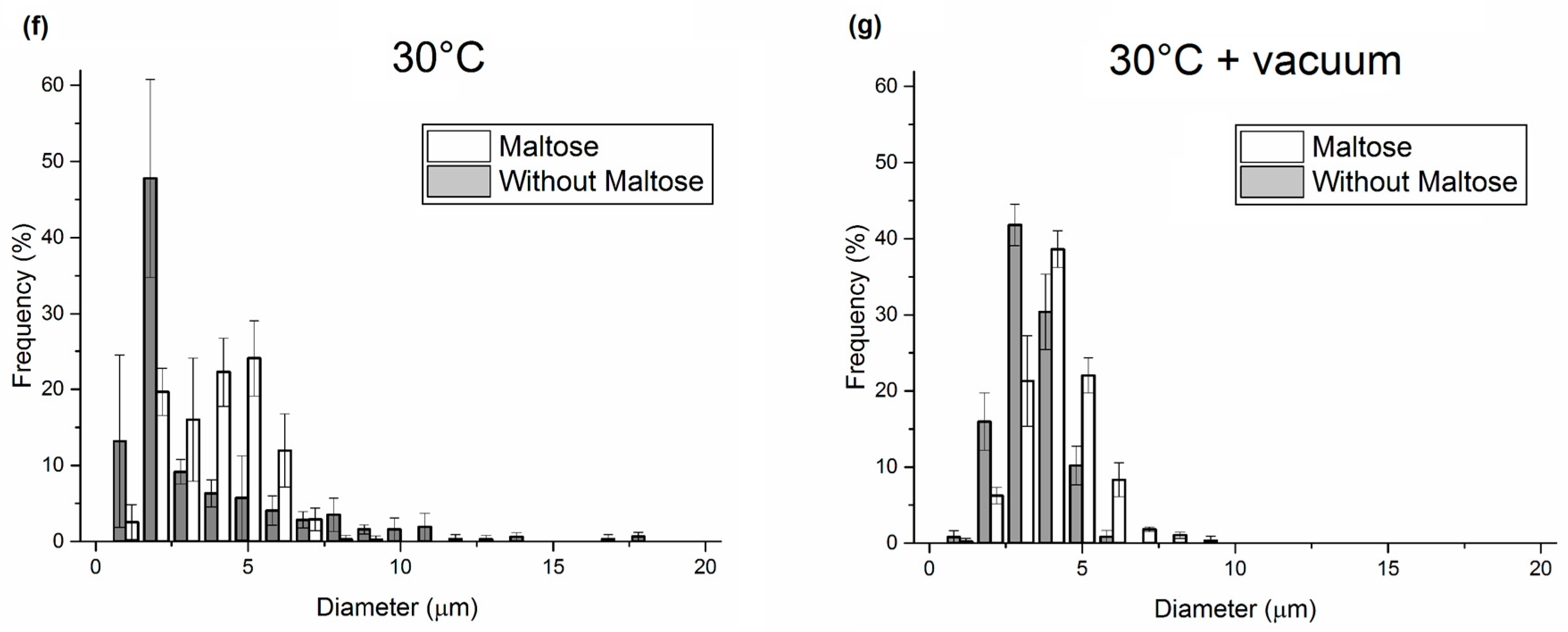
3.1. Analysis of PLGA Matrix Porosity
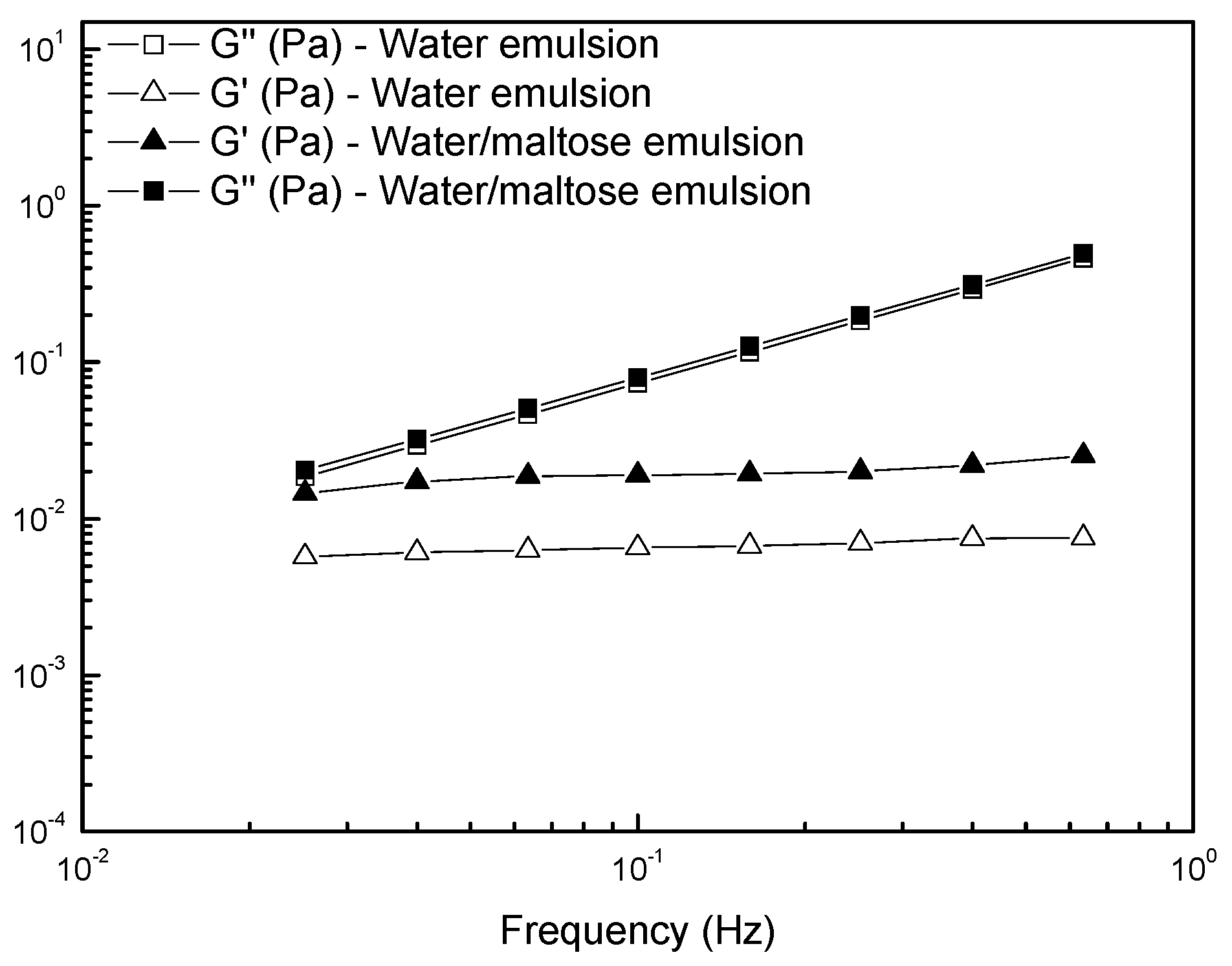
3.2. Interfacial Tension and Rheological Analysis of the Emulsions
- emulsion containing no maltose: (1.5 × 10−6)2 × (1150 − 964) = 4.2 × 10−10 Kg/m; and
- emulsion containing maltose: (2 × 10−6)2 × (1150 − 1098) = 2.1 × 10−10 Kg/m,
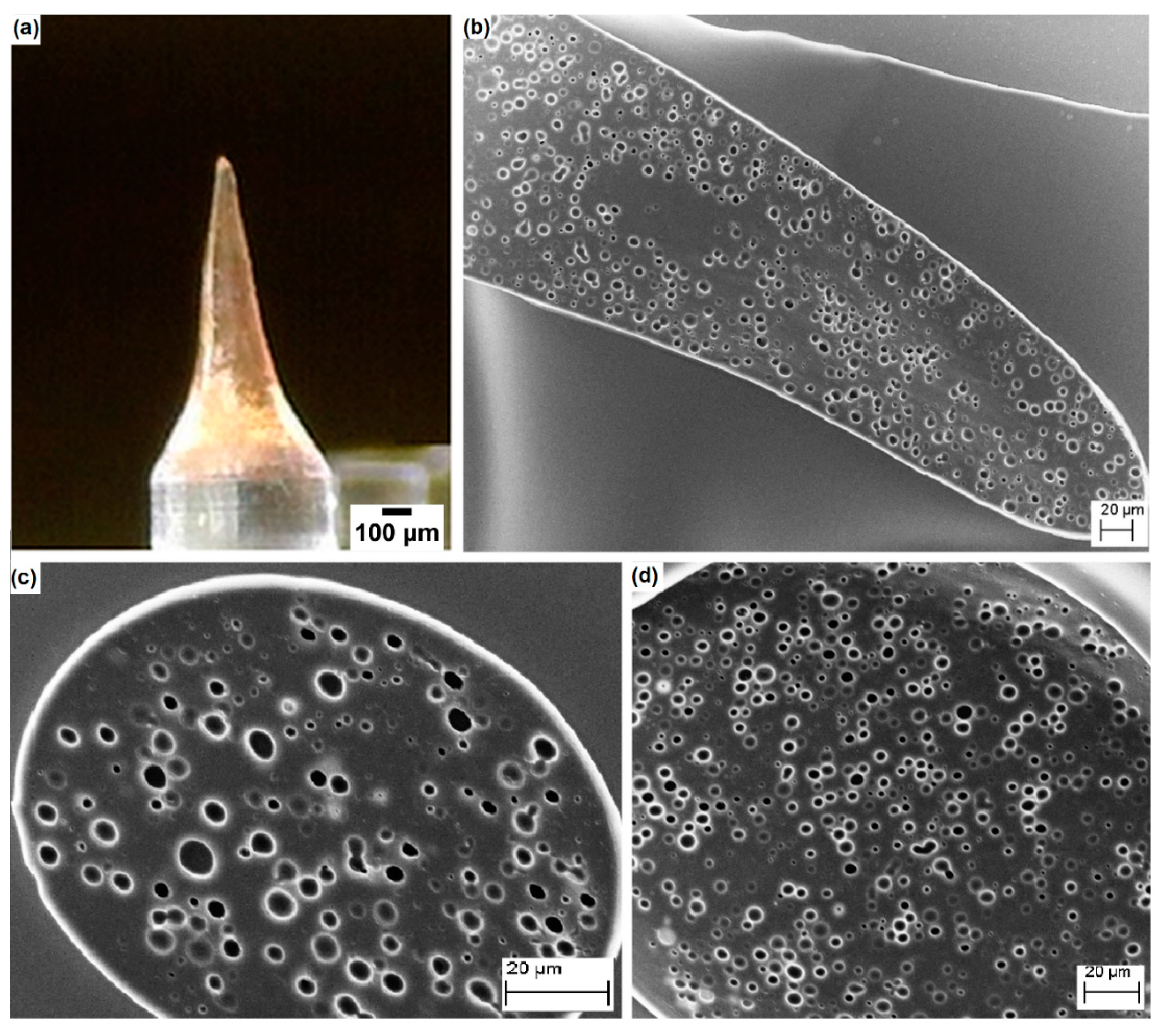
3.3. Application of the Proposed Method in the Field of Porous Polymer Microneedles
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Parveen, S.; Misra, R.; Sahoo, S.K. Nanoparticles: A boon to drug delivery, therapeutics, diagnostics and imaging. Nanomed. Nanotechnol. 2012, 8, 147–166. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.L.; Rosalia, R.A.; Varypatakia, E.; Sibuea, S.; Ossendorp, F.; Jiskoot, W. Poly-(lactic-co-glycolic-acid)-based particulate vaccines: Particle uptake by dendritic cells is a key parameter for immune activation. Vaccine 2015, 33, 847–854. [Google Scholar] [CrossRef] [PubMed]
- McCall, R.L.; Sirianni, R.W. PLGA Nanoparticles formed by single- or double-emulsion with vitamin ETPGS. J. Vis. Exp. 2013, 82, e51015. [Google Scholar] [CrossRef]
- Chen, R.R.; Mooney, D.J. Polymeric growth factor delivery strategies for tissue engineering. Pharm. Res. 2003, 20, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Saltzman, W.M.; Olbricht, W.L. Building drug delivery into tissue engineering. Nat. Rev. Drug Discov. 2002, 1, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Langer, R. Tissue engineering. Mol. Ther. 2000, 1, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Van de Weert, M.; Jorgensen, L.; Horn Moeller, E.; Frokjaer, S. Factors of importance for a successful delivery system for proteins. Expert Opin. Drug Deliv. 2005, 2, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Makadia, H.K.; Siegel, S.J. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- De Alteriis, R.; Vecchione, R.; Attanasio, C.; De Gregorio, M.; Porzio, M.; Battista, E.; Netti, P.A. A method to tune the shape of protein-encapsulated polymeric microspheres. Sci. Rep. 2015, 5, 12634. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Chung, H.J.; Park, T.G. Biodegradable polymeric microspheres with “open/closed” pores for sustained release of human growth hormone. J. Control. Release 2006, 112, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.R.; Kuo, C.J.; Yang, C.Y.; Shaw, S.Y.; Wu, Y.J. Preparation of Macroporous Biodegradable PLGA Scaffolds for Cell Attachment with the Use of Mixed Salts as Porogen Additives. J. Biomed. Mater. Res. 2002, 63, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Le Droumaguet, B.; Lacombe, R.; Ly, H.B.; Guerrouache, M.; Carbonnier, B.; Grande, D. (Engineering functional doubly porous PHEMA-based materials. Polymer 2014, 55, 373–379. [Google Scholar] [CrossRef]
- Chen, H.B.; Hollinger, E.; Wang, Y.Z.; Schiraldi, D.A. Facile fabrication of poly(vinyl alcohol) gels and derivative aerogels. Polymer 2014, 55, 380–384. [Google Scholar] [CrossRef]
- Silverstein, M.S. Emulsion-templated porous polymers: A retrospective perspective. Polymer 2014, 55, 304–320. [Google Scholar] [CrossRef]
- Zhang, H.; Cooper, A.I. Synthesis and applications of emulsion-templated porous materials. Soft Matter 2005, 1, 107–113. [Google Scholar] [CrossRef]
- He, H.; Li, W.; Lamson, M.; Zhong, M.; Konkolewicz, D.; Hui, C.M.; Yaccato, K.; Rappold, T.; Sugar, G.; David, N.E.; et al. Porous polymers prepared via high internal phase emulsion polymerization for reversible CO2 capture. Polymer 2014, 55, 385–394. [Google Scholar] [CrossRef]
- Whang, K.; Thomas, C.H.; Healy, K.E.; Nuber, G. A novel method to fabricate a bioabsorbable scaffold. Polymer 1995, 36, 837–842. [Google Scholar] [CrossRef]
- Caldwell, S.; Johnson, D.W.; Didsbury, M.P.; Murray, B.A.; Wu, J.J.; Przyborski, S.A.; Cameron, N.R. Degradable emulsion-templated scaffolds for tissue engineering from thiol–ene photopolymerisation. Soft Matter 2012, 8, 10344–10351. [Google Scholar] [CrossRef]
- Qutachi, O.; Vetsch, J.R.; Gill, D.; Cox, H.; Scurr, D.J.; Hofmann, S.; Müller, R.; Quirk, R.A.; Shakesheff, K.M.; Rahman, C.V. Injectable and porous PLGA microspheres that form highly porous scaffolds at body temperature. Acta Biomater. 2014, 10, 5090–5098. [Google Scholar] [CrossRef] [PubMed]
- Sher, P.; Ingavle, G.; Ponrathnam, S.; Pawar, A.P. Low density porous carrier drug adsorption and release study by response surface methodology using different solvents. Int. J. Pharm. 2007, 331, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bajaj, N.; Xu, P.; Ohn, K.; Tsifansky, M.D.; Yeo, Y. Development of highly porous large PLGA microparticles for pulmonary drug delivery. Biomaterials 2009, 30, 1947–1953. [Google Scholar] [CrossRef] [PubMed]
- Alshamsan, A. Nanoprecipitation is more efficient than emulsion solvent evaporation method to encapsulate cucurbitacin I in PLGA nanoparticles. Saudi Pharm. J. 2013, 22, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Fu, X.; Luo, F.; Huang, Q. Effects of maltose on stability and rheological properties of orange oil-in-water emulsion formed by OSA modified starch. Food Hydrocolloids 2013, 32, 79–86. [Google Scholar] [CrossRef]
- Koroleva, M.Y.; Yurtov, E.V. Effect of ionic strength of dispersed phase on Ostwald ripening in water-in-oil emulsions. Colloid J. 2003, 65, 40–43. [Google Scholar] [CrossRef]
- Schmidts, T.; Dobler, D.; Schlupp, P.; Nissing, C.; Garn, H.; Runkel, F. Development of multiple W/O/W emulsions as dermal carrier system for oligonucleotides: Effect of additives on emulsion stability. Int. J. Pharm. 2010, 398, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kim, J.D.; Lee, C.Y.; Her, S.; Jung, H. A high-capacity, hybrid electro-microneedle for in-situ cutaneous gene transfer. Biomaterials 2011, 32, 7705–7710. [Google Scholar] [CrossRef] [PubMed]
- Vecchione, R.; Coppola, S.; Esposito, E.; Casale, C.; Vespini, V.; Grilli, S.; Ferraro, P.; Netti, P.A. Electro-Drawn Drug-Loaded Biodegradable Polymer Microneedles as a Viable Route to Hypodermic Injection. Adv. Funct. Mater. 2014, 24, 3515–3523. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Set 1: Water Phase Composition 180 mg/mL Lecithin | Sample Set 2: Water Phase Content 80% | ||
|---|---|---|---|
| Water Content | Consolidation Process | Water Phase Composition | Consolidation Process |
| 30% | Room temperature | 180 mg/mL lecithin | 30 °C |
| 30% | 50 °C | ||
| 60% | Room temperature | 180 mg/mL lecithin | 30 °C + vacuum |
| 60% | 50 °C | ||
| 80% | Room temperature | 180 mg/mL lecithin and 360 mg/mL maltose | 30 °C |
| 80% | 50 °C | ||
| 100% | Room temperature | 180 mg/mL lecithin and 360 mg/mL maltose | 30 °C + vacuum |
| 100% | 50 °C | ||
| Phase | Density (g/mL) | Interfacial Tension (mN/m) |
|---|---|---|
| PLGA/DMC 25% w/v | 1.15 | - |
| Pure water | 1 | 7.2 ± 0.25 |
| Water/maltose | 1.13 | 8.5 ± 0.38 |
| Water/lecithin | 0.964 | 4.7 ± 0.3 |
| Water/lecithin/maltose | 1.098 | 7.3 ± 0.6 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esposito, E.; Ruggiero, F.; Vecchione, R.; Netti, P.A. Room Temperature Consolidation of a Porous Poly(lactic-co-glycolic acid) Matrix by the Addition of Maltose to the Water-in-Oil Emulsion. Materials 2016, 9, 420. https://doi.org/10.3390/ma9060420
Esposito E, Ruggiero F, Vecchione R, Netti PA. Room Temperature Consolidation of a Porous Poly(lactic-co-glycolic acid) Matrix by the Addition of Maltose to the Water-in-Oil Emulsion. Materials. 2016; 9(6):420. https://doi.org/10.3390/ma9060420
Chicago/Turabian StyleEsposito, Eliana, Flavia Ruggiero, Raffaele Vecchione, and Paolo Antonio Netti. 2016. "Room Temperature Consolidation of a Porous Poly(lactic-co-glycolic acid) Matrix by the Addition of Maltose to the Water-in-Oil Emulsion" Materials 9, no. 6: 420. https://doi.org/10.3390/ma9060420
APA StyleEsposito, E., Ruggiero, F., Vecchione, R., & Netti, P. A. (2016). Room Temperature Consolidation of a Porous Poly(lactic-co-glycolic acid) Matrix by the Addition of Maltose to the Water-in-Oil Emulsion. Materials, 9(6), 420. https://doi.org/10.3390/ma9060420