Quantum Dots and Their Multimodal Applications: A Review

Abstract
:
Index
- Introduction
- Structure of Quantum Dots
- 2.1.
- Core Structure
- 2.1.1.
- Size versus Density of States
- 2.1.2.
- Phases and Phase Transitions
- 2.1.3.
- Doping in Quantum Dots
- 2.1.4.
- Alloying of Quantum Dots
- 2.2.
- Surface Structure
- 2.2.1.
- Surface Passivation
- 2.2.1.1.
- Organically Capped Quantum Dots
- 2.2.1.2.
- Inorganically Passivated Quantum Dots
- 2.2.1.2.1.
- Epitaxial Growth
- 2.2.1.2.2.
- Non-epitaxial Growth
- 2.2.1.3.
- Multi-Shell Structure
- 2.2.2.
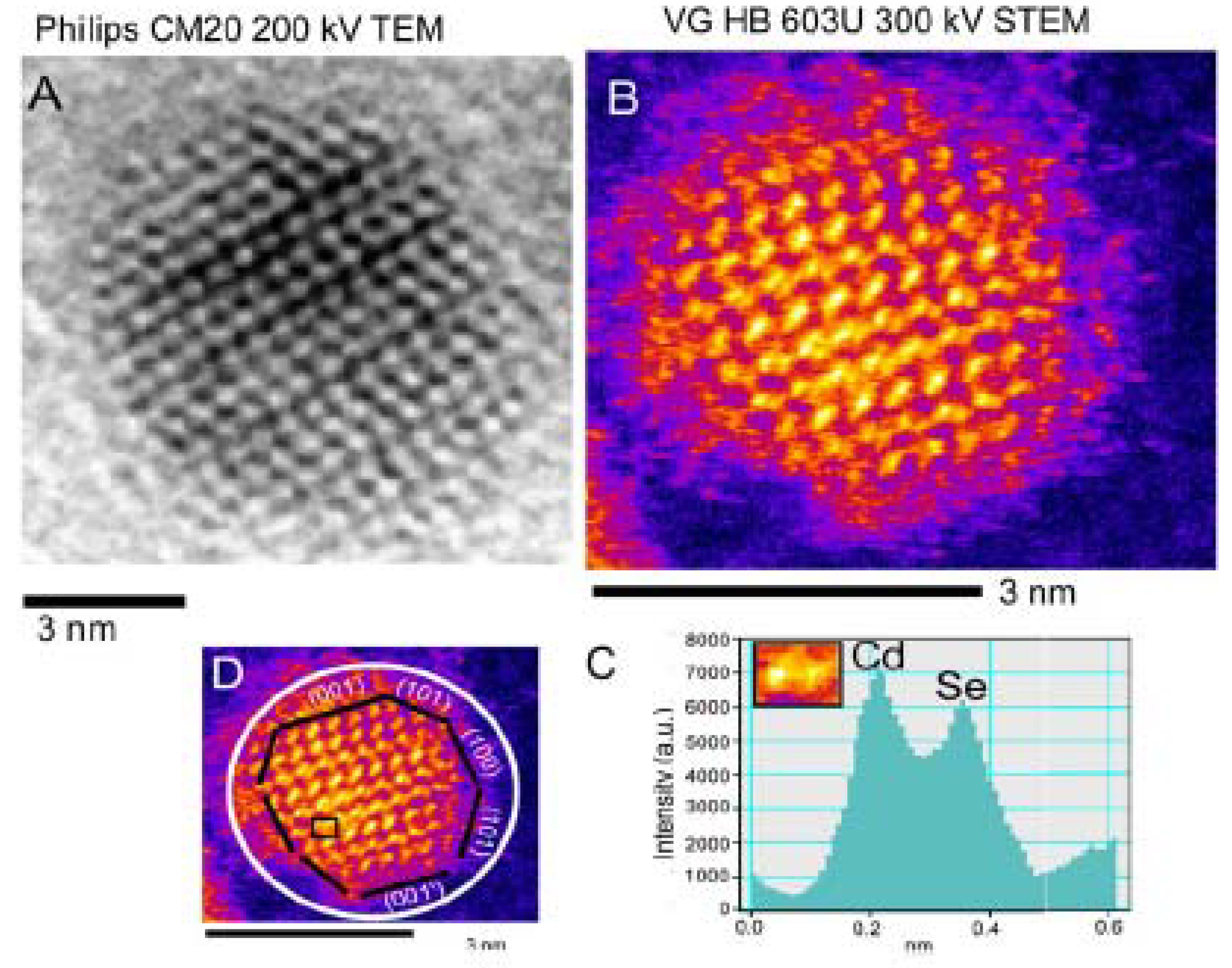
- Characterization of Shell Structures
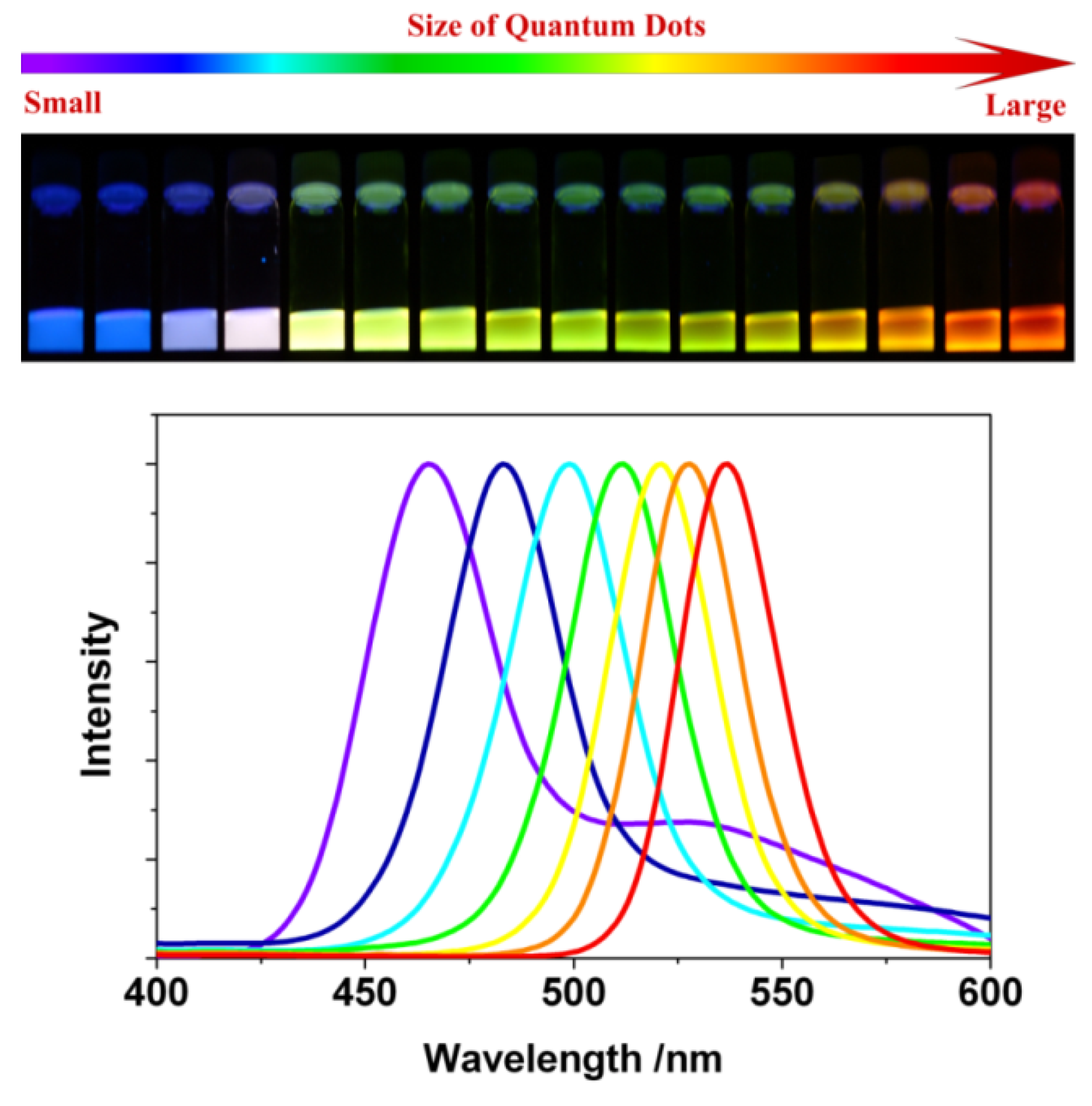
- Properties
- 3.1.
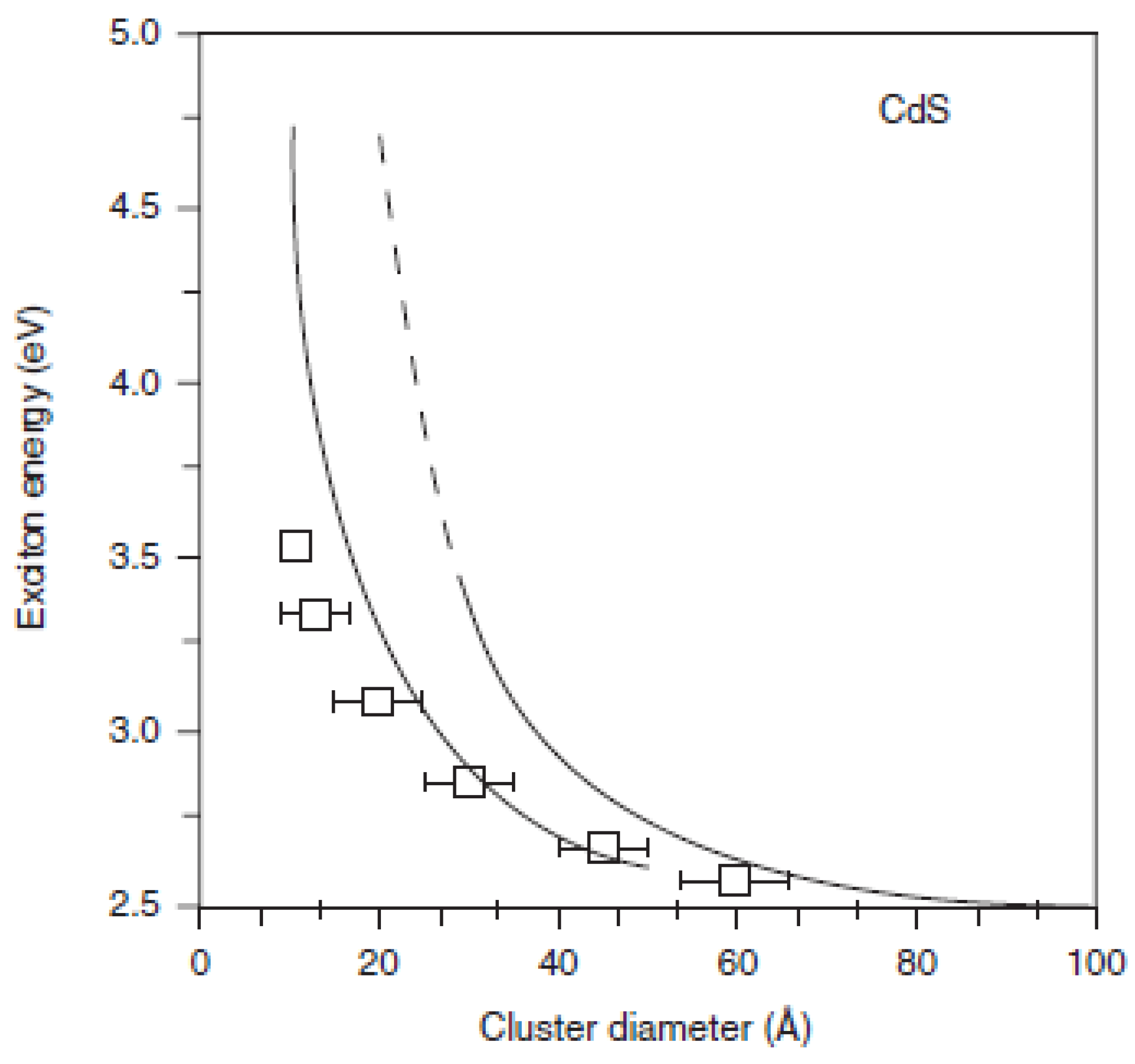
- Quantum Confinement Effects and Band-Gap
- 3.1.1.
- Effective Mass Approximation Model
- 3.1.2.
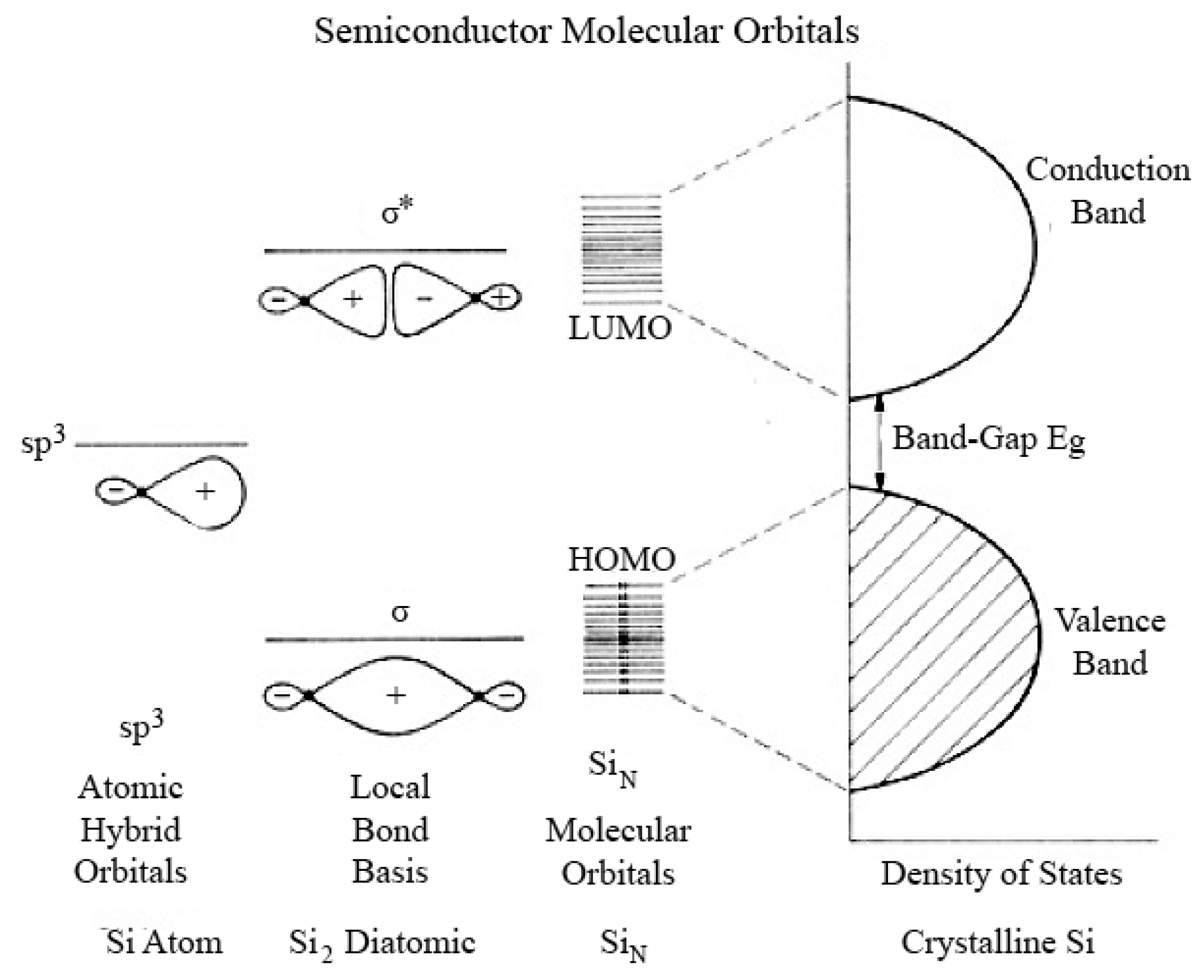
- Linear Combination of Atomic Orbital Theory – Molecular Orbital Theory
- 3.2.
- Luminescence Properties
- 3.2.1.
- Radiative Relaxation
- 3.2.1.1.
- Band-Edge Emission
- 3.2.1.2.
- Defect Emission
- 3.2.1.3.
- Activator Emission
- 3.2.2.
- Quantum Yield of Quantum Dots
- 3.2.2.1.
- Reported Quantum Yield
- 3.2.2.2.
- Change of Quantum Yield under Ultraviolet Irradiation
- 3.2.3.
- Non-radiative Process in Quantum Dots
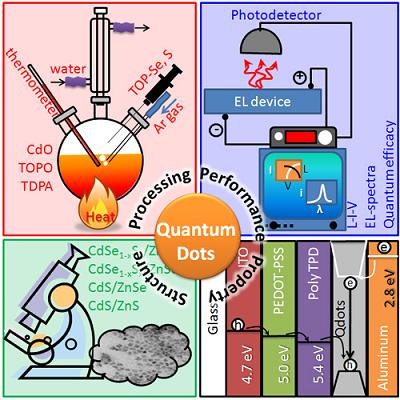
- Synthesis Processes
- 4.1.
- Top-Down Synthesis Processes
- 4.2.
- Bottom-Up Approach
- 4.2.1.
- Wet-Chemical Methods
- 4.2.1.1.
- Sol-gel Process
- 4.2.1.2.
- Microemulsion Process
- 4.2.1.3.
- Hot-Solution Decomposition Process
- 4.2.1.4.
- Other Synthesis Processes
- 4.2.2.
- Vapor-Phase Methods
- Application
- 5.1.
- Quantum Dots for Electroluminescence Device Fabrication
- 5.2.
- Downconversion of Blue or Ultraviolet Light
- 5.3.
- Quantum Dots in Solar Cell Device Fabrication
- 5.3.1.
- Quantum Dot Sensitized Solar Cell
- 5.3.2.
- Quantum Dot Dispersed Solar Cell
- 5.4.
- Quantum Dots in Other Optoelectronic Devices
- 5.5.
- Application of Quantum Dots in Bioimaging Applications
- 5.5.1.
- Fluorescence for Bioimaging
- 5.5.2.
- Use of Fluorescence Resonance Energy Transfer in Bioimaging
- 5.5.3.
- Surface Enhanced Raman Spectroscopy
- 5.5.4.
- Radio-Opaque and Paramagnetic Properties
- 5.5.5.
- Magnetic Resonance-based Bioimaging
- Perspective
1. Introduction
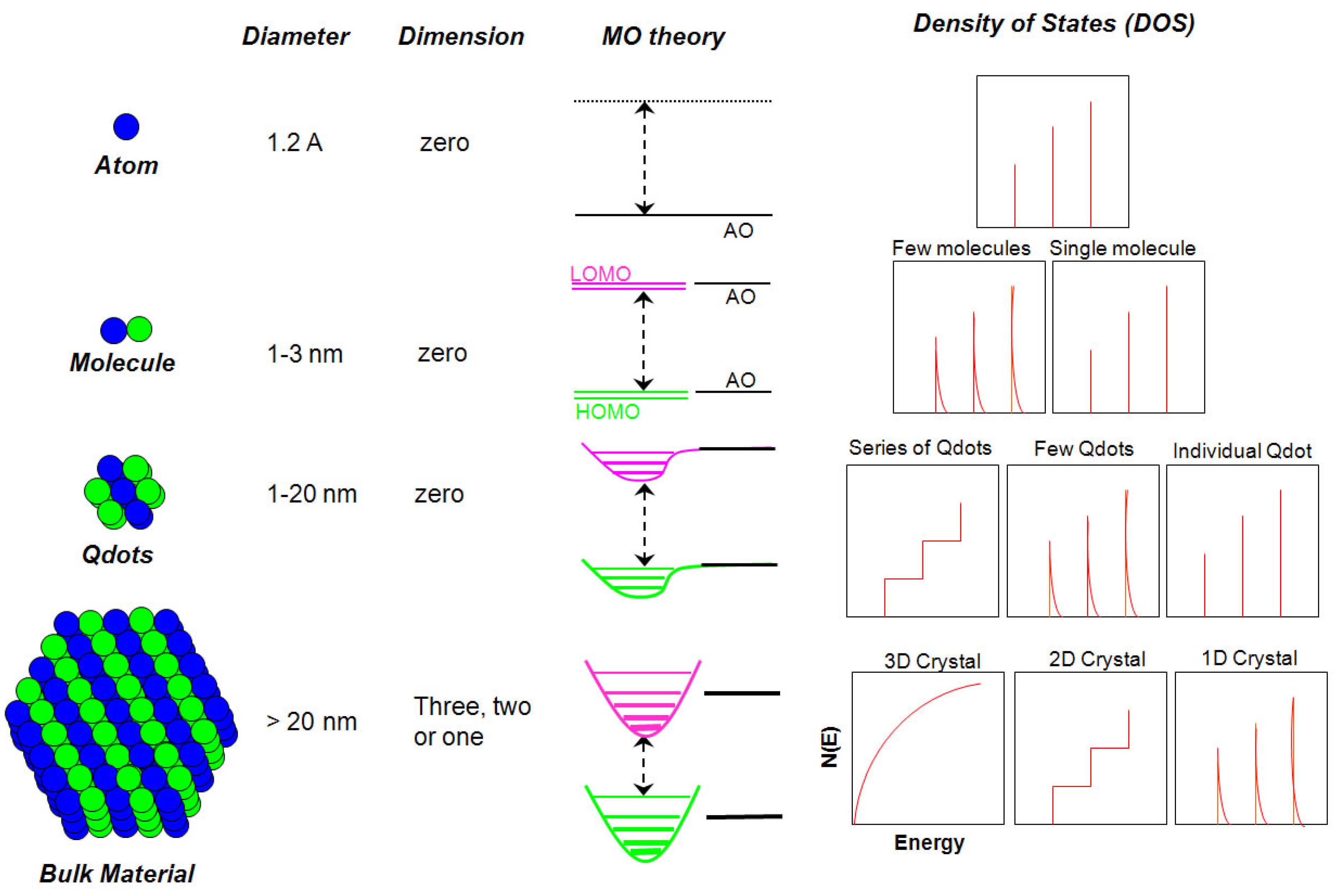
2. Structure of Quantum dots
2.1. Core Structure
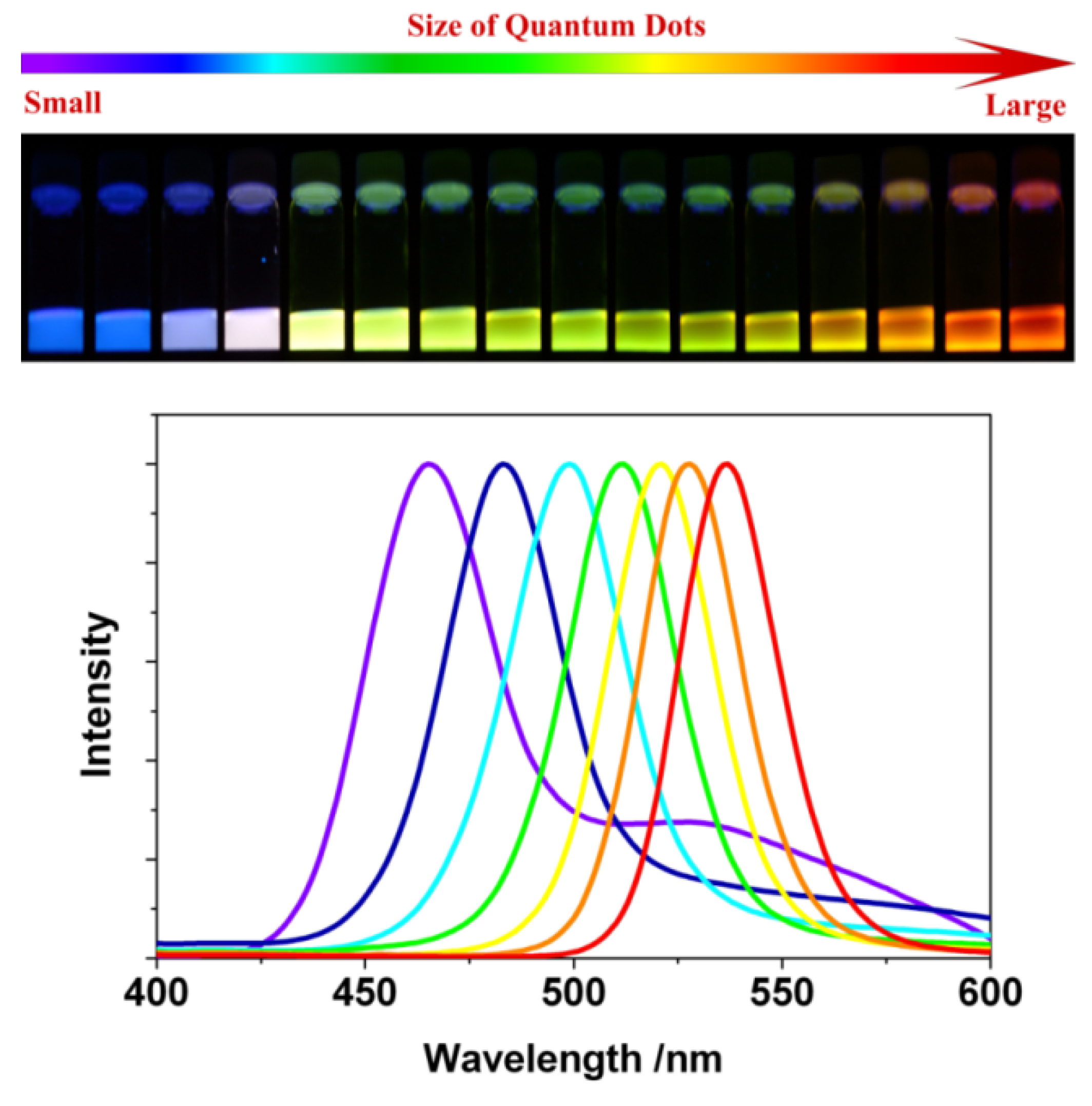
2.1.1. Size versus Density of States
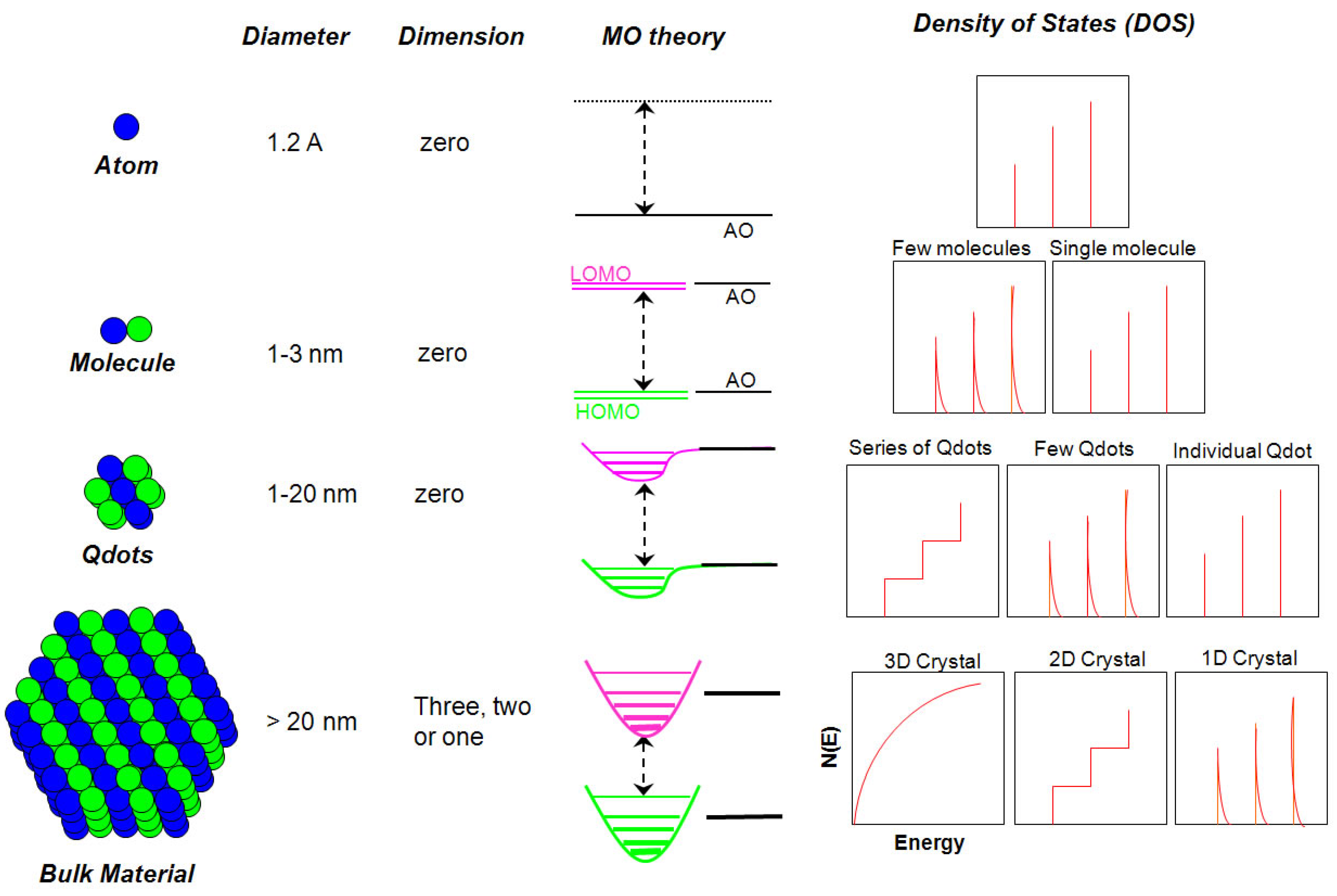
2.1.2. Phases and Phase Transitions
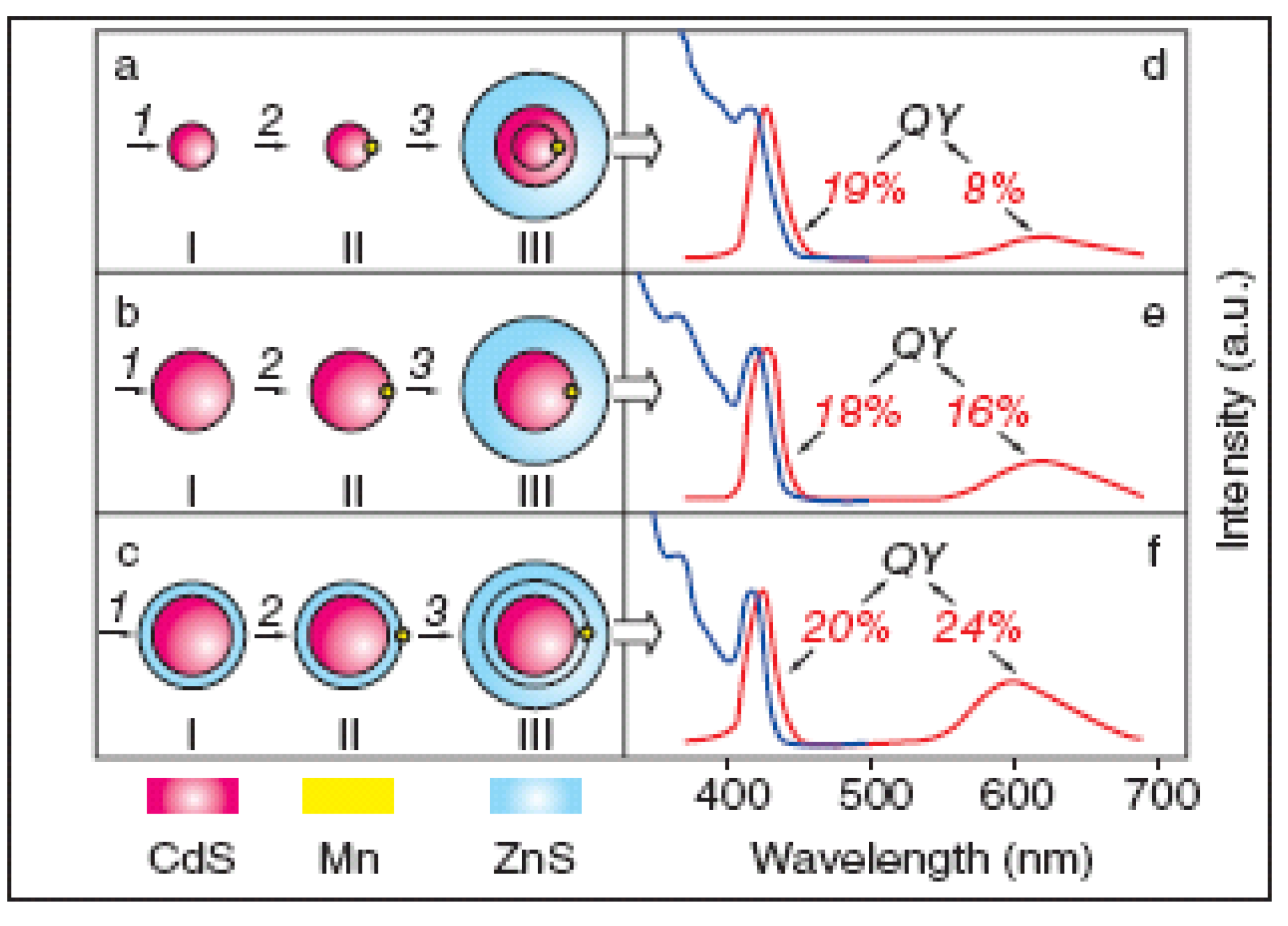
2.1.3. Doping in Quantum Dots
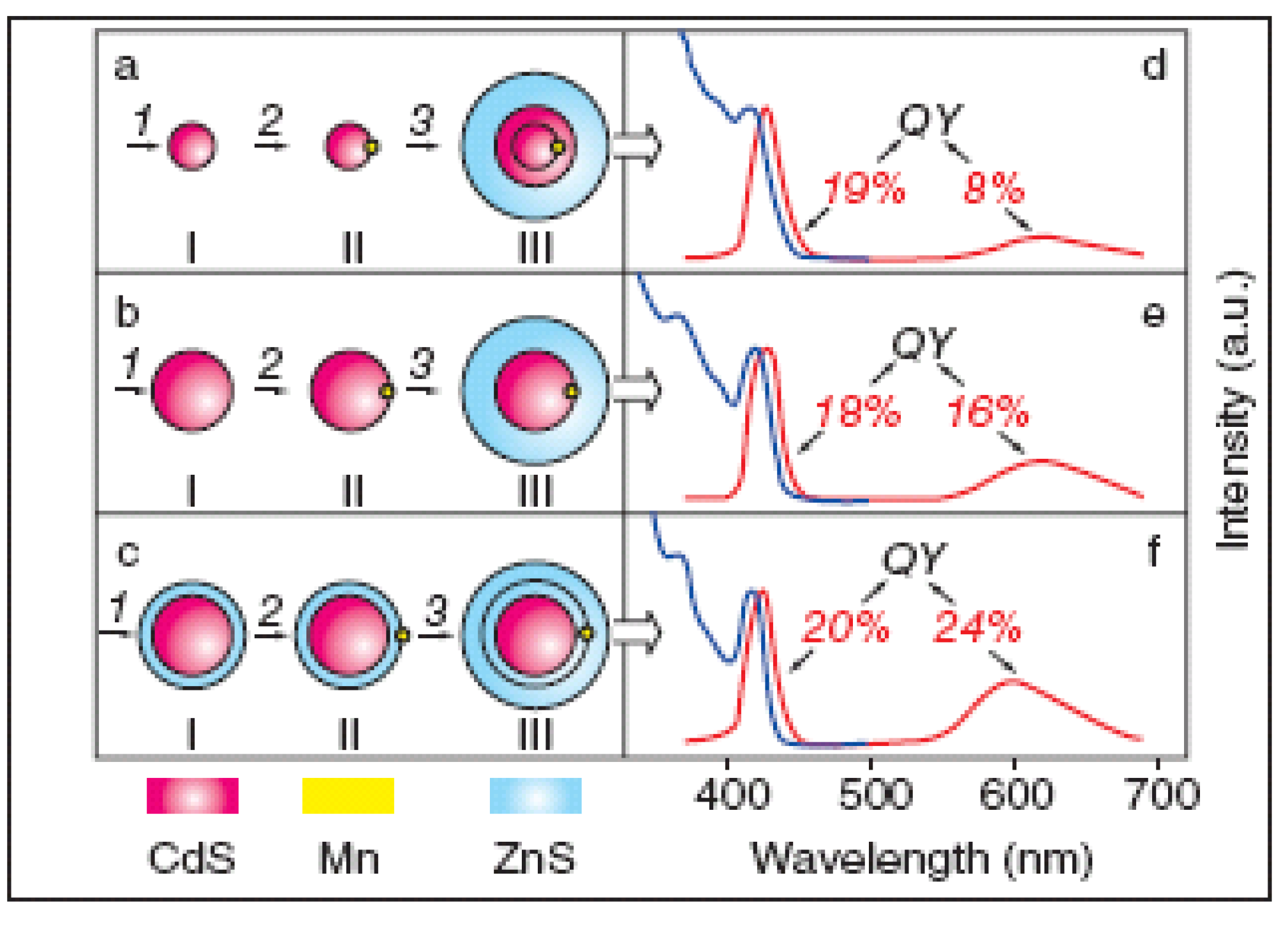
2.1.4. Alloying of Quantum Dots

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Semiconductor/ Qdot | Value of ‘x’ in Qdot composition | Calculated Particle size Diameter /nm | Band-gap (eV) |
|---|---|---|---|
| ZnS | bulk | 3.7 | |
| 2.7 | 4.1 | ||
| CdS | bulk | 2.45 | |
| 2.6 | 2.9 | ||
| HgS | bulk | ~0.0 | |
| ZnxCd1-xS | 0.14 | 2.6 | 3.0 |
| 0.14 | 3.2 | 3.0 | |
| 0.14 | 3.5 | 3.0 | |
| 0.14 | 4.3 | 2.95 | |
| 0.15 | 2.7 | 3.0 | |
| 0.25 | 4.0 | 3.05 | |
| 0.34 | 3.7 | 3.15 | |
| 0.44 | 4.7 | 3.5 | |
| 0.61 | 3.9 | 4.0 | |
| HgxCd1-xS | 0.0025 | 4.0 | 4.5 |
| 0.005 | 4.0 | 4.45 | |
| 0.05 | 4.0 | 4.4 | |
| 0.01 | 4.0 | 4.35 | |
| 0.2 | 4.0 | 3.8 | |
| 0.5 | 4.0 | 3.25 | |
| 0.75 | 4.0 | 3.15 |
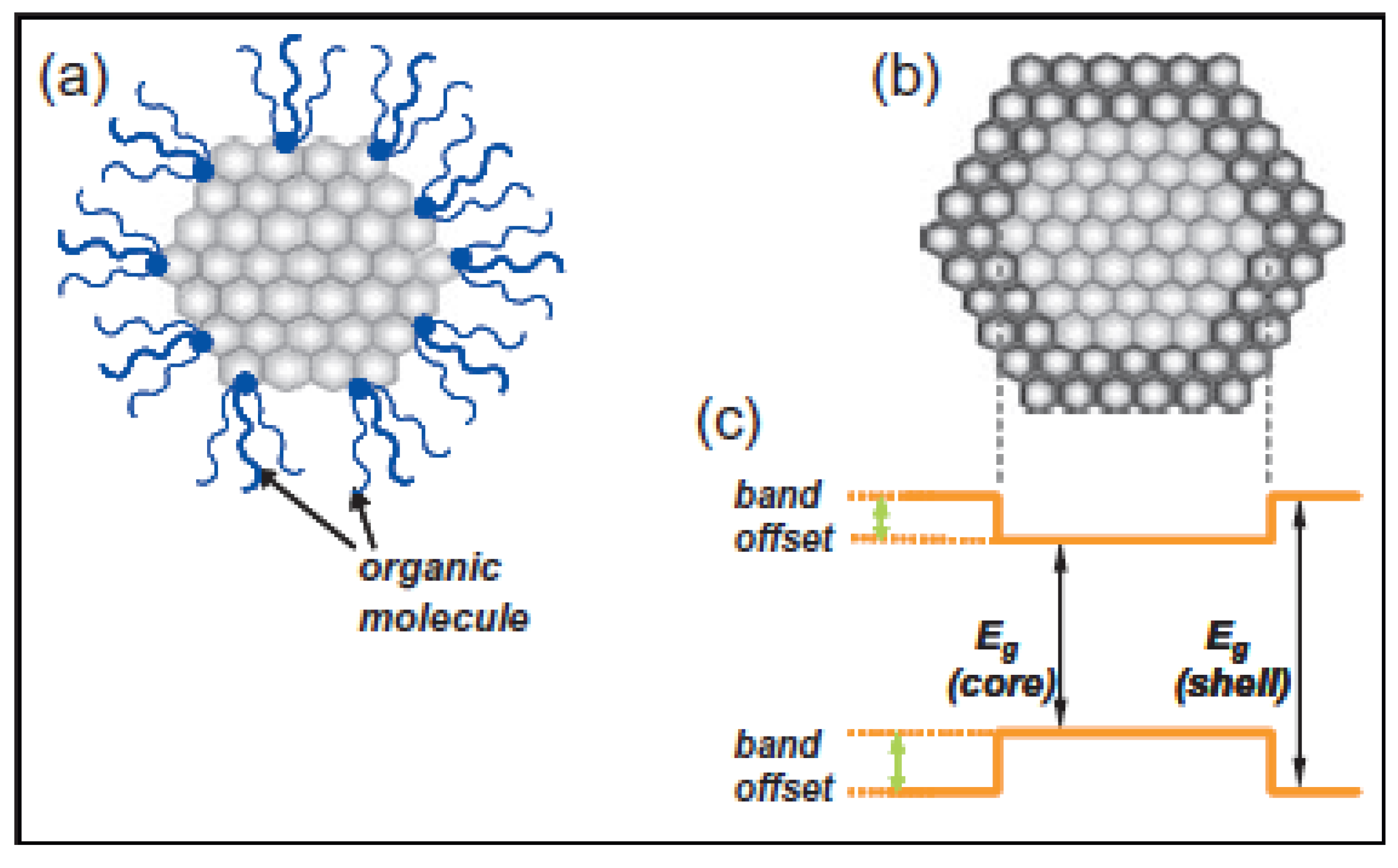
2.2. Surface Structure
2.2.1. Surface Passivation
2.2.1.1. Organically Capped Quantum Dots
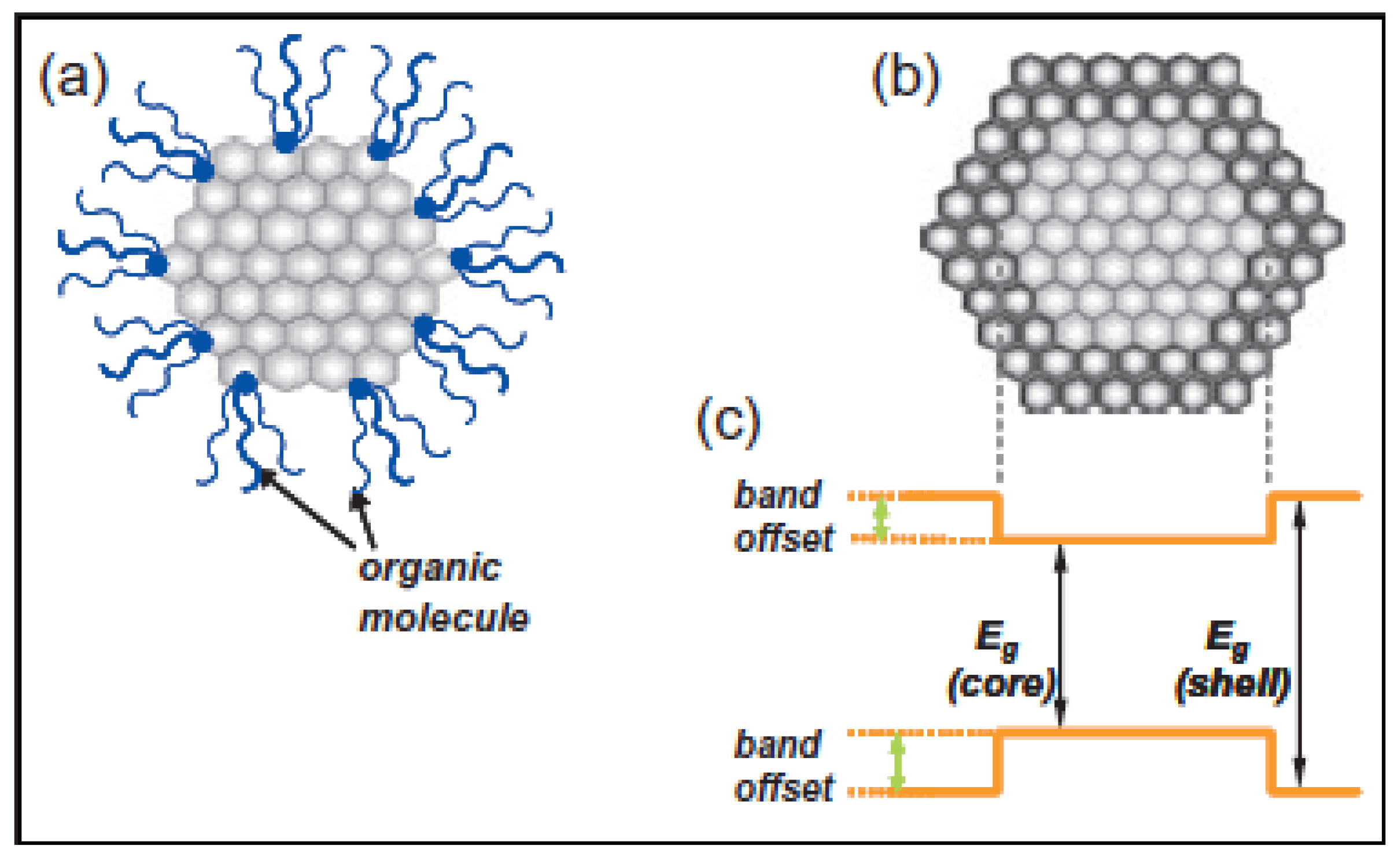
2.2.1.2. Inorganically Passivated Quantum Dots
2.2.1.2.1. Epitaxial Growth
2.2.1.2.2. Non-epitaxial Growth
2.2.1.3. Multi-Shell Structure
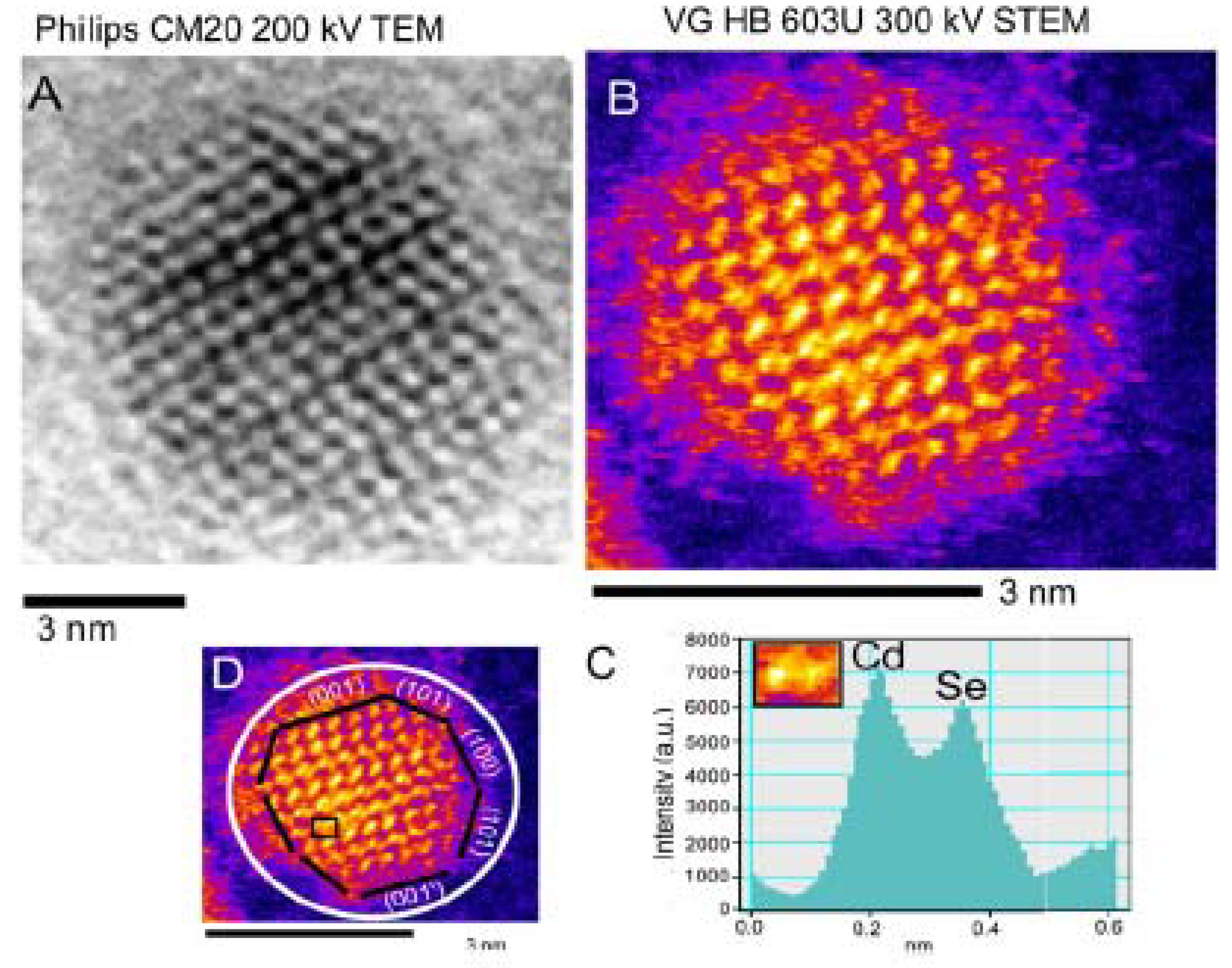
2.2.2. Characterization of Shell Structures
3. Properties
3.1. Quantum Confinement Effects and Band-Gap
3.1.1. Effective Mass Approximation Model
3.1.2. Linear Combination of Atomic Orbital Theory–Molecular Orbital Theory
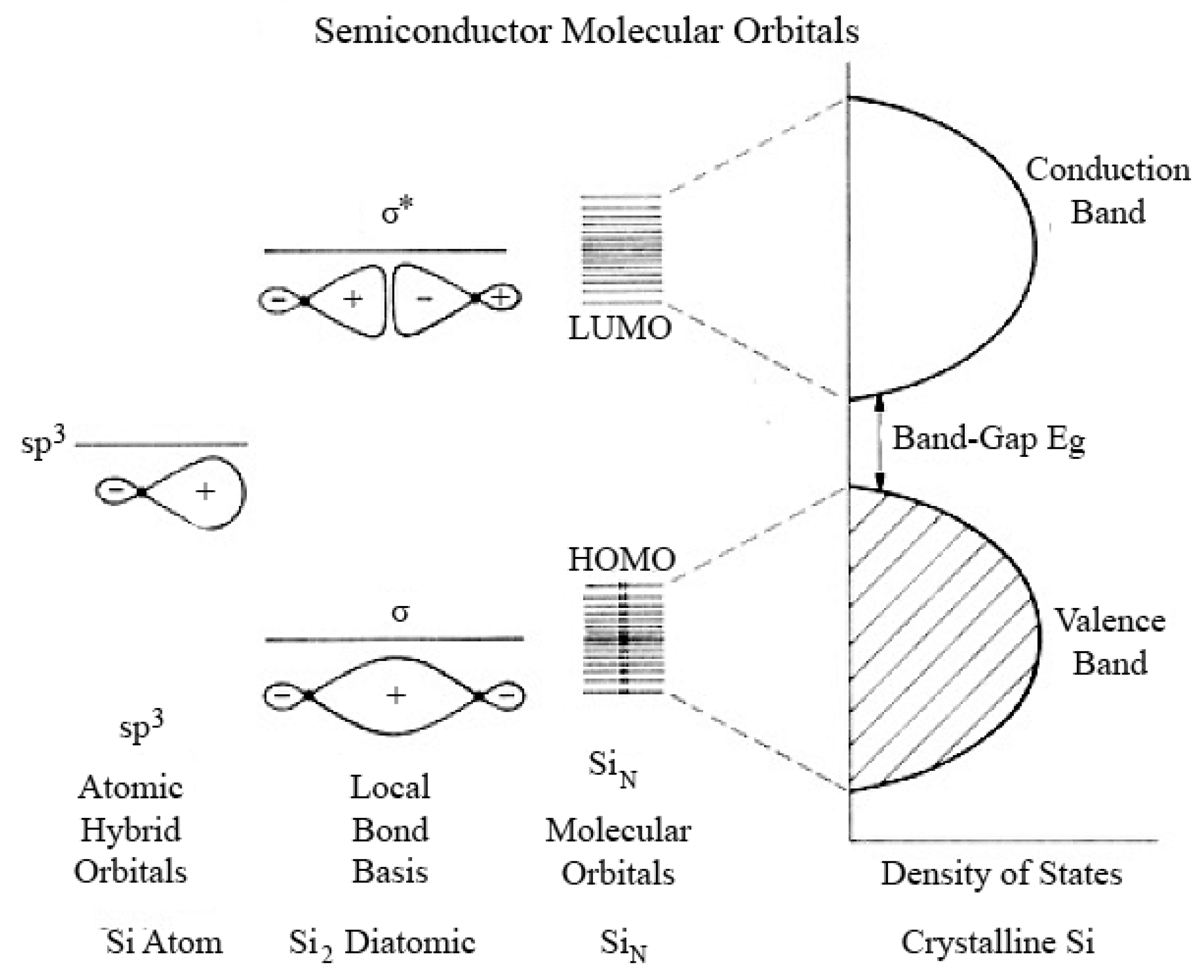
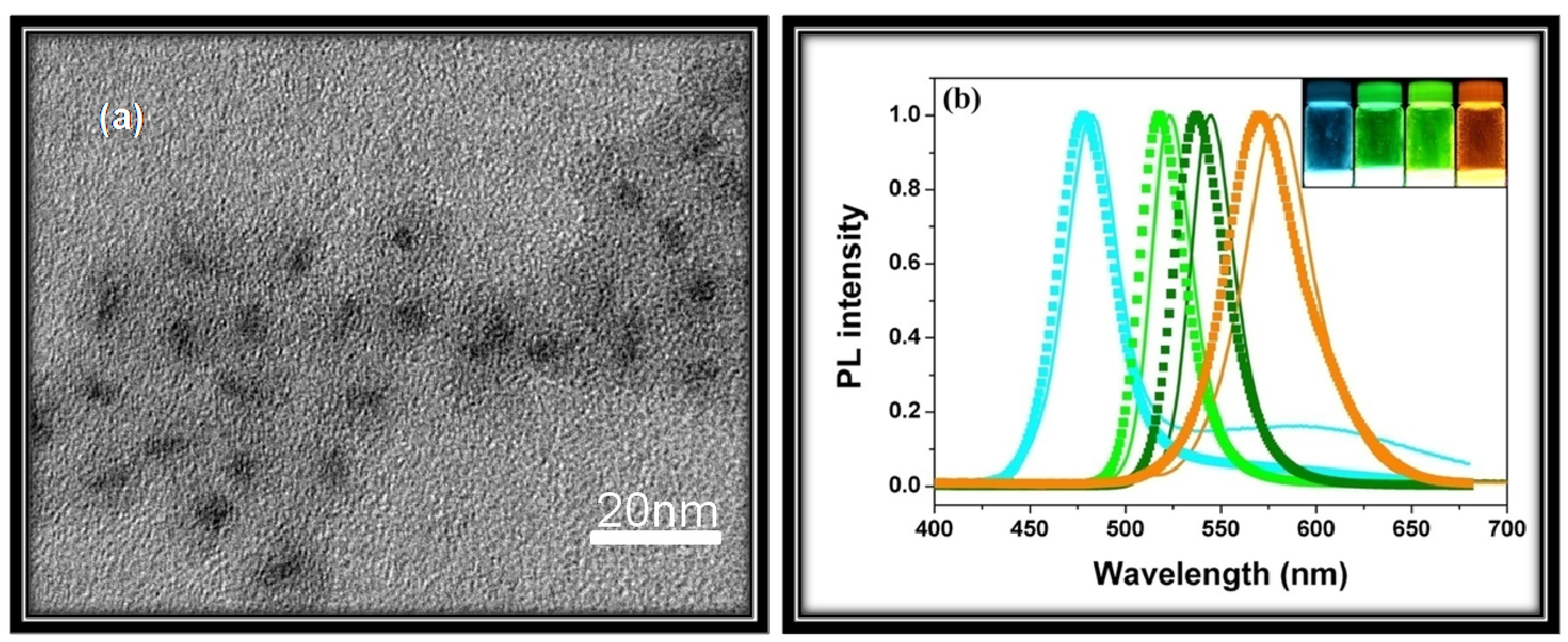
3.2. Luminescence Properties
3.2.1. Radiative Relaxation
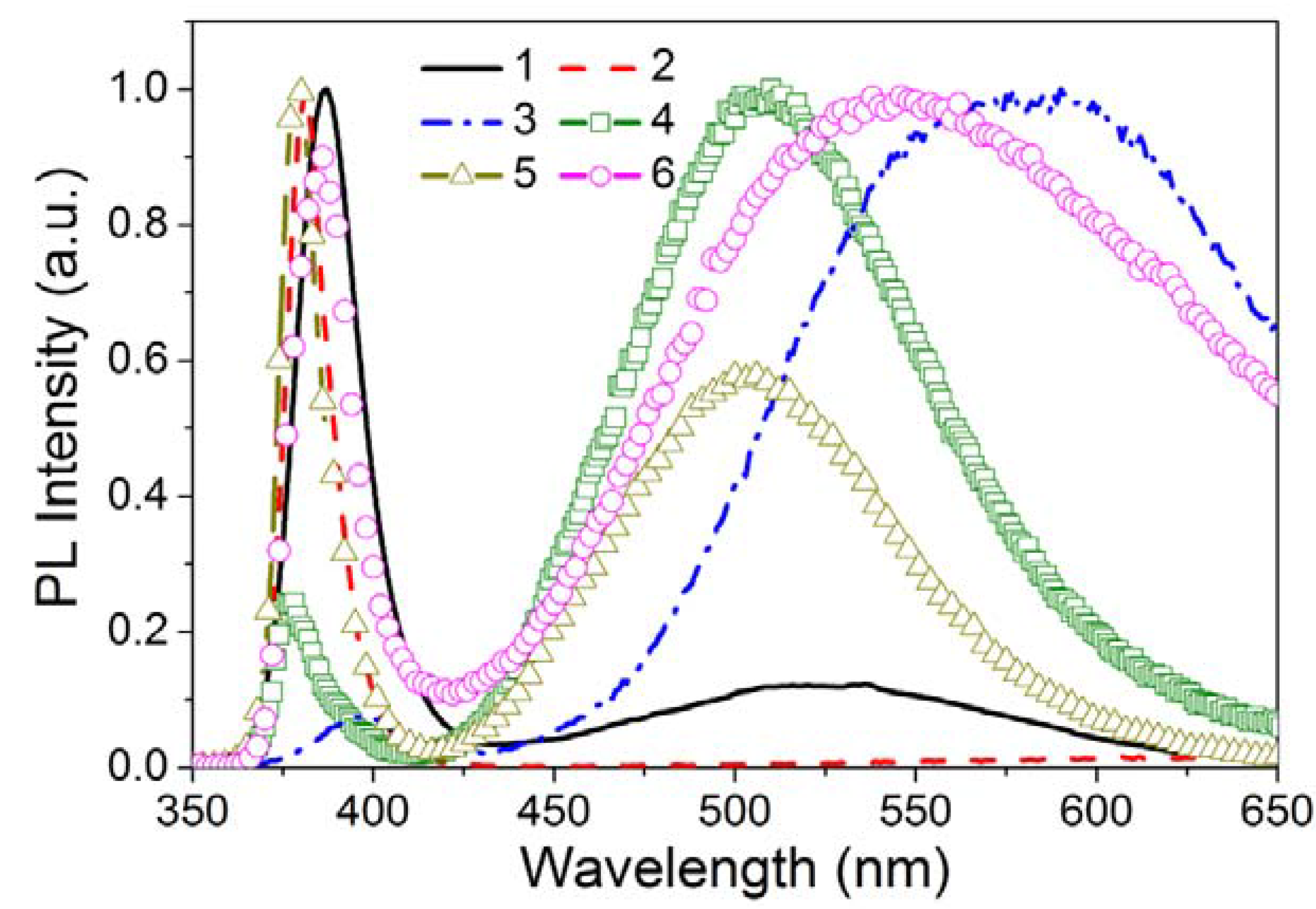
3.2.1.1. Band-Edge Emission
3.2.1.2. Defect Emission
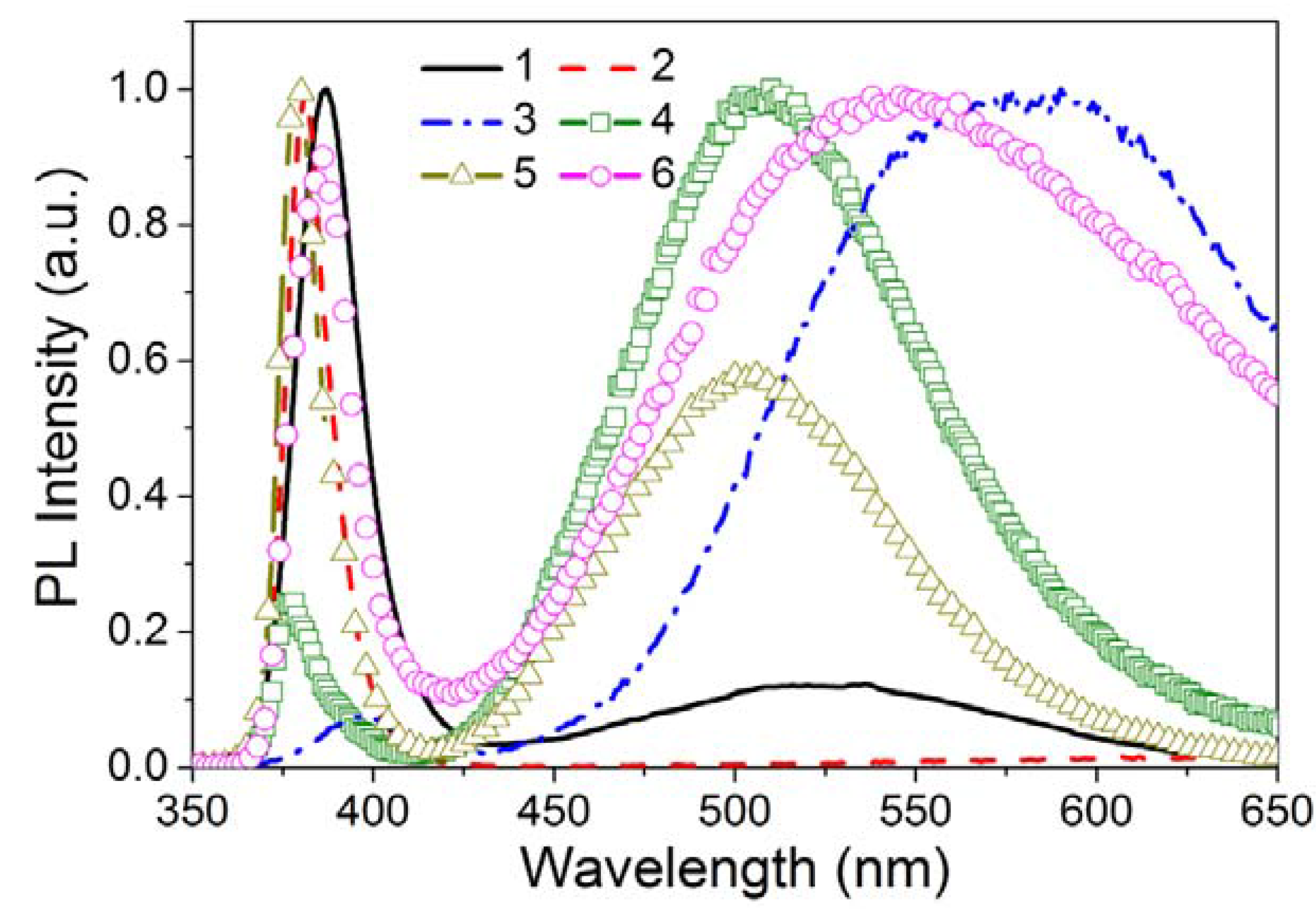
3.2.1.3. Activator Emission
3.2.2. Quantum Yield of Quantum Dots
3.2.2.1. Reported Quantum Yield
| Quantum Dot | Size (nm) | Emission | Quantum Yield | Standard (QY) | Specification | Ref |
|---|---|---|---|---|---|---|
| CdSe/ZnS | 2.7–3 | Excitation 470 nm Emission range: 480–850 nm | 50% | Rhodamine 560 in ethanol | Shell thickness: ~0.6nm | [103] |
| CdSe | 4.2 | 20% | Bare | [104] | ||
| CdSe/ZnS | 4.2 | 50% | 1.5 monolayer of ZnS | [104] | ||
| CdSe/CdS | 2.3 3.0 | 59% 84% | 2.1 MLr CdS 1.8 MLr CdS | [70] | ||
| ZnSe | 4.3–6 | 360–420 nm | 20–50% | [160] | ||
| CdSe/ZnS | 2.0 2.6 4.6 5.6 | O.D. 0.1 | 36% 49% 30% 27% | Rhodamine 590, 610, 640 | [161] | |
| CdSe/ZnS | 3.7 | 66% | 1.6 MLr ZnS | [162] | ||
| ZnSe:Mn | 2.7–6 | 22% at RT 75% at 50K | [163] | |||
| CdSe | 7.5 | O.D. 0.1 | 85% | Coumarin 540 (62% @ 458nm), Rhodamin 6G (95% @ 528nm), 3B (50% @ 550nm), 640 (100% @ 570 nm), LD 690 (63% @616 nm) | As synthesized | [164] |
| CdSe | 7.5 | O.D. 0.1 | 85% | Coumarin 540 (62% @ 458nm), Rhodamin 6G (95% @ 528nm), 3B (50% @ 550nm), 640 (100% @ 570 nm), LD 690 (63% @616 nm) | As synthesized | [164] |
| PbSe | 4–5 | 6–20% | [165] | |||
| CdSe | 2–8 | excitation: 400 nm; OD: ≤ 0.1) | 50–80% | Rhodamin B in ethanol (90% @ 400 nm) | [166] | |
| CdSe CdSe/CdS CdSe/CdS/ZnCdS CdSe/CdS/ZnCdS/ZnS | 3.8 5.2 7.6 8.9 | 30% 60% 65% 80% | Bare CdS: 2ML ZnCdS: 2ML ZnS: 2.5 ML | [89] | ||
| CdSe/ZnS ZnSe/CdSe/ZnS | 90% (550–650 nm) 70% (510–560 nm) | [167] | ||||
| CdSe CdSe/CdS CdSe/CdS/ZnS | 4.0 5.5 6.8 | Same OD with standard | 16% 38% 75% | Rhodamine 6G (95%) | Bare CdS: 2ML ZnS: 2ML | [168] |
| CdSe CdSe/SiO2 | OD: 0.01 Excitation: 350 nm (B) 450 nm (G) 500 (R) | 22% (bare 523 & 581 nm) 82% (542 nm) | 9,10-diphenyl-anthracene in cyclohexane (90% @ 350nm); Fluorescein in 0.1 M NaOH (95% @450 nm); Rhodamin 6G in methanol(95% @ 500 nm) | Shell thickness: ~6nm | [72] |
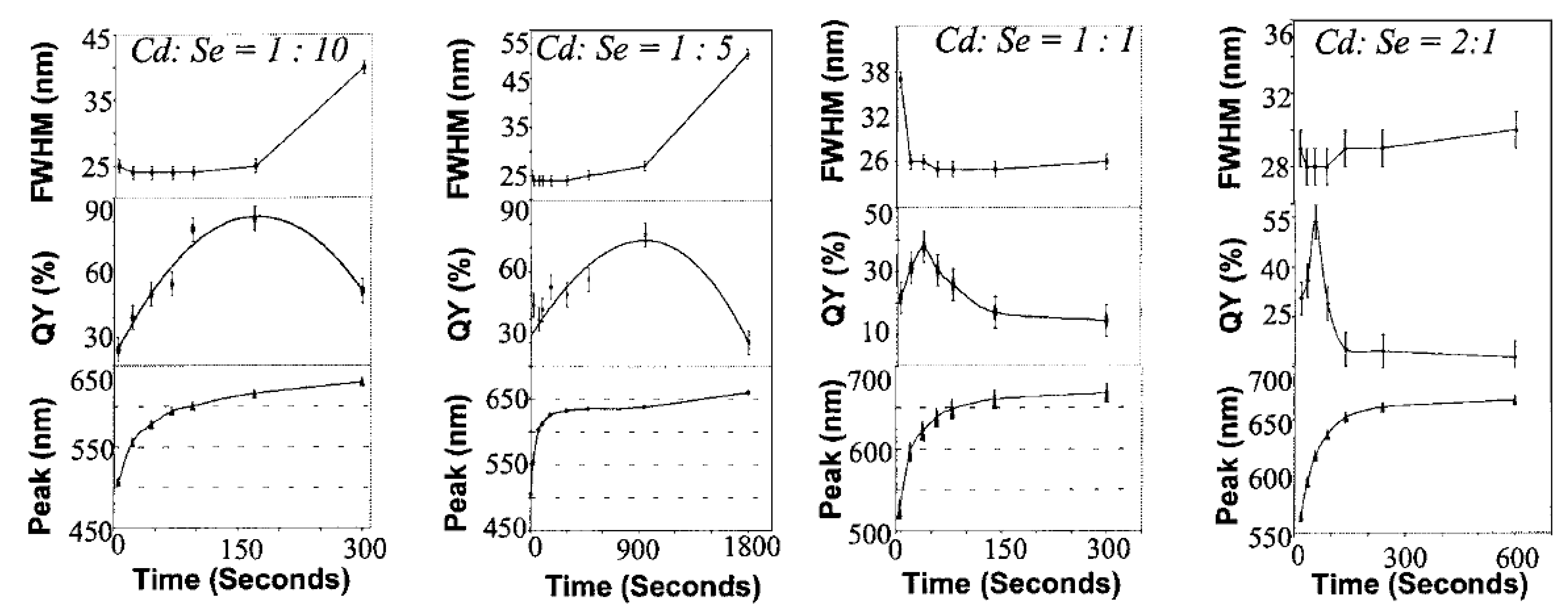
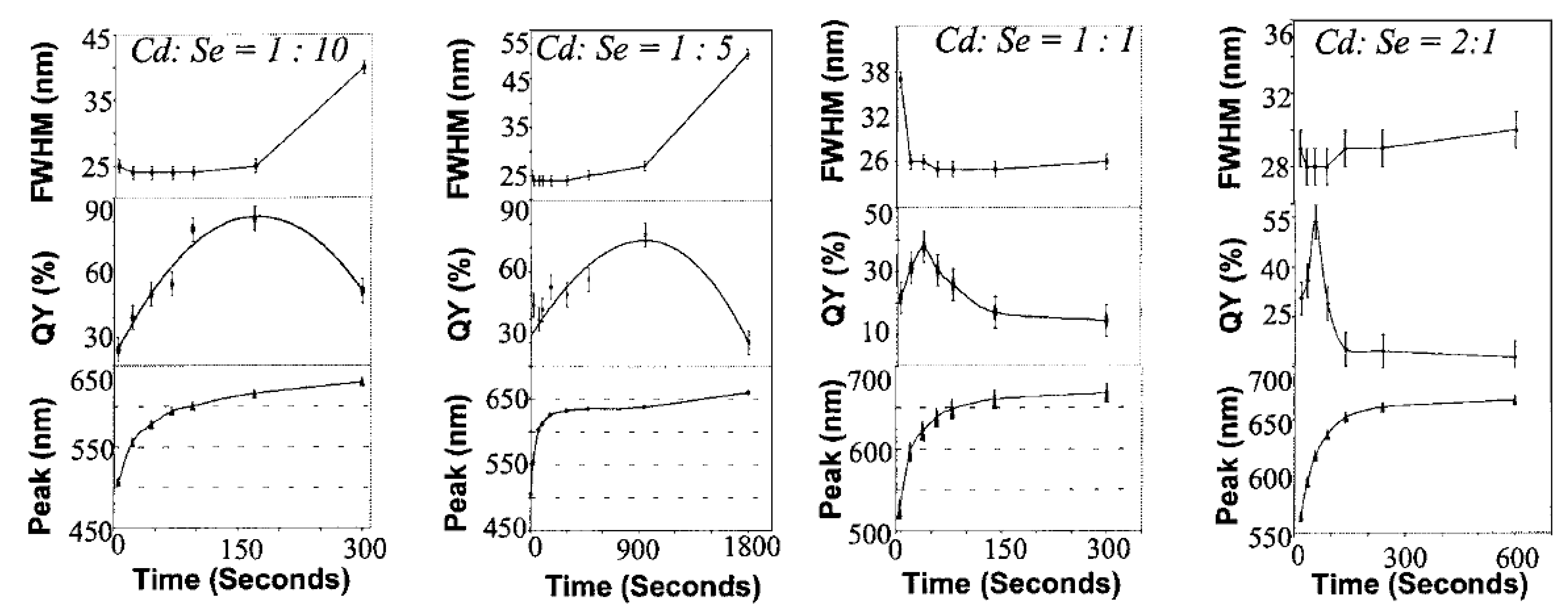
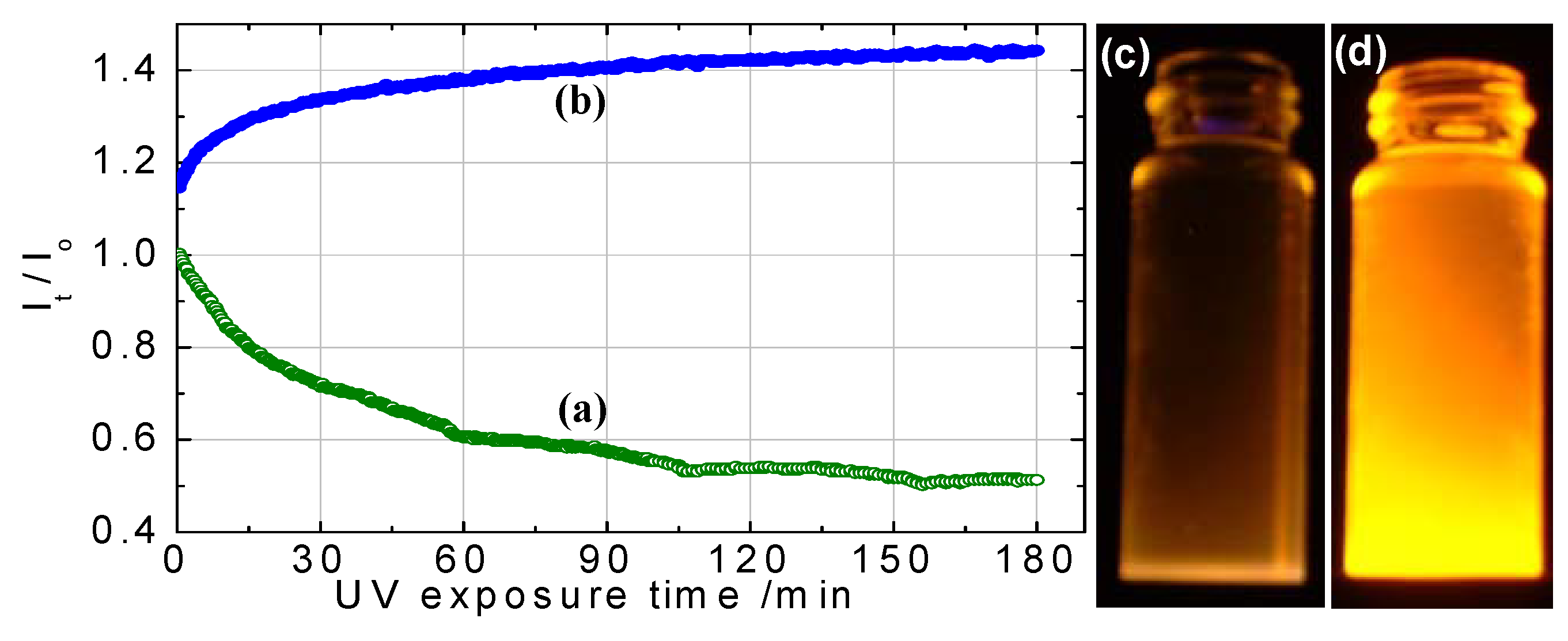
3.2.2.2. Change of Quantum Yield under Ultraviolet Irradiation

3.2.3. Non-radiative Process in Quantum Dots
4. Synthesis Processes
4.1. Top-Down Synthesis Processes
4.2. Bottom-up Approach
4.2.1. Wet-Chemical Methods
4.2.1.1. Sol-Gel Process
4.2.1.2. Microemulsion Process
4.2.1.3. Hot-Solution Decomposition Process
| Year | Qdots | Precursor | Process parameters | Particle Size (nm) | Ref |
|---|---|---|---|---|---|
| 1990 | GaAs | GaCl3, (TMS)3As in Quinoline | 240 °C for 3days; flame anneal at 450 °C | 2.4 | [200] |
| 1990 | ZnS, ZnSe, CdS, CdSe, CdTe, HgTe | M(ER)2; R: n-butyl phenyl; E: S, Se, Te; M: Cd, Zn, Hg and/or phosphine complexes; Co.Sol.: DEPE | DEPE and M(ER)2 reacted, (Temp. range: 250–300 °C) | 2.5–5 nm | [201] |
| 1993-1998 | CdS, CdSe, CdTe | Me2Cd, silylchalconides, Phosphine chalconides; Co.sol: TOPO & TOP/TBP | 300–350 °C at 1 atm at Ar (TOPO degassing); 230–260 °C (growth temp.) | 1.2–11.5 | [14,101,202] |
| 1994 | GaAs | GaCl3/ GaI3, diglyme, As, toluene, Na-K alloy | As, Na-K alloy mixture refluxed to 100 °C in Ar for 2 days; GaCl3/GaI3 diglyme mixture added, heated from 0 °C to RT to 111 °C. for 2 days | 6–10 | [203] |
| 1995 | InP, GaP, GaInP2 | Mixture of chloro-indium/gallium oxalate (GaCl3 for GaP) and (TMS)3P in CH3CN ; Co.sol: TOPO & TOP | 270–360 °C at airless condition for 3 days; Qdots dispersed in methanol | 2.6–4.6 (InP), 3 (GaP), 6.5 (GaInP2) | [204] |
| 1996 | InP, InAs | InCl3, TOPO, (TMS)3P/(TMS)3As | InCl3 & TOPO heat at 100 °C for 12 h, (TMS)3P added, after 3hr heated to 265 °C for 6 days | 2–6 | [205,206] |
| 1996 | CdSe/ZnS | Me2Cd,Me2 Zn, Se, (TMS)2S, Co.sol: TOPO, TOP | Single step synthesis Core: 350 °C at 1 atm at Ar, growth: 310 °C Shell: 300 °C | 2.7–4 | [103] |
| 1997 | CdSe/ZnS | Me2Cd, Me2 Zn, Se, (TMS)2S, Co.sol: TOPO, TOP | Two step synthesis (airless) Core growth: 290–300 °C Shell growth: 140 °C for 2.3 nm & 220°C for 5.5 nm | 2.3–5.5 | [104] |
| 1997 | CdSe/CdS | Me2Cd, Se, (TMS)2S, Co.sol: TOPO, TBP | Two step process: Core: 300 °C; Shell: 100 °C | 2.5–4 | [70] |
| 1998 | ZnSe | Me2Zn, Se, HDA, TOP | HDA dried & degassed at 150 °C for hrs in vacuum and heated to 310 °C at 1 atm in Ar; Core growth with Zn & Se precursor at 270 °C. | 4.3–6 nm | [160] |
| 1996–1999 | InAs/InP InAs/CdSe | (TMS)3As, Indium (III) chloride, TOP (TMS)3P, Me2Cd; TBP-Se | Two-step Process (airless) Core growth: 260 °C; Shell: dropwise addition; 260 °C | 2.5–6 nm (InAs); 1.7 (core/shell) | Core [202,206] Core/shell [207] |
| 2000 | CdSe | Me2Cd, Se, TBP, TOPO, HPA | TOPO (+HPA 1.5–3 wt%) degassed at 360 °C (or 310 °C, 280 °C); Core growth: 300 °C (or 280 °C or 250 °C) | ~6nm | [208,209] |
| 2001 | ZnSe:Mn | Me2Mn, Et2Zn, TOP, Se, HDA | Dimethyl Mn, TOP, Se, Diethyl Zn mixture added to HDA at 310 °C in N2. Growth: 240–300 °C | 2.7–6.3 | [163] |
| 2001- 2003 | CdSe/ZnS | Me2Cd, Se, TOP, TOPO, HDA, (TMS)2S, Me2Zn | Two Step: Core: reaction & growth: 270–310 °C; Shell: slow addition of Zn & S precursor at 180–220 °C | 4.5–5 nm | [162,210] |
| 2001 | CdSe | Scheme 1: Cd(Ac)2,, SA/ TOPO; 2: Cd(Ac)2, SA; 3: CdCO3, SA/TOPO; 4: CdCO3, LA/TOPO; 5: CdO, SA/TOPO; 6: Cd(Ac)2, tech TOPO; 7: CdO, TDPA/TOPO | Solvent & Cd-precursor heated to 250–360 °C at Ar; TOP-Se or TBP-Se injected; Growth temp: 200–320 °C (if DDA involve, temp: ~220 °C | 2–25nm | [191] |
| 2001 | CdS, CdSe, CdTe | CdO, TOPO, HPA/TDPA, S, Se, Te & TOP | One pot: CdO, HPA/TDPA heated 300 °C; Core with chalconide precursor: reaction: 270 °C, & growth 250 °C | 2–8 nm | [211] |
| 2002 | CdSe | CdO, Se, TOPO, TBP, HDA, ODA, SA | CdO & SA, heated to 150 °C in Ar; after CdO dissolution, cool to RT; TOPO & HAD added & heated to 320 °C in Ar; TBP-Se added, Growth 290 °C | [164] | |
| 2003 | PbS | PbO, OA, (TMS)2S, TOP | PbO dissolved in oleic acid at 150 °C in Ar; (TMS)2S & TOP injected | 5nm | [197] |
| 2003 | CdSeS | CdO, OA, TOA, Se, S, TOP | CdO+ OA+TOA heated at 300 °C in N2, TOP-S, TOP-Se injected | ~5nm | [26] |
| 2005 | PbSe | Pb-acetate trihydrate, OA, Se, TOP | Single Step: Pb acetate + Co.sol degassed at 100–120 °C at 300–500 mTorr for 2h; reaction and growth: 140 °C | 5 nm | [212] |
| 2006 | CdSe | CdO, OA, TOA, C8SH or C18SH, | CdO + OA + TOA heated at 300 °C; TOA + C8SH or C18SH injected | 3, 4, 6 nm | [213] |
4.2.1.4. Other Synthesis Processes
4.2.2. Vapor-Phase Methods
5. Application
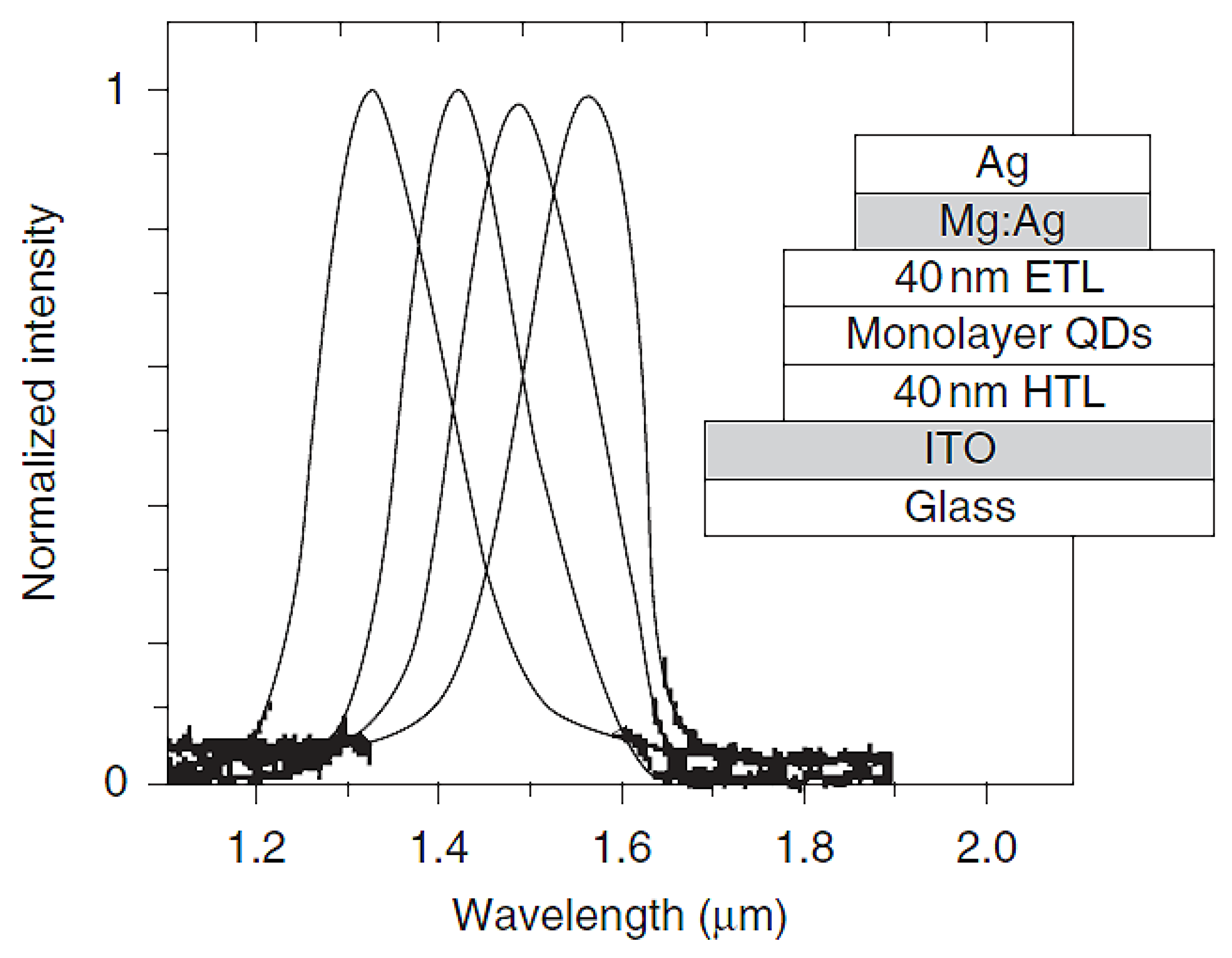
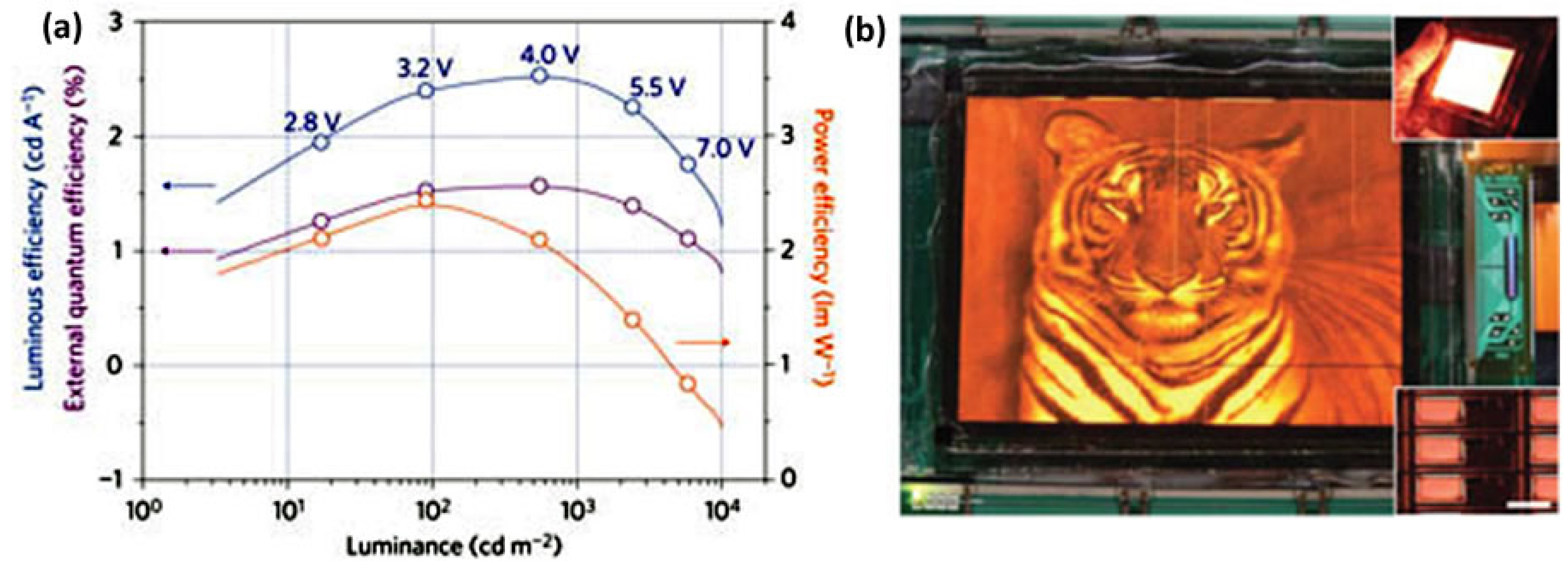
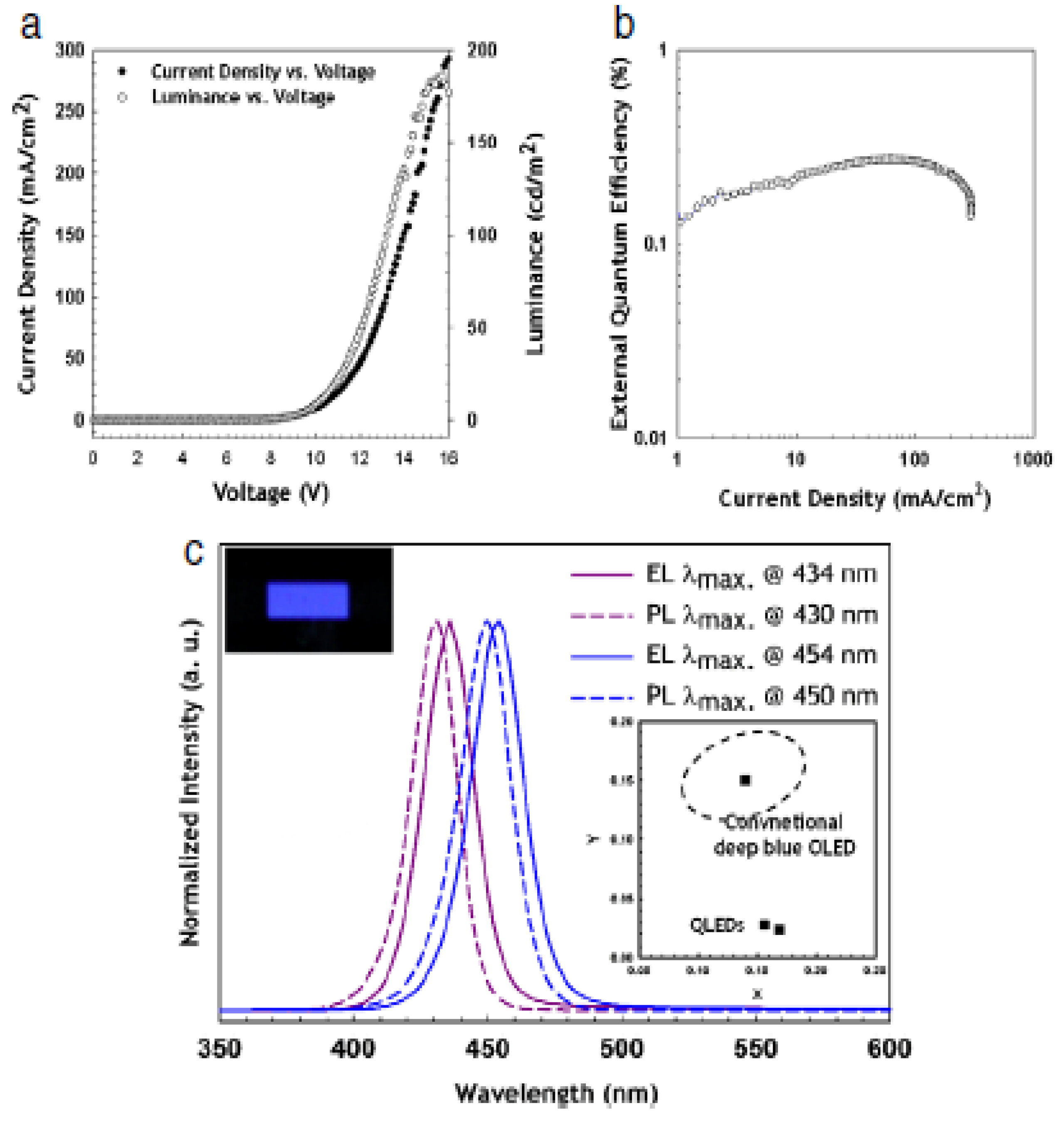
5.1. Quantum Dots for Electroluminescence Device Fabrication
| Organics | Conduction Band (eV) | Valence Band (eV) | Reference |
|---|---|---|---|
| Alq3 | 3.1 | 5.8 | [242,243] |
| CBP | 2.9 | 6.0 | [244] |
| PBD | 2.6 | 6.1 | [245] |
| PCBM | 4.0 | 6.5 | [246] |
| PPV | 2.5 | 5.1 | [247,248] |
| PVK | 2.2 | 5.3 | [245] |
| TAZ | 3.0 | 6.5 | [249] |
| TFB | 2.2 | 5.4 | [250] |
| TPBI | 2.7 | 6.2 | [244,251] |
| TPD | 2.1 | 5.4 | [242] |
| Poly TPD | 2.3/2.5 | 5.2/5.4 | [244,252] |
| Qdots | Conduction Band (eV) | Valence Band (eV) | Particle Size (nm) | Emission | Ref. |
|---|---|---|---|---|---|
| CdSe | 4.4 | 6.5 | 5 | [248] | |
| CdSe/CdS | 4.4 | 6.5 | 4.6 | [251] | |
| CdSe/CdS | 4.7 | 6.8 | 4 | [253] | |
| CdSe/CdS/ZnS | 4.8 | 6.8 | 6.8 | 600 | [250] |
| CdSe/ZnS | 4.4 | 6.5 | [247] | ||
| CdSe/ZnS | 4.3 | 6.5 | 550 nm | [242] | |
| CdSe/ZnS | 4.8 | 6.5 | [245] | ||
| CdSe/ZnS | 4.6 (CdSe) | 6.8 (CdSe) | 5.8 | [249] | |
| CdSe/ZnS | 4.7 | 6.7 | [254] | ||
| CdSe/ZnS/CdS | 3.9 | 6.0 | 3–8.3 | G, Y, O, R | [252] |
| Materials | Work function (eV) |
|---|---|
| Al | 4.1 |
| Ag | 4.6 |
| Ca | 2.9 |
| ITO | 4.7 |
| LiF/Al | 2.8 |
| Mg | 3.7 |
| PEDOT:PSS | 5 |
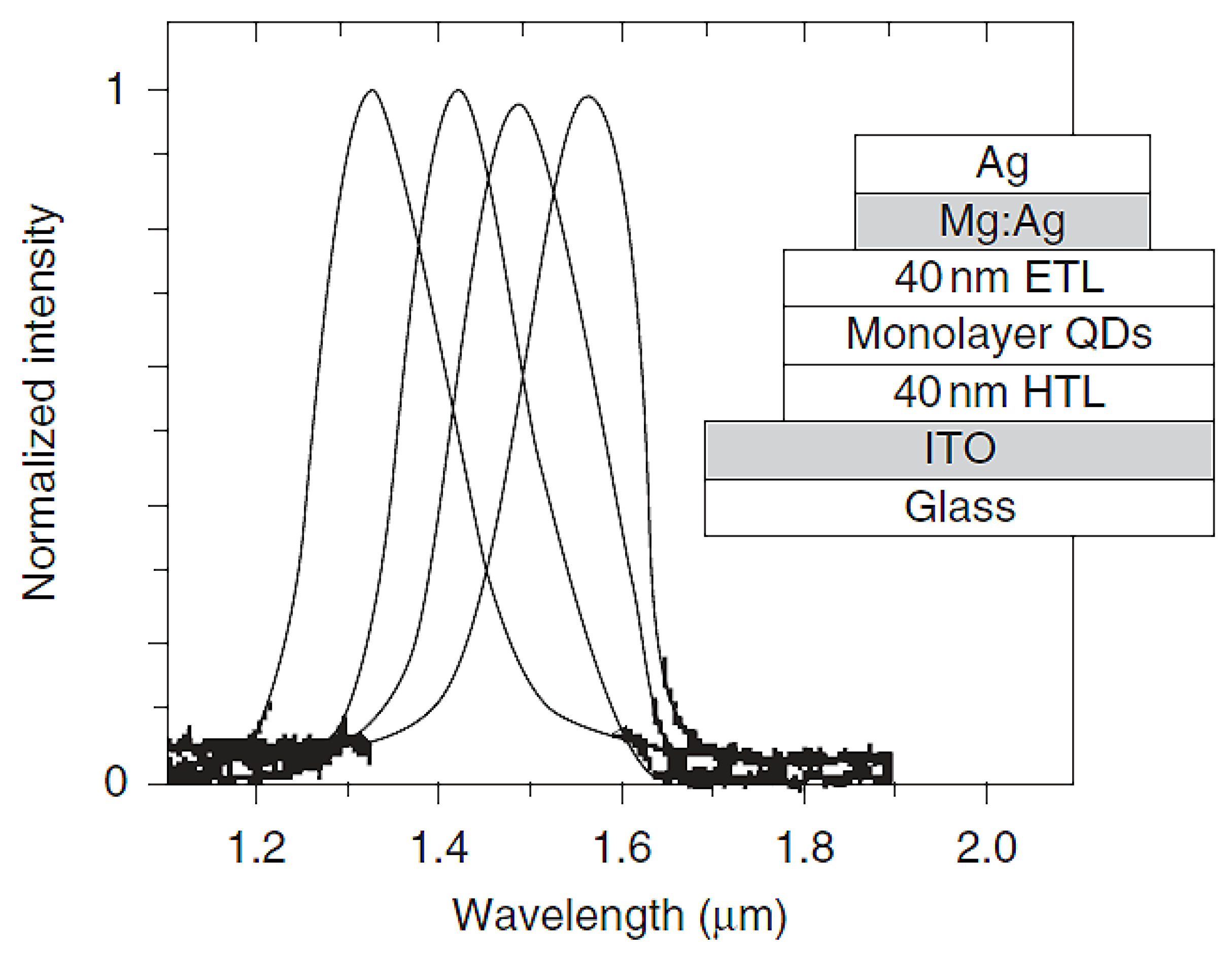
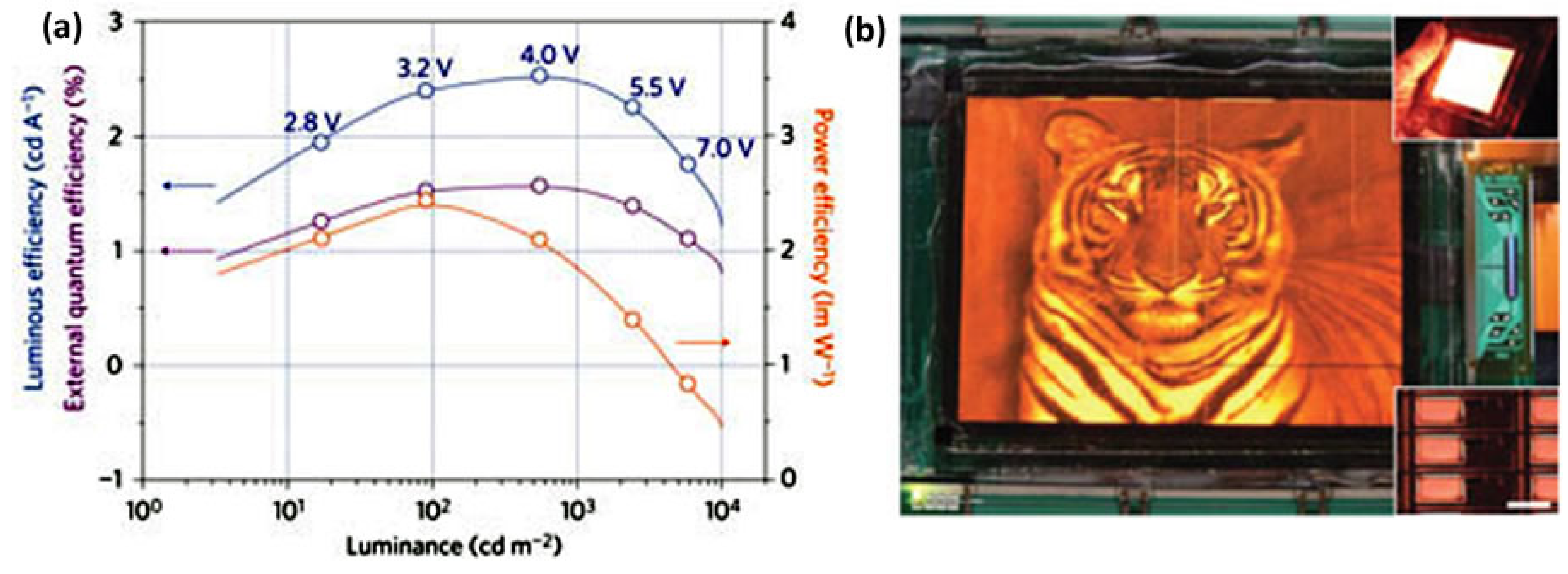
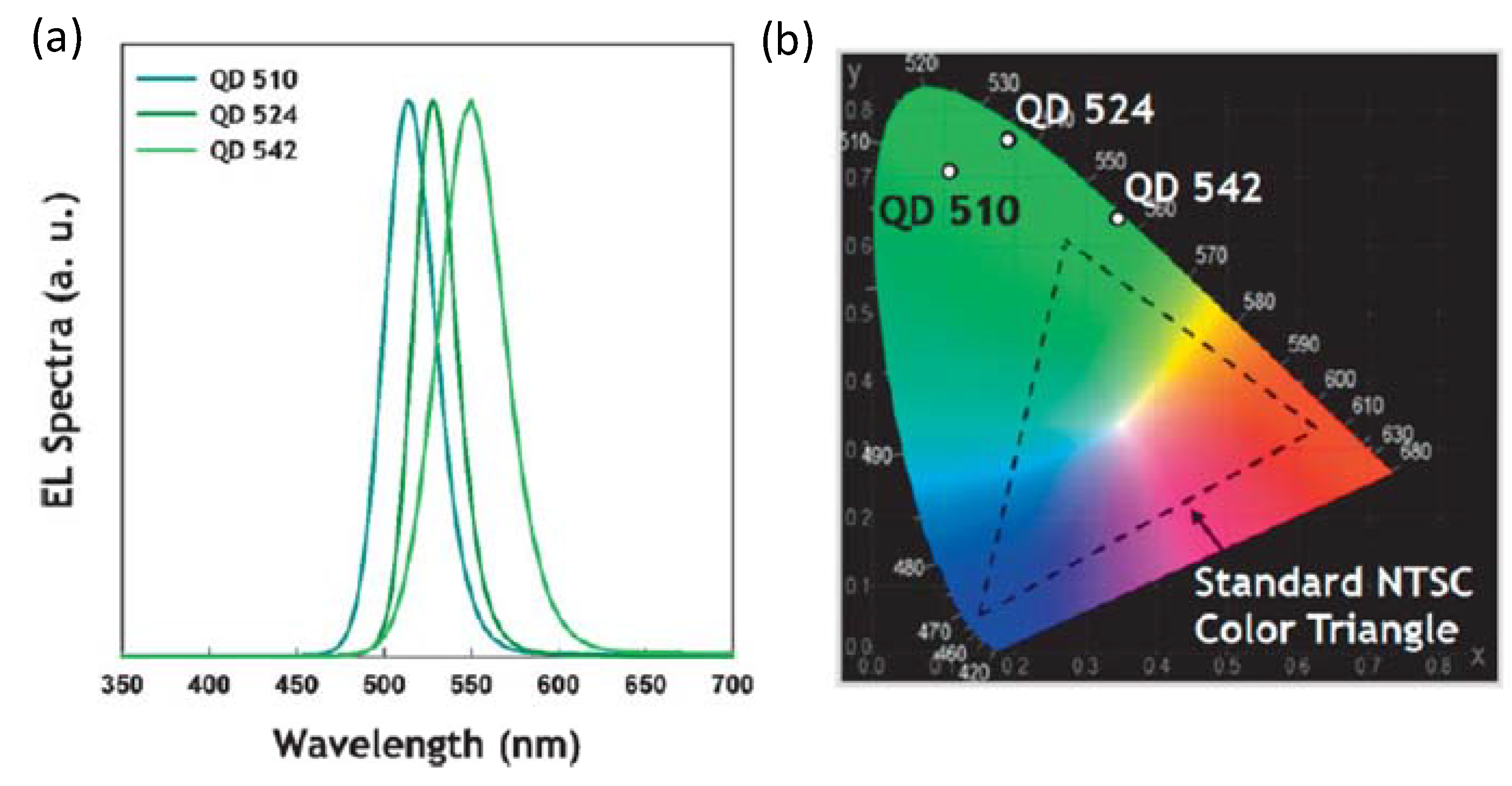
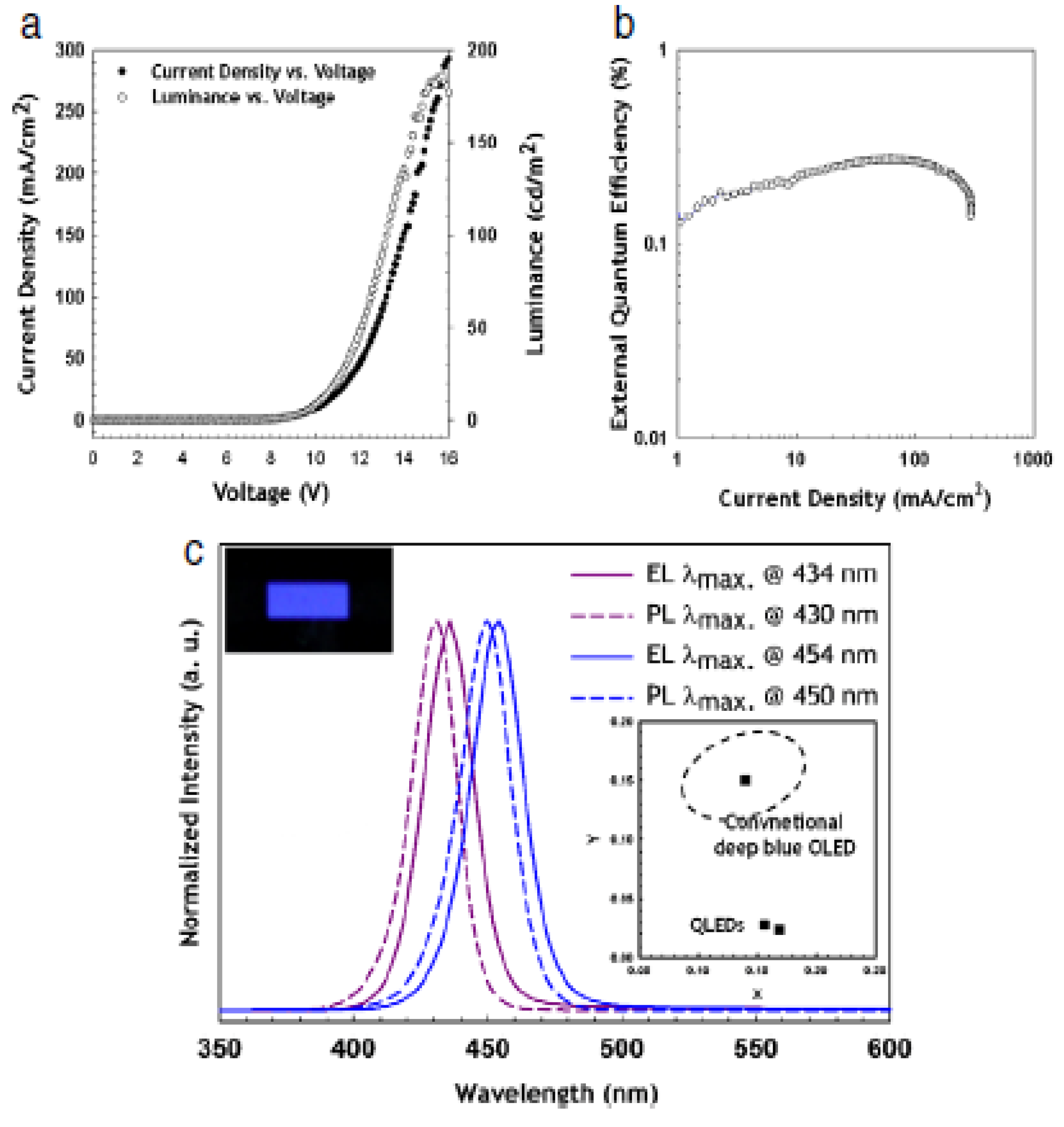
| Emitting Wavelength (nm) | Vturn-on (V) | Lmax (nit) | LE (cd/A) | PE (lm/W) | EQE (%) | Device Structure | Ref. |
|---|---|---|---|---|---|---|---|
| ~610 | 4 | 100 | -- | -- | 0.001–0.01% | ITO//CdSe//PPV//Mg & ITO//PPV//CdSe//Mg | [256] |
| 520, 550, 610 | -- | -- | -- | -- | 0.0005% | ITO//(mixture PVK – t-Bu-PBD- CdSe)//Al | [262] |
| 560 | 4 | 600 | 0.2% | ITO//PPV//CdSe/CdS// MgAg | [253] | ||
| 600 | 3–3.5 | 0.1% | ITO//PPV (multilayer) //CdSe//Al | [247] | |||
| 620 | 3.5 | 0.01–0.2% | ITO//PPV//(CdSe in block co-polymer)//Al | [248] | |||
| 550–650 | 2.3–3.5 | 0.1% | ITO//(mixture of PDDA & CdTe)//Al | [269] | |||
| 560 | ~3.5 | 2000 | 1.9 | -- | 0.52% | ITO//TPD//CdSe/ZnS//Alq3//Mg:Ag ITO//TPD//CdSe/ZnS//TAZ//Alq3//Mg:Ag | [242] |
| ~580 | 13 | 0.005% | ITO//(mixture of CdSeS & PDBD)//TAZ//Alq3//LiF//Al | [26] | |||
| 1300–1400 | 15 | 0.5% | ITO//PEDOT//(Mixture of InAs/ZnSe & MEHPPV or F6BT)//Ca:Al | [264] | |||
| 540–635 | 3.5 | -- | -- | -- | 1.1% | ITO//TPD//CdSe/ZnS(monolayer)//TAZ//Alq3//Mg:Ag//Ag | [249] |
| 1330–1560 | ~3 | 0.001% | ITO//TPD or αNPD//PbSe//Alq3//BCP// Mg:Ag//Ag | [165] | |||
| 1000–1500 | 1.2%* | ITO//(mixture of PbS & MEHPPV or CNPPV)//Mg//Ag | [197] | ||||
| ~610 | 5 | 500 | 0.2% | ITO//PVK//CdSe/ZnS//bu-PBD//Al | [245] | ||
| 615 | 3 | 7000 | 2.0 | 1.0 | 2% | ITO//TPD//CdSe//Alq3//Mg:Ag//Ag | [212] |
| 573–619 | 0.001–0.1% | Au//pGaN//CdSe/ZnS//nGaN//In | [254] | ||||
| 1590 | ~1.2 | 0.02% | ITO//PEDOT:PSS//HgTe// Al | [220] | |||
| 610 | 4 | 1000 | 1.2% | ITO//PS-TPD-PFCB//TCTA-BVB//CdSe/CdS//TPBI//Ca//Ag | [251] | ||
| 625 | 3000 | 0.18% | ITO//NiO//CdSe/ZnS// Alq3//Ag:Mg//Ag | [243] | |||
| 520 | 2.5 | 0.5% | ITO//CBP//CdZnSe/CdZnS//TAZ//Alq3//Mg:Ag//Ag | [27] | |||
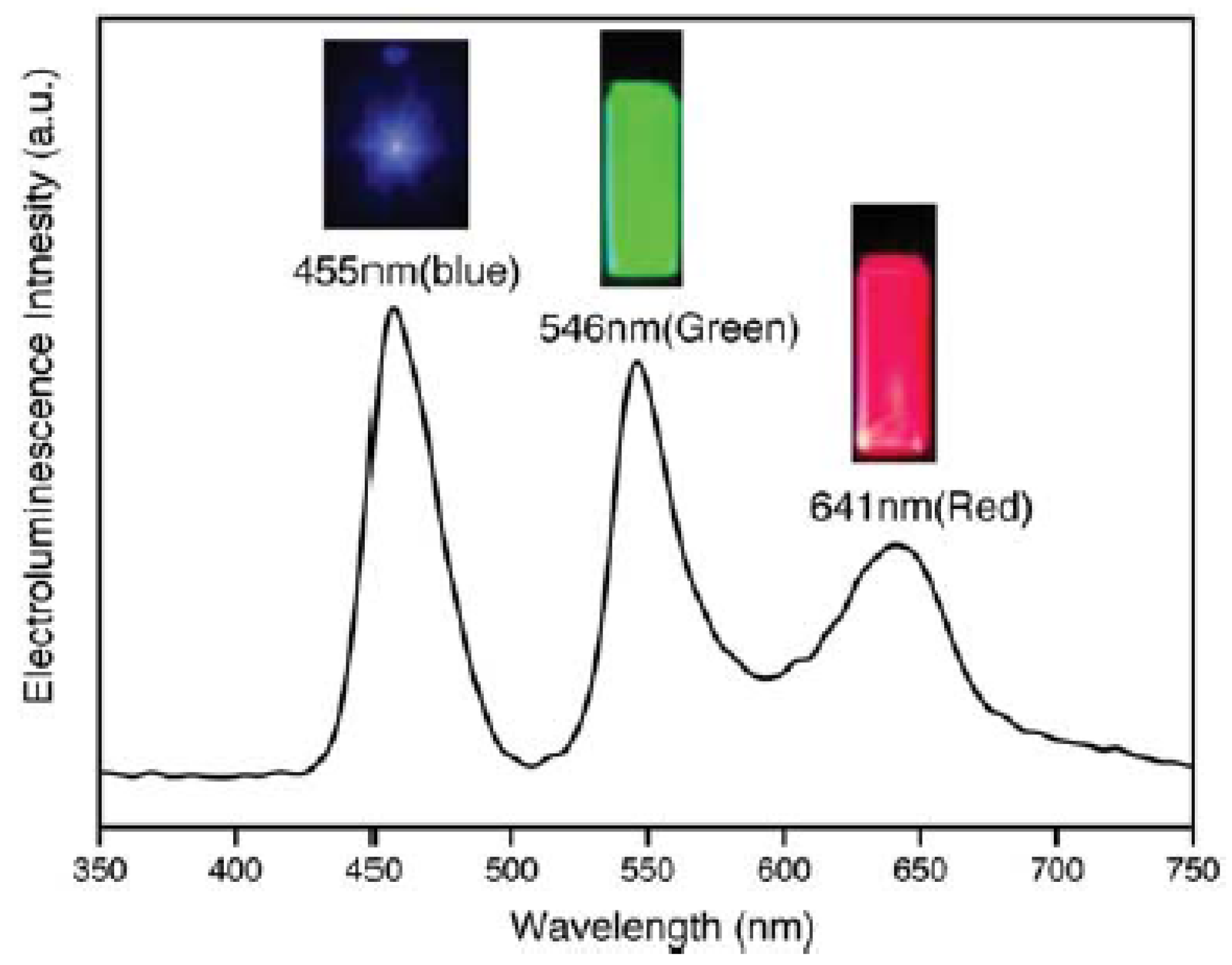
| ~440 (B) ~545 (G) ~610 (R) | 2–3 | 830 (W) | 0.9 (W) | 0.57 (W) | 0.35% (B) 0.65% (G) 1.6% (R) 0.36% (W) | ITO//PEDOT:PSS//TPD//Qdots//TAZ//Alq3//Mg/Ag//Ag | [167] |
| 517 (G) 546 (Y) 589 (O) 600 (R) | 4 (G), 5 (Y), 3 (O), 3 (R) | 3700 (G), 4470 (Y), 3200 (O), 9064 (R) | 1.1–2 (G-R) | <1.1 | ITO//PEDOT-PSS//poly-TPD//CdSe/ZnS or CdSe/CdS/ZnS//Alq3//Ca/Al | [252] | |
| 460 | 2.5 | 1,600 | 0.5 | 0.5 | 0.06 | ITO//PEDOT:PSS//poly-TPD//CdS/ZnS//Al | [267] |
| 638 | 3.8 | 1950 | 0.1% | ITO//NiO//ZnCdSe//ZnO:SnO2//Ag | [257] | ||
| ~600 | 1.9 | 12,380 | 1.67 | ITO//PEDOT:PSS//TFB//CdSe/CdS/ZnS//TiO2//Al | [250] | ||
| 434–450 | 5 | 150 | -- | -- | 0.1–0.3 | ITO//PEDOT:PSS//poly-TPD:CBP//CdZnS/ZnS// TPBI//LiF//Al | [244] |
| 510, 524, 542 | 3.5 | 16,000 | 6.0 | -- | 1.4 | ITO//PEDOT:PSS//poly-TPD//CdSe/ZnS//TPBI//LiF //Al | [268] |
5.2. Downconversion of Blue or Ultraviolet Light
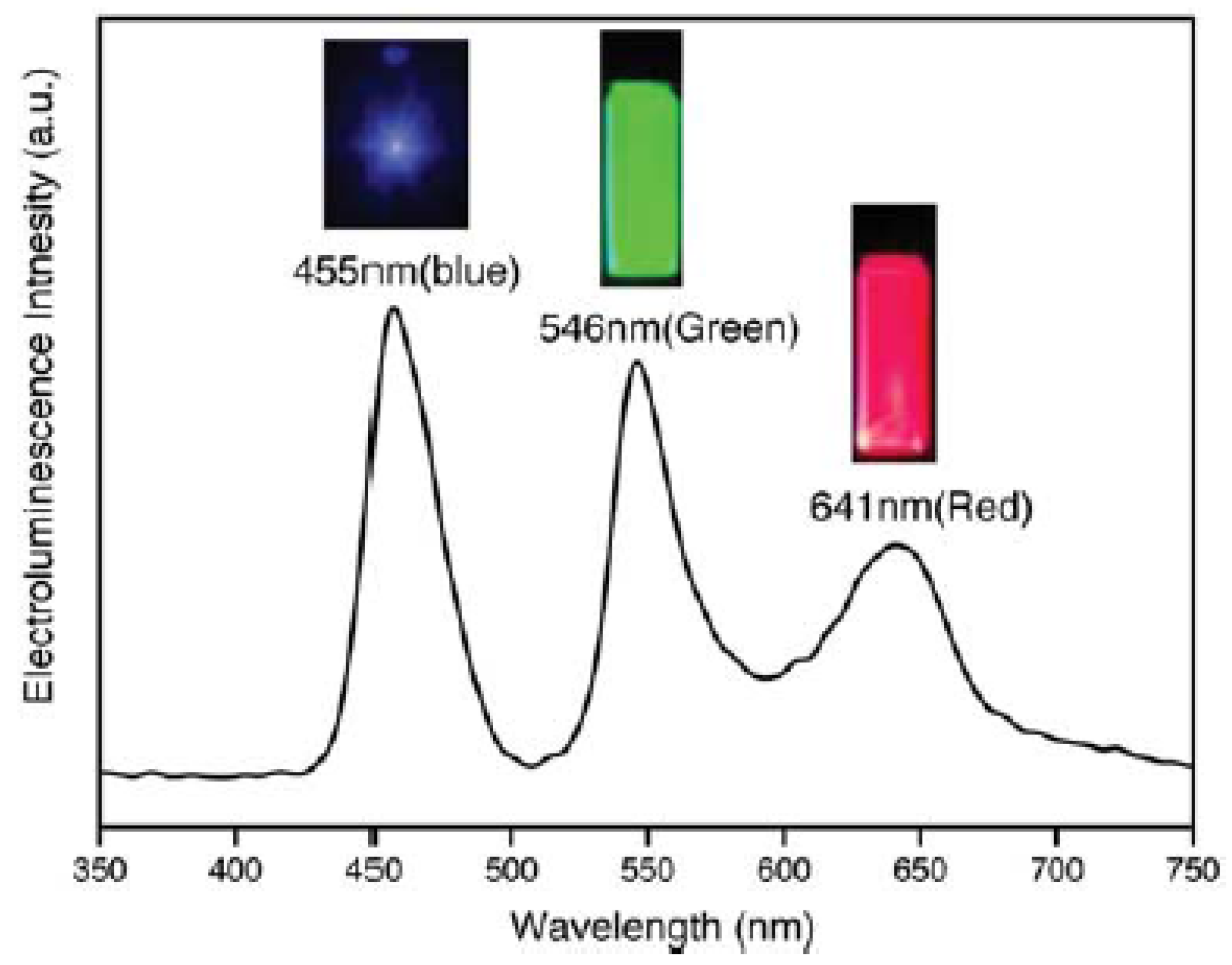
| Year | Source light | Qdots | Matrix | Emitted light | Ref. |
|---|---|---|---|---|---|
| 2000 | UV (Hg lamp), Blue GaN Commercial LED | CdSe/ZnS (2.0, 2.6, 4.6, 5.6 nm) | Polyauryl-methacrylate | UV: Blue, orange, red; Blue: red (590 nm) | [161] |
| 2005 | InGaN (near UV) | ZnSe (TOPO & Stearic acid coated) | organics coated ZnSe (10 wt%) dispersed in epoxy resin | White; CIE (0.38, 0.41) Conversion efficiency: 30% relative to RGB commercial phosphors | [196] |
| 2006 | InGaN (455 nm) | CdSe/ZnSe (G); CdSe/ZnSe (R) CdSe/ZnSe (Y) | TOPO-coated CdSe/ZnSe dispersed in silicone | White, CIE: (0.33, 0.33), CRI: 91 with R& G; White, CIE: (0.32, 0.33), CRI: 50 with Y; Efficiency: 15-30 lm/W | [270] |
| 2007 | 390 nm UV LED | CdSe/CdS/ZnS | 2wt% Qdot in chloroform & epoxy resin at 1:1 (vol); Thermally cured | Red (620 nm) | [168] |
| 2007 | InGaN/GaN (440 nm, 452 nm) | CdSe/ZnS (440-452 | Qdots blended with resin; 400 -1700 μm (Qdot density: 3.04-140 nanomoles/1ml resin) | White; with 453 nm & CdSe/ZnS (540, 500, 580 & 520 nm): CIE (0.24, 0.33), CRI: 71 | [271] |
| 2008 | InGaN/GaN (blue/green) | CdSe/ZnS (620nm, R) & Au particles (for surface Plasmon enhanced emission) | 5 wt% Qdots and 0.05 wt% Au in toluene spin-coated on LED (thickness ~200 nm) | White: (0.27, 0.24); Conversion efficiency ~53% | [272] |
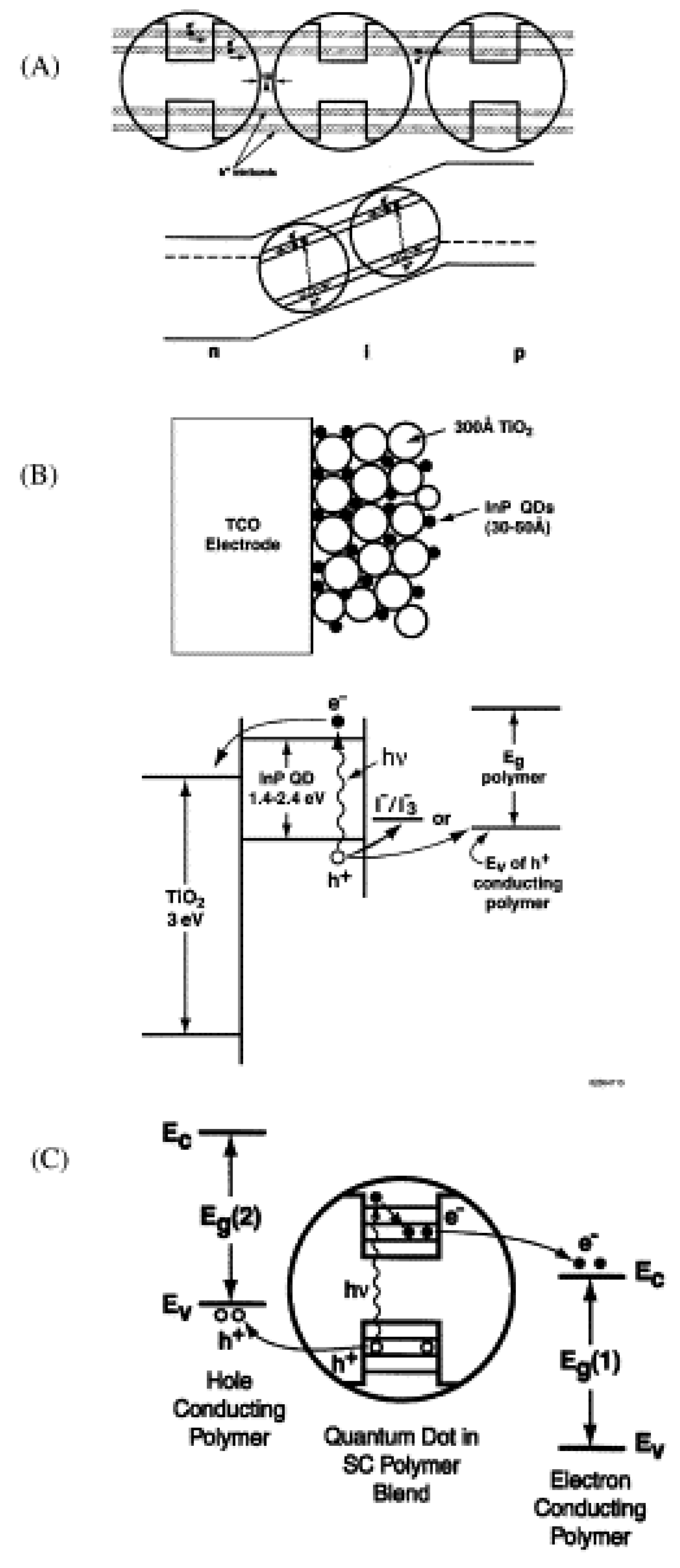
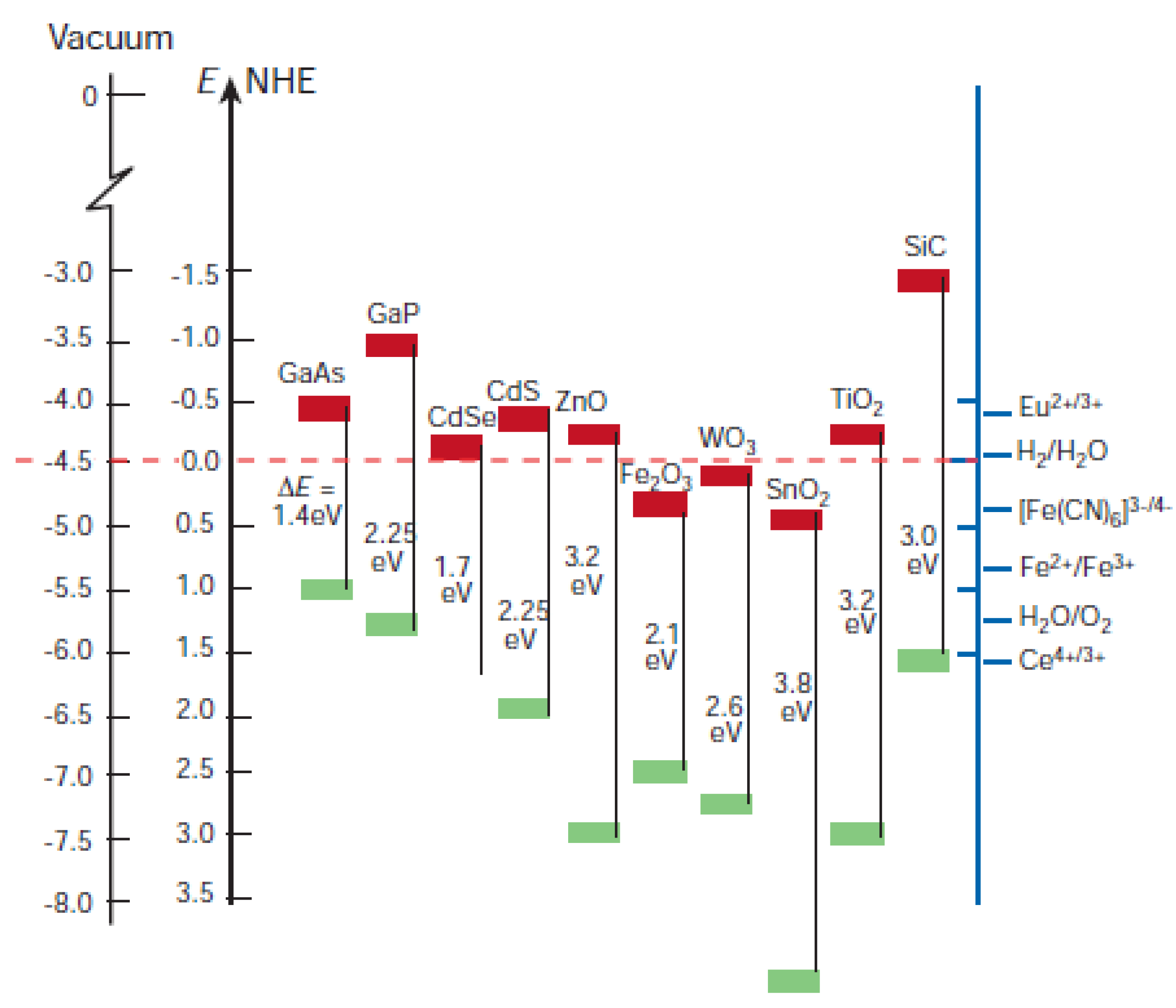
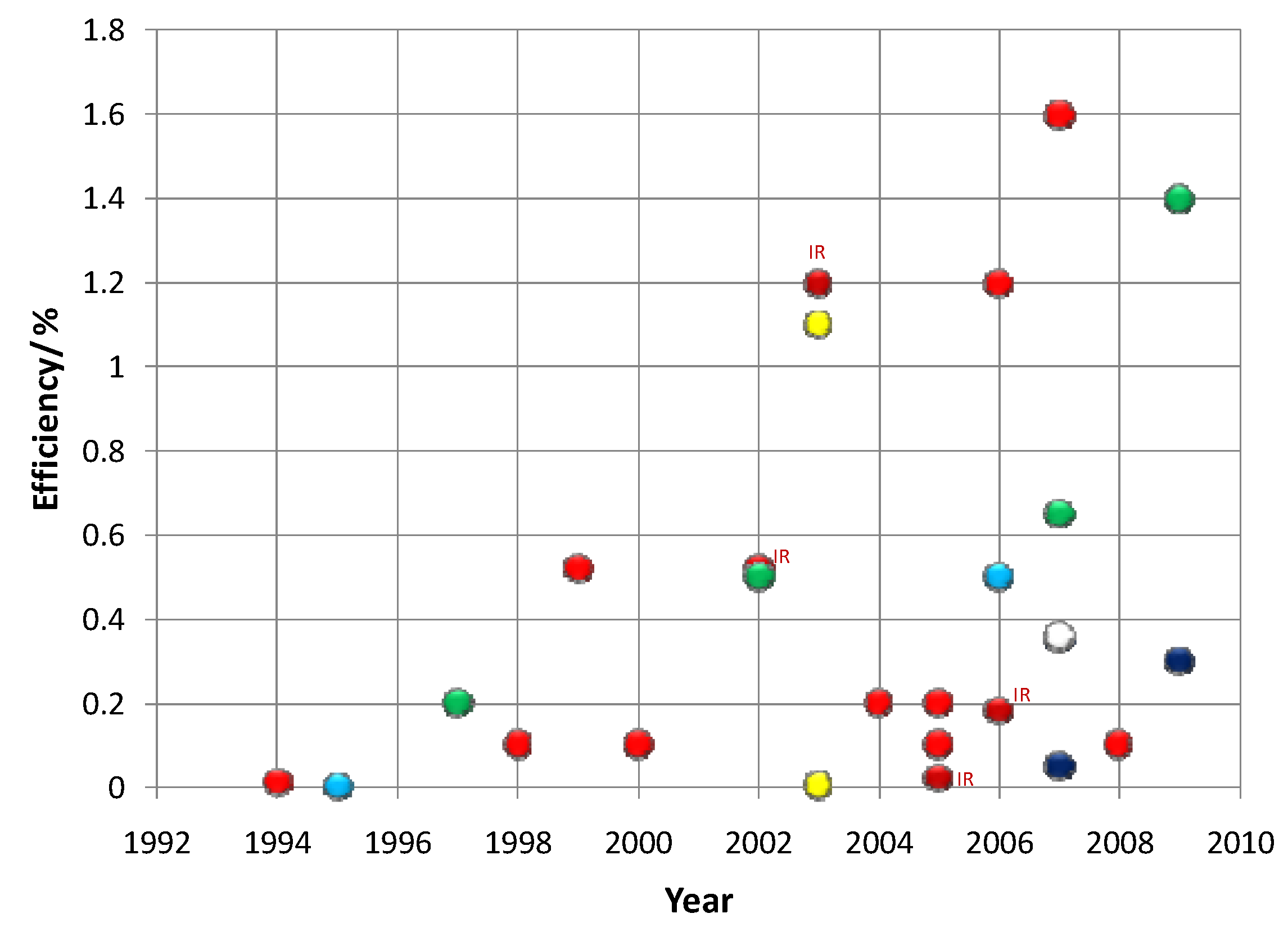
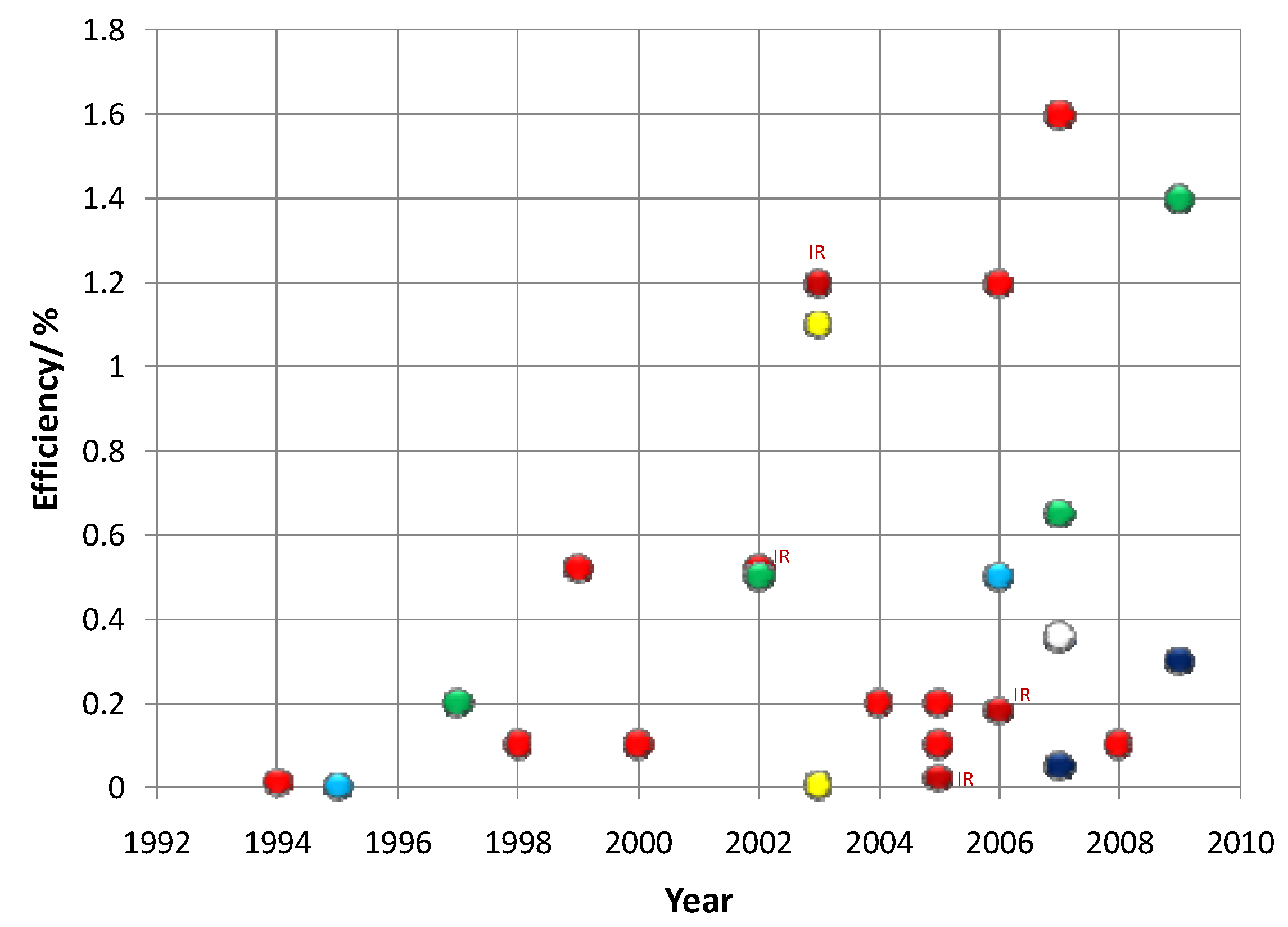
5.3. Quantum Dots in Solar Cell Device Fabrication
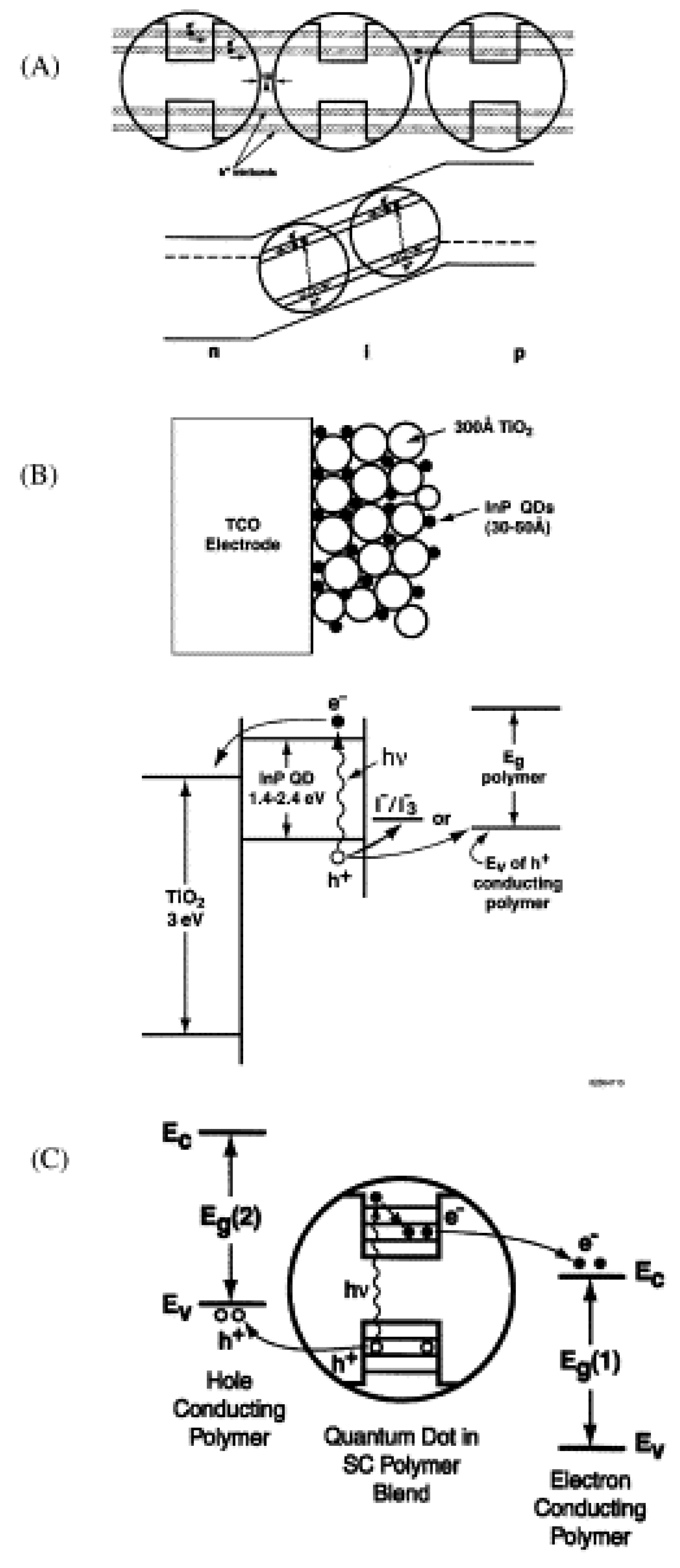
5.3.1. Quantum Dot Sensitized Solar Cell
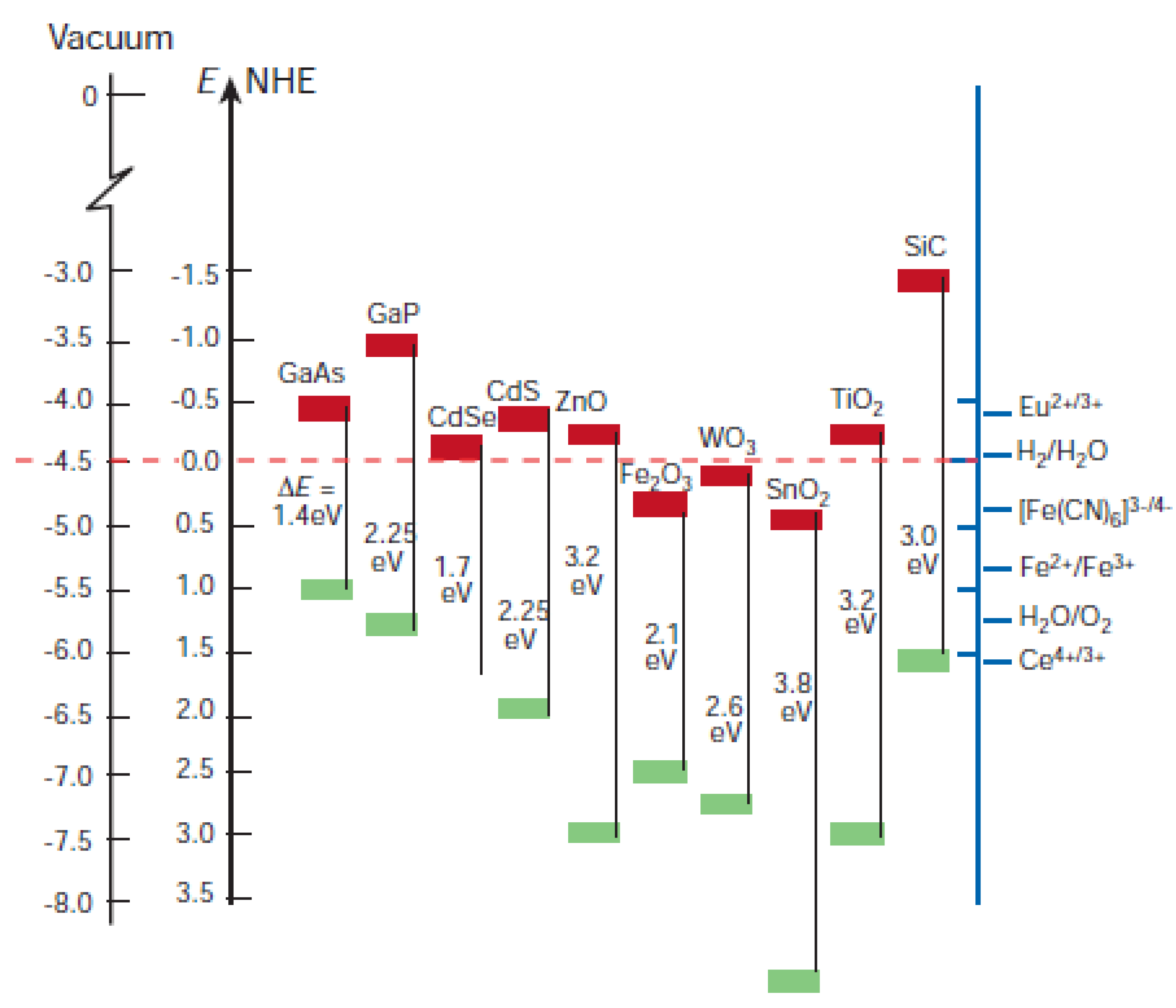
| Year | Qdots | Results | Conversion Efficiency % | Device Structure | Ref |
|---|---|---|---|---|---|
| 1994 | CdS: 4nm PbS: 5nm | JSC: ~1.9 mA/cm2 VOC: 1 V FF: 0.45 EQE: 80% (@ 460 nm) | ITO//TiO2//CdS or PbS | [293] | |
| 1998 | InP: 6.5 nm | FF: 0.685 | 1.5% (400–800 nm) | TiO2//InP | [305] |
| 2002 | PbS: 7 nm | VOC: 0.24V EQE: 45% | (at AM 1.5) 0.49% | SnO2:F//TiO2//PbS//Spiro-OMeTAD, Au | [296] |
| 2006 | CdSe CdS ~5nm | JSC: 10.5 mA/cm2 VOC: 0.66 V FF: 39.5 | 2.8% | TiO2//CdSe or CdS (self-assembled) | [300] |
| 2007 | CdS (4–6 nm) | JSC: 3.44 mA/cm2 VOC: 0.657 V FF: 0.6 | 1.35% | TiO2//CdS (Self-assembled) | [298] |
| 2007 | CdSe | JSC: 7.51 mA/cm2 VOC: 0.71 V FF: 0.5 | 2.7% | TiO2//F//CdSe//F//ZnS | [301] |
| 2008 | CdS | JSC: 7.82 mA/cm2 VOC: 1.27 V FF: 0.578 | 2.8% | TiO2 (nanotubes)//CdS | [302] |
| 2009 | CdS CdSe | FF: 0.49 VOC: 0.5137 V JSc: 16.8 mA/cm2 | 4.22% (AM 1.5) | TiO2//CdS(3)//CdSe(4)//ZnS, Au | [304] |
5.3.2. Quantum Dot Dispersed Solar Cell
| Year | Qdot Size | Results | Conversion Efficiency % (at AM 1.5 G) | Device Structure | Ref |
|---|---|---|---|---|---|
| 1997 | 5nm | VOC: 0.5 V FF: 0.26 EQE: 12% | 0.2 (monochromatic illumination at 514 nm) | ITO// MEHPPV: CdSe or CdS//Al | [307] |
| 2002 | CdSe or CdS Dot: 7nm Rod: 7 X 60 nm | VOC: 0.7V FF: 0.4 EQE: 55% (20% Qdots) | 1.7 | ITO//CdSe: P3HT//Al | [309] |
| 2004 | CdSe: Rod: 7 x 30 nm | 1.5 | ITO//PEDOT:PSS//P3HT-functionalized CdSe //Al | [317] | |
| 2006 | PbS: 4nm | VOC: 1 ISC: -0.13 FF: 0.28 | 0.7 | ITO//PEDOT:PSS//PbS:MEHPPV//Al | [318] |
5.4. Quantum Dots in Other Optoelectronic Devices
5.5. Application of Quantum Dots in Bioimaging Applications
| Qdots | Purpose | Imaging Techniques | Emission/ Size of Qdots | Ref |
|---|---|---|---|---|
| CdSe/CdS/SiO2 | Mouse fibroblast cell imaging | In vitro Fluorescence | 550 nm & 630 nm | [347] |
| CdSe/ZnS | Biological detection/ sensing | In vitro fluorescence | 1–4 nm | [348] |
| CdSe/ZnS/SiO2 | Phagokinetic track imaging | In vitro fluorescence | 554 nm & 626 nm | [349] |
| CdSe/ZnS | Tumor vasculature and lung endothelium imaging | In vitro and in vivo fluorescence | <10 nm | [350] |
| CdTe/CdSe | Cancer cell lymph nodes imaging | In vivo Fluorescence | NIR | [351] |
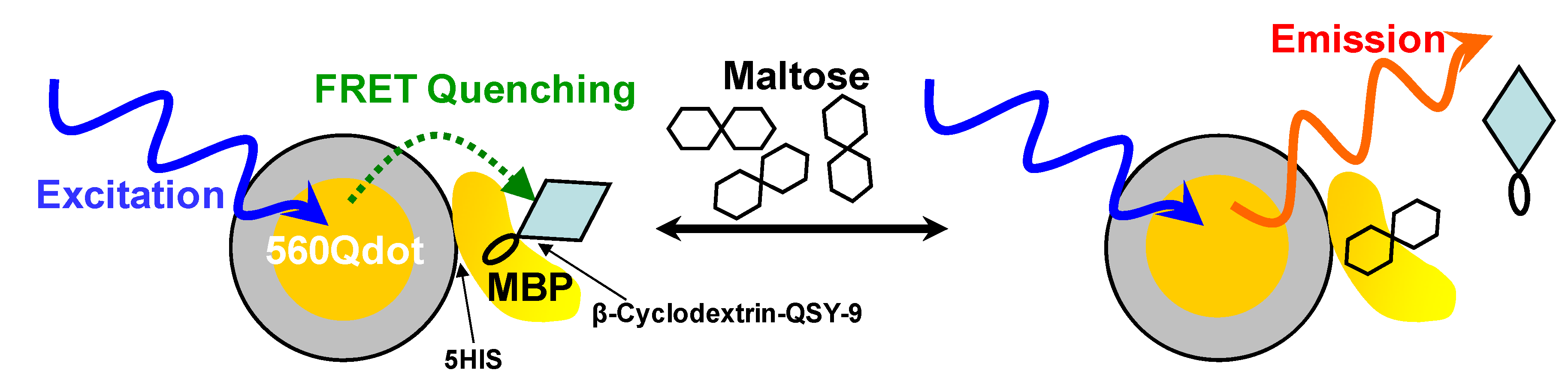
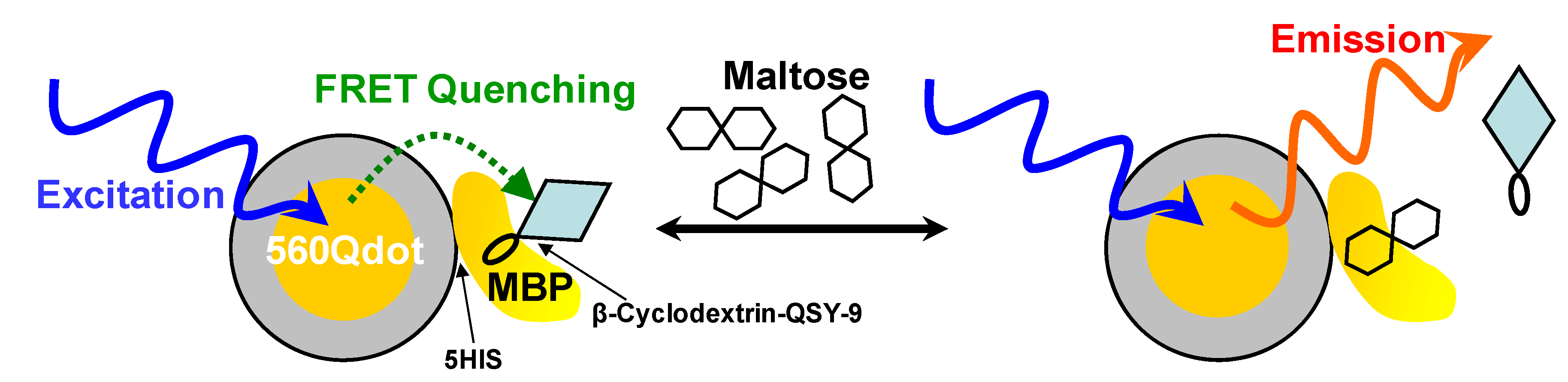
| CdSe/ZnS | Maltose binding Protein | In vitro FRET | 560 nm | [352] |
5.5.1. Fluorescence for Bioimaging

5.5.2. Use of Fluorescence Resonance Energy Transfer in Bioimaging
5.5.3. Surface Enhanced Raman Spectroscopy
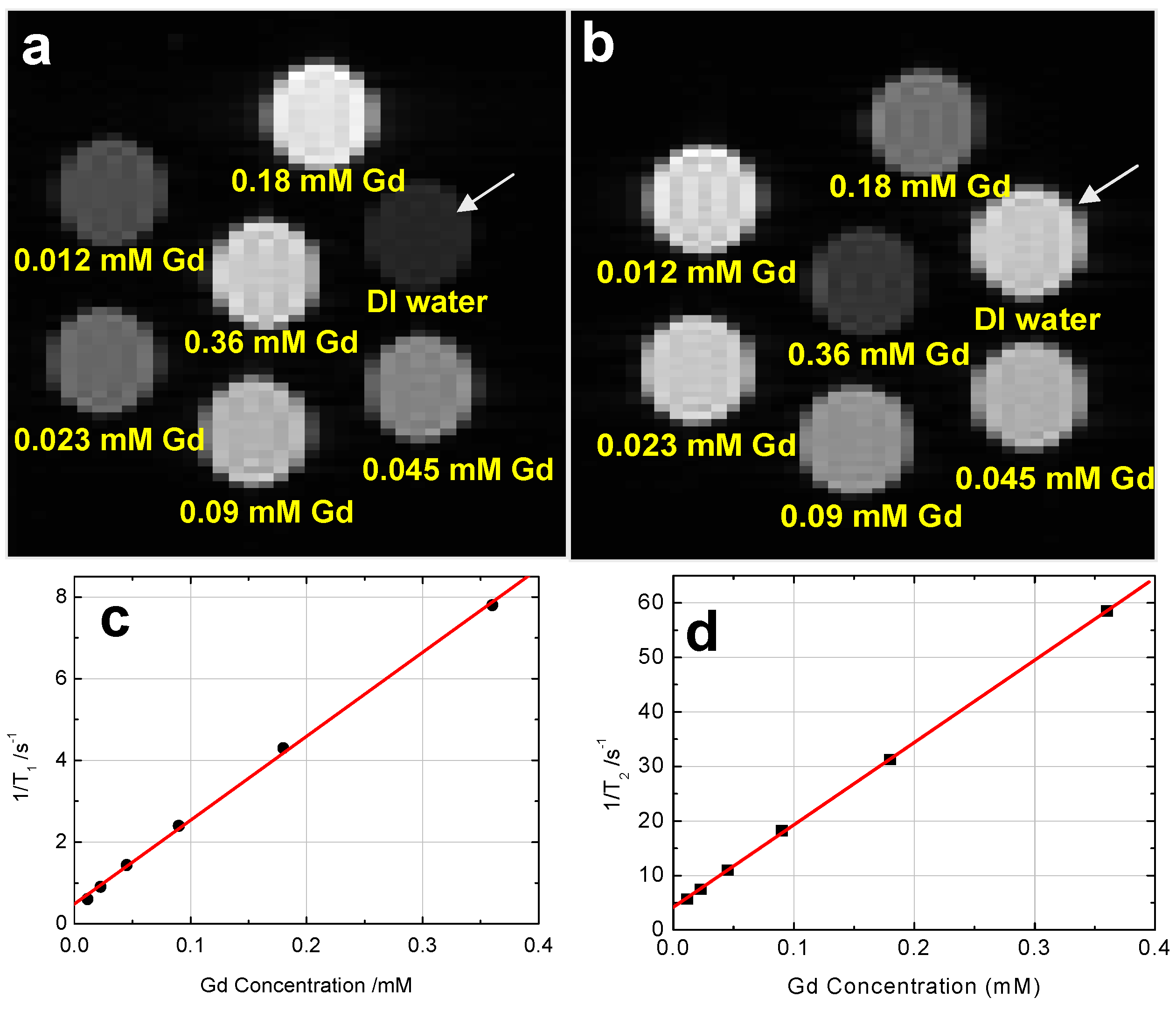
5.5.4. Radio-Opacity and Paramagnetic Properties
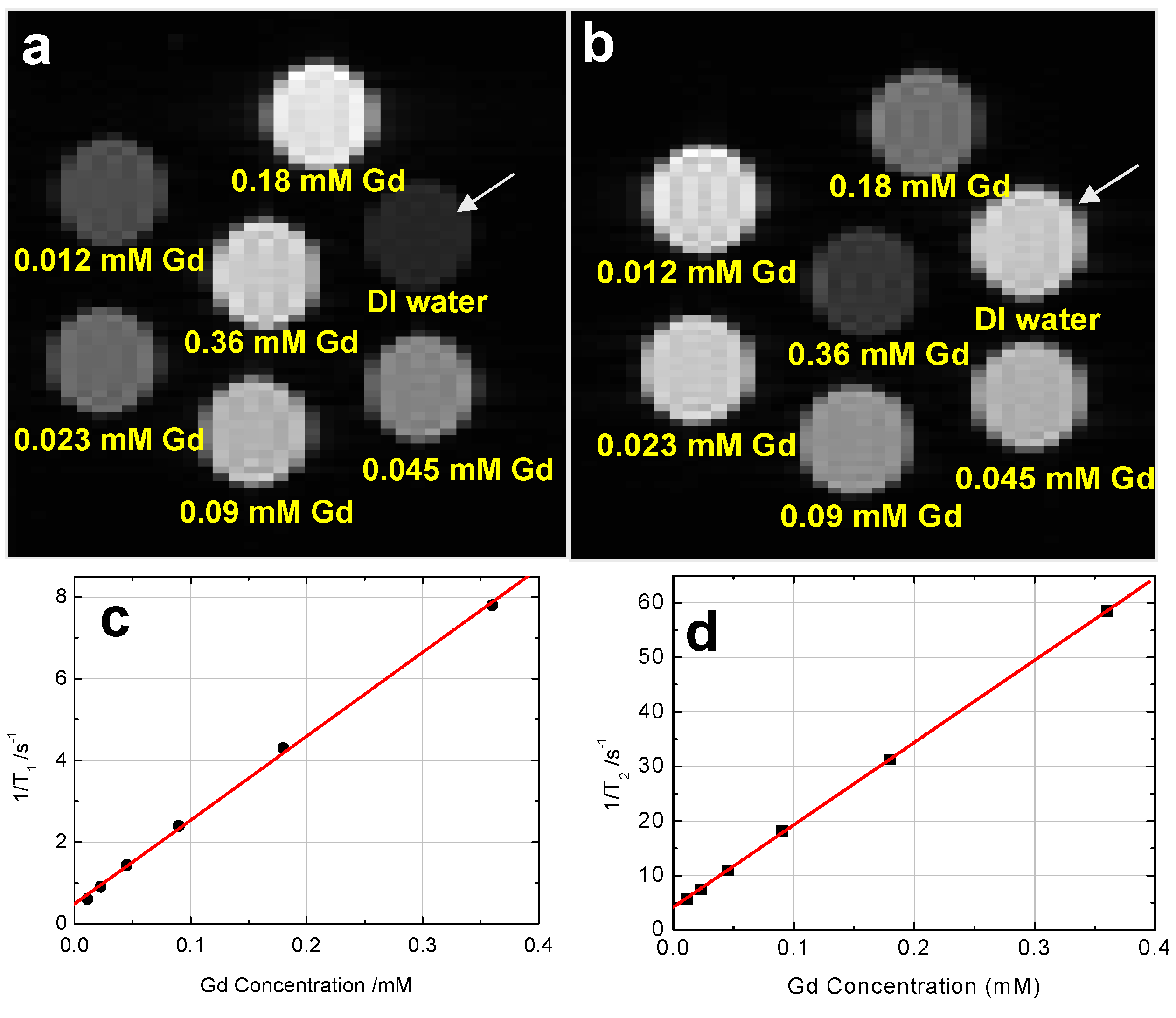
5.5.5. Magnetic Resonance-based Bioimaging
6. Perspective
Abbreviations
| Ac | Acetate |
| ADF | Annular dark field |
| ALE | Atomic layer epitaxy |
| Alq3 | Tris-(8-hydroxyquinoline) aluminum |
| AM | Air mass |
| AO | Atomic orbital |
| AOT | Aerosol OT |
| B | Blue |
| BCP | Bathocuproine |
| BSA | Bovine serum albumin |
| CBP | 4,4’,N,N’-diphenylcarbazole |
| CBP | 4,4’-N,N’-dicarbazolyl-biphenyl |
| CIE | International commission on illumination or Commission Internationale de l’Eclairage |
| CNPPP | Poly(2-(6-cyano-6’-methylheptyloxy)- 1,4- phenylene) |
| Co.sol | Coordinating solvent |
| CRI | Color rendering index |
| CT | Computer tomography |
| CTAB | Cetyl trimethyl-ammonium bromide |
| CV | Cyclic voltametry |
| CVD | Chemical vapor deposition |
| dBSA | Denatured bovine serum albumin |
| DDA | Dodecylamine |
| DEPE | 1,2-bis(diethyl-phosphino)-ethane |
| DI | deionized |
| DMPA | 2,2 –dimethoxy-2-phenylacetophenone |
| DOS | Density of states |
| DSC | Dye-sensitized solar cell |
| Ea | Electron affinity |
| EELS | Electron energy loss spectroscopy |
| EGDMA | Ethylene glycol domethacrylate |
| EL | Electroluminescence |
| EMA | Effective mass approximation |
| EML | Emitting layer |
| EQE | External quantum efficiency |
| ERFR | Epidermal growth factor receptor |
| ESR | Electron spin resonance |
| Et | Ethyl |
| ETL | Electron transport layer |
| F6BT | Poly[(9,9-dihexylfluorenyl-2,7-diyl)-co-(1,4-{benzo-[2,1’,3]thiadiazole})] |
| FF | Fill factor |
| FIB | Focused ion beam |
| FRET | Fluorescence resonance energy transfer |
| FvdM | Frank-van der Merwe mode |
| FWHM | Full width at half maximum |
| G | Green |
| HAADF | High-angle annular dark field |
| HDA | Hexadecylamine |
| HOMO | Highest occupied molecular orbital |
| HPA | Hexyl-phosphonic acid |
| HRTEM | High resolution transmission electron microscopic |
| HTL | Hole transport layer |
| Ip | Ionization potential |
| IR | Infrared |
| IR | Infrared |
| ISC | Short-circuit current |
| ITO | Indium tin oxide |
| LA | Lauric acid |
| LCAO | Linear combination of atomic orbital |
| LE | Luminous efficiency |
| LED | Light emitting devices |
| Lmax | Maximum luminance value |
| LUMO | Lowest unoccupied molecular orbital |
| MA | Methacrylic acid |
| MBE | Molecular beam epitaxy |
| MBE | Molecular beam epitaxy |
| MBP | Maltose binding protein |
| MDMO-PPV | Poly[2-methoxy-5-(3’,7’-dimethyloctyloxy)-1,4-phenylene vinylene] |
| Me | Methyl |
| MEHPPV | Poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene] |
| ML | Monolayer |
| MMA | Methylmethacrylate |
| MO | Molecular orbital |
| MPA | Marcaptopropionic acid |
| MR | Magnetic resonance |
| MRI | Magnetic resonance imaging |
| MWNT | Multi-wall carbon nanotubes |
| NIR | Near infrared |
| NMR | nuclear magnetic resonance |
| O | Orange |
| OA | Oleic acid |
| OD | Optical density |
| ODA | Octadecylamine |
| ODE | 1-octadecene |
| OLED | Organic light emitting diode |
| OMeTAD | 2,2’,7,7’-tetrakis(N,N-di-p-methoxyphenyl-amine)9,9’-spirobifluorene |
| P3HT | Poly-3(hexylthiophene) |
| PCBM | [6,6]-phenyl C61 butyric acid methyl ester |
| PE | Power efficiency |
| PEDOT | Poly~3,4-ethylenedioxythiophene |
| PEDOT:PSS | Poly(3,4-ethylene-dioxy-thiophene) : poly(styrene-sulfonate) |
| PEG | Polyethylene glycol |
| PFBD | Poly(9,9’-dioctylfluorene-co-N-(4-butylphenyl)diphenylamine |
| PFCB | Perfluorocyclobutane |
| PL | Photoluminescence |
| PLE | Photoluminescence excitation |
| PP | Sodium polyphosphate |
| PPV | Poly(phenylene vinylene) |
| PPV | Poly(phenylene vinylene) |
| PS | Polystyrene |
| PSS | Polystyrene sulfonate |
| PVA | Poly(vinylalcohol) |
| PVB | Poly(vinybutryral) |
| PVD | Physical vapor deposition |
| PVK | Poly(vinyl-carbazole) |
| PVK | Polyvinyl carbazole |
| Qdot | Quantum dot |
| QLED | Quantum dots-based light emitting diodes |
| QW | Quantum well |
| QWQD | Quantum well quantum dot |
| QY | Quantum yield or quantum yield |
| R | Red |
| RD | Rhodamine |
| RIE | Reactive ion etching |
| SA | Stearic acid |
| SDS | Sodium dodecyl sulphate |
| SERS | Surface enhanced Raman spectroscopy |
| SK | Stranski-Krastonow mode |
| SPR | Surface plasmon resonance |
| STEM | Scanning transmission electron microscopy |
| SWNT | Single-wall carbon nanotubes |
| TAZ | 3-(4-Biphenylyl)- 4-phenyl-5-tert-butylphenyl-1, 2, 4-triazole |
| TBP | Tri-n-butyl phosphine |
| t-Bu-PBD | 2-(4-biphenylyl)-5-(4-tert-butylphenyl)-1,3,4 oxadiazole |
| TDPA | Tetradecylphosphonic acid |
| Tech-TOPO | Technical grade tri-n-octyl-phophine oxide |
| TEM | Transmission electron microscopy |
| TFB | Poly[(9,9-dioctylfluorenyl-2,70diyl)-co-(4-4’-(N-(4-sec-butylphenyl)) diphenylamine)] |
| TMS | trimethyl-silyl |
| TOA | trioctyl amine |
| TOP | Tri-n-octyl-phosphine |
| TOPO | Tri-n-octyl phosphene oxide |
| TPBI | 1,3,5-tris(N-phenylbenzimidazole-2-yl)-benzene |
| TPD | N, N’-diphenyl-N, N’-bis(3-methylphenyl)-(1, 1’-biphenyl)-4, 4’-diamine |
| UV | Ultraviolet |
| VM | Volmer-Weber mode |
| VOC | Open circuit voltage |
| vol. | By volume |
| Vturn-on | Turn-on voltage of the device |
| W | White |
| XAFS | X-ray absorption fine structure |
| XPS | X-ray photoelectron spectroscopy |
| XRD | X-ray diffraction (XRD) |
| Y | Yellow |
| αNPD | 4,4-bis[N-(1-naphyl)-N-phenylamino]biphenyl |
Acknowledgement
References
- Henglein, A. Small-particle research−physicochemical properties of extremely small colloidal metal and semiconductor particles. Chem. Rev. 1989, 89, 1861–1873. [Google Scholar] [CrossRef]
- Trindade, T.; O'Brien, P.; Pickett, N.L. Nanocrystalline semiconductors: Synthesis, properties, and perspectives. Chem. Mater. 2001, 13, 3843–3858. [Google Scholar] [CrossRef]
- Kuchibhatla, S.; Karakoti, A.S.; Bera, D.; Seal, S. One dimensional nanostructured materials. Prog. Mater. Sci. 2007, 52, 699–913. [Google Scholar] [CrossRef]
- Bera, D.; Kuiry, S.C.; Seal, S. Synthesis of nanostructured materials using template-assisted electrodeposition. JOM 2004, 56, 49–53. [Google Scholar] [CrossRef]
- Alivisatos, A.P. Perspectives on the physical chemistry of semiconductor nanocrystals. J. Phys. Chem. 1996, 100, 13226–13239. [Google Scholar] [CrossRef]
- Bera, D.; Qian, L.; Holloway, P.H. Phosphor Quantum Dots; John WIley & Sons, Ltd: West Sussex, UK, 2008. [Google Scholar]
- Walter, P.; Welcomme, E.; Hallegot, P.; Zaluzec, N.J.; Deeb, C.; Castaing, J.; Veyssiere, P.; Breniaux, R.; Leveque, J.L.; Tsoucaris, G. Early use of PbS nanotechnology for an ancient hair dyeing formula. Nano Lett. 2006, 6, 2215–2219. [Google Scholar] [CrossRef] [PubMed]
- Rocksby, H.P. Color of selenium ruby glasses. J. Soc. Glass Technol. 1932, 16, 171. [Google Scholar]
- Ekimov, A.I.; Onushchenko, A.A. Quantum size effect in 3-dimensional microscopic semiconductor crystals. JETP Lett. 1981, 34, 345–349. [Google Scholar]
- Efros, A.L.; Efros, A.L. Interband absorption of light in a semiconductor sphere. Sov. Phys. Semicond. 1982, 16, 772–775. [Google Scholar]
- Rossetti, R.; Ellison, J.L.; Gibson, J.M.; Brus, L.E. Size effects in the excited electronic states of small colloidal CdS crystallites. J. Chem. Phys. 1984, 80, 4464–4469. [Google Scholar] [CrossRef]
- Spanhel, L.; Haase, M.; Weller, H.; Henglein, A. Photochemistry of colloidal semiconductors .20. Surface modification and stability of strong luminescing CdS particles. J. Am. Chem. Soc. 1987, 109, 5649–5655. [Google Scholar] [CrossRef]
- Kortan, A.R.; Hull, R.; Opila, R.L.; Bawendi, M.G.; Steigerwald, M.L.; Carroll, P.J.; Brus, L.E. Nucleation and growth of CdSe on ZnS quantum crystallite seeds, and vice versa, in inverse micelle media. J. Am. Chem. Soc. 1990, 112, 1327–1332. [Google Scholar] [CrossRef]
- Murray, C.B.; Norris, D.J.; Bawendi, M.G. Synthesis and characterization of nearly monodisperse CdE (E = S, Se, Te) semiconductor nanocrystallites. J. Am. Chem. Soc. 1993, 115, 8706–8715. [Google Scholar] [CrossRef]
- Grieser, F.; Furlong, D.N.; Scoberg, D.; Ichinose, I.; Kimizuka, N.; Kunitake, T. Size-quantized semiconductor cadmium chalcogenide particles in Langmuir-Blodgett-films. J. Chem. Soc. Faraday Trans. 1992, 88, 2207–2214. [Google Scholar] [CrossRef]
- Sundaram, M.; Chalmers, S.A.; Hopkins, P.F.; Gossard, A.C. New quantum sructures. Science 1991, 254, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, R.; Ploog, K. Frequency and density dependent radiative recombination processes in III-V semiconductor quantum-wells and superlattices. Adv. Phys. 1991, 40, 535–623. [Google Scholar] [CrossRef]
- Alivisatos, A.P. Semiconductor clusters, nanocrystals, and quantum dots. Science 1996, 271, 933–937. [Google Scholar] [CrossRef]
- Klimov, V.I.; Mikhailovsky, A.A.; Xu, S.; Malko, A.; Hollingsworth, J.A.; Leatherdale, C.A.; Eisler, H.J.; Bawendi, M.G. Optical gain and stimulated emission in nanocrystal quantum dots. Science 2000, 290, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Artemyev, M.V.; Woggon, U.; Wannemacher, R.; Jaschinski, H.; Langbein, W. Light trapped in a photonic dot: Microspheres act as a cavity for quantum dot emission. Nano Lett. 2001, 1, 309–314. [Google Scholar] [CrossRef]
- Graham-Rowe, D. From dots to devices. Nat. Photonics 2009, 3, 307–309. [Google Scholar] [CrossRef]
- Coe-Sullivan, S. Quantum dot developments. Nat. Photonics 2009, 3, 315–316. [Google Scholar] [CrossRef]
- Bera, D.; Qian, L.; Holloway, P.H. Semiconducting Quantum Dots for Bioimaging; Informa Heathcare: New York, NY, USA, 2009; Vol. 191. [Google Scholar]
- Yoffe, A.D. Low-dimensional systems–quantum-size effects and electronic-properties of semiconductor microcrystallites (zero-dimensional systems) and some quasi-2-dimensional systems. Adv. Phys. 1993, 42, 173–266. [Google Scholar] [CrossRef]
- Yoffe, A.D. Semiconductor quantum dots and related systems: Electronic, optical, luminescence and related properties of low dimensional systems. Adv. Phys. 2001, 50, 1–208. [Google Scholar] [CrossRef]
- Jang, E.; Jun, S.; Pu, L. High quality CdSeS nanocrystals synthesized by facile single injection process and their electroluminescence. Chem. Comm. 2003, 2964–2965. [Google Scholar] [CrossRef]
- Steckel, J.S.; Snee, P.; Coe-Sullivan, S.; Zimmer, J.R.; Halpert, J.E.; Anikeeva, P.; Kim, L.A.; Bulovic, V.; Bawendi, M.G. Color-saturated green-emitting QD-LEDs. Angew. Chem. Int. Ed. 2006, 45, 5796–5799. [Google Scholar] [CrossRef]
- Yang, H.S.; Holloway, P.H.; Ratna, B.B. Photoluminescent and electroluminescent properties of Mn-doped ZnS nanocrystals. J. Appl. Phys. 2003, 93, 586–592. [Google Scholar] [CrossRef]
- Tolbert, S.H.; Herhold, A.B.; Johnson, C.S.; Alivisatos, A.P. Comparison of quantum confinement effects on the electronic absorption-spectra of direct and indirect gap semiconductor nanocrystals. Phys. Rev. Lett. 1994, 73, 3266–3269. [Google Scholar] [CrossRef] [PubMed]
- Tolbert, S.H.; Alivisatos, A.P. The wurtzite to rock-salt structural transformation in CdSe nanocrystals under high-pressure. J. Chem. Phys. 1995, 102, 4642–4656. [Google Scholar] [CrossRef]
- Woggon, U. Optical Properties of Semiconductor Quantum Dots; Springer-Verlag: Berlin, Germay, 1997; Volume 136. [Google Scholar]
- Bryan, J.D.; Gamelin, D.R. Doped semiconductor nanocrystals: Synthesis, characterization, physical properties, and applications. Prog. Inorg. Chem. 2005, Volume 54, 47–126. [Google Scholar]
- Erwin, S.C.; Zu, L.J.; Haftel, M.I.; Efros, A.L.; Kennedy, T.A.; Norris, D.J. Doping semiconductor nanocrystals. Nature (London) 2005, 436, 91–94. [Google Scholar] [CrossRef]
- Norris, D.J.; Efros, A.L.; Erwin, S.C. Doped nanocrystals. Science 2008, 319, 1776–1779. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, N.; Goorskey, D.; Thessing, J.; Peng, X.G. An alternative of CdSe nanocrystal emitters: Pure and tunable impurity emissions in ZnSe nanocrystals. J. Am. Chem. Soc. 2005, 127, 17586–17587. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.H.; Zhao, J.H.; Bi, J.F.; Wang, W.Z.; Ji, Y.; Wu, X. G.; Xia, J.B. Cr-doped InAs self-organized diluted magnetic quantum dots with room-temperature ferromagnetism. Chin. Phys. Lett. 2007, 24, 2118–2121. [Google Scholar] [CrossRef]
- Bhargava, R.N. Doped nanocrystalline materials–Physics and applications. J. Lumin. 1996, 70, 85–94. [Google Scholar] [CrossRef]
- Stowell, C.A.; Wiacek, R.J.; Saunders, A.E.; Korgel, B.A. Synthesis and characterization of dilute magnetic semiconductor manganese-doped indium arsenide nanocrystals. Nano Lett. 2003, 3, 1441–1447. [Google Scholar] [CrossRef]
- Beaulac, R.; Schneider, L.; Archer, P.I.; Bacher, G.; Gamelin, D.R. Light-induced spontaneous magnetization in doped colloidal quantum dots. Science 2009, 325, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Radovanovic, P.V.; Gamelin, D.R. Electronic absorption spectroscopy of cobalt ions in diluted magnetic semiconductor quantum dots: Demonstration of an isocrystalline core/shell synthetic method. J. Am. Chem. Soc. 2001, 123, 12207–12214. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.L.; Zhang, X.Q.; Mao, H.B.; Wang, J.Q.; Zhu, Z.Q.; Jing, W.P. Photoluminescence and XPS investigations of Cu2+-doped ZnS quantum dots capped with polyvinylpyrrolidone. Phys. Status Solidi B 2009, 246, 2333–2336. [Google Scholar] [CrossRef]
- Kim, J.U.; Kim, Y.K.; Yang, H. Reverse micelle-derived Cu-doped Zn1-xCdxS quantum dots and their core/shell structure. J. Colloid. Interface Sci. 2010, 341, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Murugadoss, A.; Chattopadhyay, A. Tuning photoluminescence of ZnS nanoparticles by silver. Bull. Mater. Sci. 2008, 31, 533–539. [Google Scholar] [CrossRef]
- Fujii, M.; Yamaguchi, Y.; Takase, Y.; Ninomiya, K.; Hayashi, S. Photoluminescence from impurity codoped and compensated Si nanocrystals. Appl. Phys. Lett. 2005, 87(211919), 1–3. [Google Scholar]
- Orlinskii, S.B.; Schmidt, J.; Baranov, P.G.; Hofmann, D.M.; Donega, C.D.; Meijerink, A. Probing the wave function of shallow Li and Na donors in ZnO nanoparticles. Phys. Rev. Lett. 2004, 92(047603), 1–4. [Google Scholar]
- Yang, H.S.; Santra, S.; Holloway, P.H. Syntheses and applications of Mn-doped II-VI semiconductor nanocrystals. J. Nanosci. Nanotechnology 2005, 5, 1364–1375. [Google Scholar] [CrossRef]
- Yang, Y.A.; Chen, O.; Angerhofer, A.; Cao, Y.C. Radial-position-controlled doping in CdS/ZnS core/shell nanocrystals. J. Am. Chem. Soc. 2006, 128, 12428–12429. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.K.; Winkler, U.; Deshmukh, N.; Borse, P.H.; Fink, R.; Umbach, E. Investigations on chemically capped CdS, ZnS and ZnCdS nanoparticles. Appl. Surf. Sci. 2001, 169, 438–446. [Google Scholar] [CrossRef]
- Wang, W.Z.; Germanenko, I.; El-Shall, M.S. Room-temperature synthesis and characterization of nanocrystalline CdS, ZnS, and CdxZn1-xS. Chem. Mater. 2002, 14, 3028–3033. [Google Scholar] [CrossRef]
- Petrov, D.V.; Santos, B.S.; Pereira, G.A.L.; Donega, C.D. Size and band-gap dependences of the first hyperpolarizability of CdxZn1-xS nanocrystals. J. Phys. Chem. B 2002, 106, 5325–5334. [Google Scholar] [CrossRef]
- Bailey, R.E.; Nie, S.M. Alloyed semiconductor quantum dots: Tuning the optical properties without changing the particle size. J. Am. Chem. Soc. 2003, 125, 7100–7106. [Google Scholar] [CrossRef]
- Zhong, X.H.; Han, M.Y.; Dong, Z.L.; White, T.J.; Knoll, W. Composition-tunable ZnxCd1-xSe nanocrystals with high luminescence and stability. J. Am. Chem. Soc. 2003, 125, 8589–8594. [Google Scholar] [CrossRef] [PubMed]
- Gurusinghe, N.P.; Hewa-Kasakarage, N.N.; Zamkov, M. Composition-tunable properties of CdSxTe1-x alloy nanocrystals. J. Phys. Chem. C 2008, 112, 12795–12800. [Google Scholar] [CrossRef]
- Korgel, B.A.; Monbouquette, H.G. Controlled synthesis of mixed core and layered (Zn,Cd)S and (Hg,Cd)S nanocrystals within phosphatidylcholine vesicles. Langmuir 2000, 16, 3588–3594. [Google Scholar] [CrossRef]
- Harrison, M.T.; Kershaw, S.V.; Burt, M.G.; Eychmuller, A.; Weller, H.; Rogach, A.L. Wet chemical synthesis and spectroscopic study of CdHgTe nanocrystals with strong near-infrared luminescence. Mater. Sci. Eng. B 2000, 69, 355–360. [Google Scholar] [CrossRef]
- Lee, H.; Holloway, P.H.; Yang, H. Synthesis and characterization of colloidal ternary ZnCdSe semiconductor nanorods. J. Chem. Phys. 2006, 125, 2363181–2363189. [Google Scholar]
- Zhong, X.H.; Feng, Y.Y.; Knoll, W.; Han, M.Y. Alloyed ZnxCd1-xS nanocrystals with highly narrow luminescence spectral width. J. Am. Chem. Soc. 2003, 125, 13559–13563. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Gao, X.H.; Nie, S.M. Quantum dot nanocrystals for in vivo molecular and cellular imaging. Photochem. Photobiol. 2004, 80, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Bailey, R.E.; Smith, A.M.; Nie, S.M. Quantum dots in biology and medicine. Physica E 2004, 25, 1–12. [Google Scholar] [CrossRef]
- Wang, Y.; Herron, N. Nanometer-sized semiconductor clusters–materials synthesis, quantum size effects, and photophysical properties. J. Phys. Chem. 1991, 95, 525–532. [Google Scholar] [CrossRef]
- Bang, J.; Yang, H.; Holloway, P.H. Enhanced and stable green emission of ZnO nanoparticles by surface segregation of Mg. Nanotechnology 2006, 17, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Kucur, E.; Bucking, W.; Giernoth, R.; Nann, T. Determination of defect states in semiconductor nanocrystals by cyclic voltammetry. J. Phys. Chem. B 2005, 109, 20355–20360. [Google Scholar] [CrossRef] [PubMed]
- Colvin, V.L.; Goldstein, A.N.; Alivisatos, A.P. Semiconductor nanocrystals covalently bound to metal-surfaces with self-assembled monolayers. J. Am. Chem. Soc. 1992, 114, 5221–5230. [Google Scholar] [CrossRef]
- Dabbousi, B.O.; Murray, C.B.; Rubner, M.F.; Bawendi, M.G. Langmuir-Blodgett manipulation of size-selected CdSe nanocrystallites. Chem. Mater. 1994, 6, 216–219. [Google Scholar] [CrossRef]
- Murray, C.B.; Kagan, C.R.; Bawendi, M.G. Self-organization of CdSe nanocrystallites into 3-dimensional quantum-dot superlattices. Science 1995, 270, 1335–1338. [Google Scholar] [CrossRef]
- Wang, Q.; Kuo, Y.C.; Wang, Y.W.; Shin, G.; Ruengruglikit, C.; Huang, Q.R. Luminescent properties of water-soluble denatured bovine serum albumin-coated CdTe quantum dots. J. Phys. Chem. B 2006, 110, 16860–16866. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Yang, J.; Stewart, K.M.; Kelley, S.O. DNA-passivated CdS nanocrystals: Luminescence, bioimaging, and toxicity profiles. Langmuir 2007, 23, 12783–12787. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.B.; Lou, Y.B.; Samia, A.C.; Burda, C. Coherency strain effects on the optical response of core/shell heteronanostructures. Nano Lett. 2003, 3, 799–803. [Google Scholar] [CrossRef]
- Yang, H.S.; Santra, S.; Walter, G.A.; Holloway, P.H. Gd-III-functionalized fluorescent quantum dots as multimodal imaging probes. Adv. Mater. 2006, 18, 2890–2894. [Google Scholar] [CrossRef]
- Peng, X.G.; Schlamp, M.C.; Kadavanich, A.V.; Alivisatos, A.P. Epitaxial growth of highly luminescent CdSe/CdS core/shell nanocrystals with photostability and electronic accessibility. J. Am. Chem. Soc. 1997, 119, 7019–7029. [Google Scholar] [CrossRef]
- Reiss, P.; Bleuse, J.; Pron, A. Highly luminescent CdSe/ZnSe core/shell nanocrystals of low size dispersion. Nano Lett. 2002, 2, 781–784. [Google Scholar] [CrossRef]
- Qian, L.; Bera, D.; Tseng, T.K.; Holloway, P.H. High efficiency photoluminescence from silica-coated CdSe quantum dots. Appl. Phys. Lett. 2009, 94, 073112. [Google Scholar] [CrossRef]
- Geng, B.Y.; Du, Q.B.; Liu, X.W.; Ma, J.Z.; Wei, X.W.; Zhang, L.D. One-step synthesis and enhanced blue emission of carbon-encapsulated single-crystalline ZnSe nanoparticles. Appl. Phys. Lett. 2006, 89, 033115. [Google Scholar] [CrossRef]
- Ebenstein, Y.; Mokari, T.; Banin, U. Fluorescence quantum yield of CdSe/ZnS nanocrystals investigated by correlated atomic-force and single-particle fluorescence microscopy. Appl. Phys. Lett. 2002, 80, 4033–4035. [Google Scholar] [CrossRef]
- Nazzal, A.Y.; Wang, X.Y.; Qu, L.H.; Yu, W.; Wang, Y.J.; Peng, X.G.; Xiao, M. Environmental effects on photoluminescence of highly luminescent CdSe and CdSe/ZnS core/shell nanocrystals in polymer thin films. J. Phys. Chem. B 2004, 108, 5507–5515. [Google Scholar] [CrossRef]
- Yang, H.; Holloway, P.H.; Cunningham, G.; Schanze, K.S. CdS:Mn nanocrystals passivated by ZnS: Synthesis and luminescent properties. J. Chem. Phys. 2004, 121, 10233–10240. [Google Scholar] [CrossRef] [PubMed]
- Nirmal, M.; Dabbousi, B.O.; Bawendi, M.G.; Macklin, J.J.; Trautman, J.K.; Harris, T.D.; Brus, L.E. Fluorescence intermittency in single cadmium selenide nanocrystals. Nature (London) 1996, 383, 802–804. [Google Scholar] [CrossRef]
- Balet, L.P.; Ivanov, S.A.; Piryatinski, A.; Achermann, M.; Klimov, V.I. Inverted core/shell nanocrystals continuously tunable between type-I and type-II localization regimes. Nano Lett. 2004, 4, 1485–1488. [Google Scholar] [CrossRef]
- Kim, S.; Fisher, B.; Eisler, H.J.; Bawendi, M. Type-II quantum dots: CdTe/CdSe (core/shell) and CdSe/ZinTe (core/shell) heterostructures. J. Am. Chem. Soc. 2003, 125, 11466–11467. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.S.; Holloway, P.H.; Santra, S. Water-soluble silica-overcoated CdS:Mn/ZnS semiconductor quantum dots. J. Chem. Phys. 2004, 121, 7421–7426. [Google Scholar] [CrossRef] [PubMed]
- Boldt, K.; Bruns, O.T.; Gaponik, N.; Eychmuller, A. Comparative examination of the stability of semiconductor quantum dots in various biochemical buffers. J. Phys. Chem. B 2006, 110, 1959–1963. [Google Scholar] [CrossRef] [PubMed]
- Wolcott, A.; Gerion, D.; Visconte, M.; Sun, J.; Schwartzberg, A.; Chen, S.W.; Zhang, J.Z. Silica-coated CdTe quantum dots functionalized with thiols for bioconjugation to IgG proteins. J. Phys. Chem. B 2006, 110, 5779–5789. [Google Scholar] [CrossRef] [PubMed]
- Santra, S.; Bagwe, R.P.; Dutta, D.; Stanley, J.T.; Walter, G.A.; Tan, W.; Moudgil, B.M.; Mericle, R.A. Synthesis and characterization of fluorescent, radio-opaque, and paramagnetic silica nanoparticles for multimodal bioimaging applications. Adv. Mater. 2005, 17, 2165–2169. [Google Scholar] [CrossRef]
- Santra, S.; Dutta, D.; Moudgil, B.M. Functional dye-doped silica nanoparticles for bioimaging, diagnostics and therapeutics. Food Bioprod. Process. 2005, 83, 136–140. [Google Scholar] [CrossRef]
- Empedocles, S.A.; Bawendi, M.G. Quantum-confined stark effect in single CdSe nanocrystallite quantum dots. Science 1997, 278, 2114–2117. [Google Scholar] [CrossRef] [PubMed]
- Jeang, E.H.; Lee, J.H.; Je, K.C.; Yim, S.Y.; Park, S.H.; Choi, Y.S.; Choi, J.G.; Treguer, M.; Cardinal, T. Fabrication and optical characteristics of CdS/Ag metal-semiconductor composite quantum dots. Bull. Korean Chem. Soc. 2004, 25, 934–936. [Google Scholar] [CrossRef]
- Je, K.C.; Ju, H.; Treguer, M.; Cardinal, T.; Park, S.H. Local field-induced optical properties of Ag-coated CdS quantum dots. Opt. Express 2006, 14, 7994–8000. [Google Scholar] [CrossRef] [PubMed]
- Talapin, D.V.; Mekis, I.; Gotzinger, S.; Kornowski, A.; Benson, O.; Weller, H. CdSe/CdS/ZnS and CdSe/ZnSe/ZnS core-shell-shell nanocrystals. J. Phys. Chem. B 2004, 108, 18826–18831. [Google Scholar] [CrossRef]
- Xie, R.G.; Kolb, U.; Li, J.X.; Basche, T.; Mews, A. Synthesis and characterization of highly luminescent CdSe-Core CdS/Zn0.5Cd0.5S/ZnS multishell nanocrystals. J. Am. Chem. Soc. 2005, 127, 7480–7488. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.F.; Xiao, M. Lasing action in colloidal CdS/CdSe/CdS quantum wells. Appl. Phys. Lett. 2005, 87, 173117 1. [Google Scholar]
- Qian, L.; Bera, D.; Holloway, P. Photoluminescence from ZnS/CdS:Mn/ZnS quantum well quantum dots. Appl. Phys. Lett. 2008, 92, 093103. [Google Scholar] [CrossRef]
- Santra, S.; Xu, J.S.; Wang, K.M.; Tan, W.H. Luminescent nanoparticle probes for bioimaging. J. Nanosci. Nanotech. 2004, 4, 590–599. [Google Scholar] [CrossRef]
- Santra, S.; Yang, H.; Stanley, J.T.; Holloway, P.H.; Moudgil, B.M.; Walter, G.; Mericle, R.A. Rapid and effective labeling of brain tissue using TAT-conjugated CdS:Mn/ZnS quantum dots. Chem. Comm. 2005, 3144–3146. [Google Scholar] [CrossRef]
- Santra, S.; Yang, H.S.; Holloway, P.H.; Stanley, J.T.; Mericle, R.A. Synthesis of water-dispersible fluorescent, radio-opaque, and paramagnetic CdS:Mn/ZnS quantum dots: A multifunctional probe for bioimaging. J. Am. Chem. Soc. 2005, 127, 1656–1657. [Google Scholar] [CrossRef] [PubMed]
- Holloway, P.H.; Yang, H.; Lee, H.; Seo, S.; Santra, S.; Qian, L.; Bera, D. Nanophosphor: PL, EL, and Biological Markers; Proc. Int. Display Workshops, IDW'06. Otsu, Japan, 6–8 December 2006.
- Yu, Z.H.; Guo, L.; Du, H.; Krauss, T.; Silcox, J. Shell distribution on colloidal CdSe/ZnS quantum dots. Nano Lett. 2005, 5, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Holloway, P.H. Efficient and Photostable ZnS-passivated CdS:Mn luminescent nanocrystals. Adv. Func. Mater. 2004, 14, 152–156. [Google Scholar] [CrossRef]
- Rosenthal, S.J.; McBride, J.; Pennycook, S.J.; Feldman, L.C. Synthesis, surface studies, composition and structural characterization of CdSe, core/shell and biologically active nanocrystals. Surf. Sci. Rep. 2007, 62, 111–157. [Google Scholar] [CrossRef] [PubMed]
- McBride, J.R.; Kippeny, T.C.; Pennycook, S.J.; Rosenthal, S.J. Aberration-corrected Z-contrast scanning transmission electron microscopy of CdSe nanocrystals. Nano Lett. 2004, 4, 1279–1283. [Google Scholar] [CrossRef]
- McBride, J.; Treadway, J.; Feldman, L.C.; Pennycook, S.J.; Rosenthal, S.J. Structural basis for near unity quantum yield core/shell nanostructures. Nano Lett. 2006, 6, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- Katari, J.E.B.; Colvin, V.L.; Alivisatos, A.P. X-Ray photoelectron-spectroscopy of CdSe nanocrystals with applications to studies of the nanocrystal surface. J. Phys. Chem. 1994, 98, 4109–4117. [Google Scholar] [CrossRef]
- Bera, D.; Qian, L.; Sabui, S.; Santra, S.; Holloway, P.H. Photoluminescence of ZnO quantum dots produced by a sol-gel process. Opt. Mater. 2008, 30, 1233–1239. [Google Scholar] [CrossRef]
- Hines, M.A.; Guyot-Sionnest, P. Synthesis and characterization of strongly luminescing ZnS-capped CdSe nanocrystals. J. Phys. Chem. 1996, 100, 468–471. [Google Scholar] [CrossRef]
- Dabbousi, B.O.; RodriguezViejo, J.; Mikulec, F.V.; Heine, J.R.; Mattoussi, H.; Ober, R.; Jensen, K.F.; Bawendi, M.G. (CdSe)ZnS core-shell quantum dots: Synthesis and characterization of a size series of highly luminescent nanocrystallites. J. Phys. Chem. B 1997, 101, 9463–9475. [Google Scholar] [CrossRef]
- Klimov, V.I. Mechanisms for photogeneration and recombination of multiexcitons in semiconductor nanocrystals: Implications for lasing and solar energy conversion. J. Phys. Chem. B 2006, 110, 16827–16845. [Google Scholar] [CrossRef] [PubMed]
- Brus, L.E. A simple-model for the ionization-potential, electron-affinity, and aqueous redox potentials of small semiconductor crystallites. J. Chem. Phys. 1983, 79, 5566–5571. [Google Scholar] [CrossRef]
- Steigerwald, M.L.; Brus, L.E. Semiconductor crystallites–a class of large molecules. Acc. Chem. Res. 1990, 23, 183–188. [Google Scholar] [CrossRef]
- Ogawa, S.; Hu, K.; Fan, F.R.F.; Bard, A.J. Photoelectrochemistry of films of quantum size lead sulfide particles incorporated in self-assembled monolayers on gold. J. Phys. Chem. B 1997, 101, 5707–5711. [Google Scholar] [CrossRef]
- Haram, S.K.; Quinn, B.M.; Bard, A.J. Electrochemistry of CdS nanoparticles: A correlation between optical and electrochemical band gaps. J. Am. Chem. Soc. 2001, 123, 8860–8861. [Google Scholar] [CrossRef] [PubMed]
- Kucur, E.; Riegler, J.; Urban, G.A.; Nann, T. Determination of quantum confinement in CdSe nanocrystals by cyclic voltammetry. J. Chem. Phys. 2003, 119, 2333–2337. [Google Scholar] [CrossRef]
- Bae, Y.; Myung, N.; Bard, A.J. Electrochemistry and electrogenerated chemiluminescence of CdTe nanoparticles. Nano Lett. 2004, 4, 1153–1161. [Google Scholar] [CrossRef]
- Poznyak, S.K.; Osipovich, N.P.; Shavel, A.; Talapin, D.V.; Gao, M.Y.; Eychmuller, A.; Gaponik, N. Size-dependent electrochemical behavior of thiol-capped CdTe nanocrystals in aqueous solution. J. Phys. Chem. B 2005, 109, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
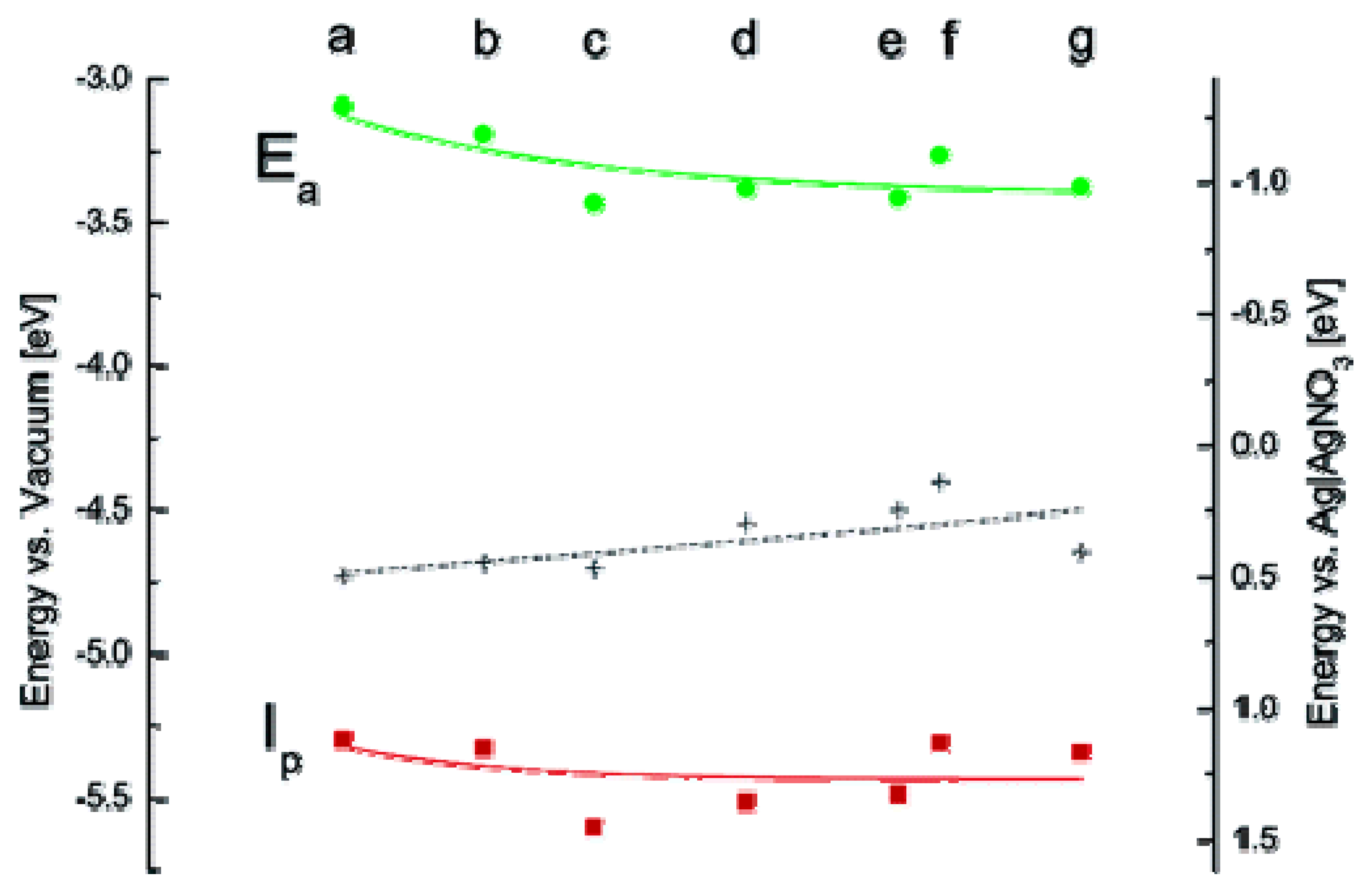
- Inamdar, S.N.; Ingole, P.P.; Haram, S.K. Determination of band structure parameters and the quasi-particle gap of CdSe quantum dots by cyclic voltammetry. Chem. Phys. Chem. 2008, 9, 2574–2579. [Google Scholar] [PubMed]
- Reiss, P.; Quemard, G.; Carayon, S.; Bleuse, J.; Chandezon, F.; Pron, A. Luminescent ZnSe nanocrystals of high color purity. Mater. Chem. Phys. 2004, 84, 10–13. [Google Scholar] [CrossRef]
- Karanikolos, G.N.; Alexandridis, P.; Itskos, G.; Petrou, A.; Mountziaris, T.J. Synthesis and size control of luminescent ZnSe nanocrystals by a microemulsion-gas contacting technique. Langmuir 2004, 20, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Bawendi, M.G.; Wilson, W.L.; Rothberg, L.; Carroll, P.J.; Jedju, T.M.; Steigerwald, M.L.; Brus, L.E. Electronic-structure and photoexcited-carrier dynamics in nanometer-size CdSe clusters. Phys. Rev. Lett. 1990, 65, 1623–1626. [Google Scholar] [CrossRef] [PubMed]
- Efros, A.L.; Rosen, M.; Kuno, M.; Nirmal, M.; Norris, D.J.; Bawendi, M. Band-edge exciton in quantum dots of semiconductors with a degenerate valence band: Dark and bright exciton states. Phys. Rev. B 1996, 54, 4843–4856. [Google Scholar] [CrossRef]
- Efros, A.L.; Rosen, M. The electronic structure of semiconductor nanocrystals. Annu. Rev. Mater. Sci. 2000, 30, 475–521. [Google Scholar] [CrossRef]
- Efros, A.L.; Rosen, M. Random telegraph signal in the photoluminescence intensity of a single quantum dot. Phys. Rev. Lett. 1997, 78, 1110–1113. [Google Scholar] [CrossRef]
- Stefani, F.D.; Knoll, W.; Kreiter, M.; Zhong, X.; Han, M.Y. Quantification of photoinduced and spontaneous quantum-dot luminescence blinking. Phys. Rev. B 2005, 72, 125304 1–7. [Google Scholar]
- Kuno, M.; Fromm, D.P.; Hamann, H.F.; Gallagher, A.; Nesbitt, D.J. Nonexponential "blinking" kinetics of single CdSe quantum dots: A universal power law behavior. J. Chem. Phys. 2000, 112, 3117–3120. [Google Scholar] [CrossRef]
- van Sark, W.; Frederix, P.; van den Heuvel, D.J.; Bol, A.A.; van Lingen, J.N.J.; Donega, C.D.; Gerritsen, H.C.; Meijerink, A. Time-resolved fluorescence spectroscopy study on the photophysical behavior of quantum dots. J. Fluoresc. 2002, 12, 69–76. [Google Scholar] [CrossRef]
- Stefani, F.D.; Zhong, X.H.; Knoll, W.; Han, M.Y.; Kreiter, M. Memory in quantum-dot photoluminescence blinking. New J. Phys. 2005, 7, 197. [Google Scholar] [CrossRef]
- Issac, A.; von Borczyskowski, C.; Cichos, F. Correlation between photoluminescence intermittency of CdSe quantum dots and self-trapped states in dielectric media. Phys. Rev. B 2005, 71, 161302 1. [Google Scholar]
- Gfroerer, T.H. Photoluminescence in analysis of surface and interfaces; John Wiley & Sons Ltd.: Chichster, UK, 2000; pp. 9209–9231. [Google Scholar]
- Djurisic, A.B.; Leung, Y.H.; Choy, W.C.H.; Cheah, K.W.; Chan, W.K. Visible photoluminescence in ZnO tetrapod and multipod structures. Appl. Phys. Lett. 2004, 84, 2635–2637. [Google Scholar] [CrossRef]
- Norberg, N.S.; Gamelin, D.R. Influence of surface modification on the luminescence of colloidal ZnO nanocrystals. J. Phys. Chem. B 2005, 109, 20810–20816. [Google Scholar] [CrossRef] [PubMed]
- Djurisic, A.B.; Leung, Y.H. Optical properties of ZnO nanostructures. Small 2006, 2, 944–961. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.M.; Lin, K.F.; Hsu, H.C.; Hsieh, W.F. Size dependence of photoluminescence and resonant Raman scattering from ZnO quantum dots. Appl. Phys. Lett. 2006, 88, 261909. [Google Scholar] [CrossRef]
- Lu, J.G.; Ye, Z.Z.; Zhang, Y.Z.; Liang, Q.L.; Fujita, S.; Wang, Z.L. Self-assembled ZnO quantum dots with tunable optical properties. Appl. Phys. Lett. 2006, 89, 023122. [Google Scholar] [CrossRef]
- Xu, P.S.; Sun, Y.M.; Shi, C.S.; Xu, F.Q.; Pan, H.B. The electronic structure and spectral properties of ZnO and its defects. Nucl. Instrum. Methods Phys. Res. Sect. B 2003, 199, 286–290. [Google Scholar] [CrossRef]
- Aleksandra, B.D.; Leung, Y. H. Optical properties of ZnO nanostructures. Small 2006, 2, 944–961. [Google Scholar] [CrossRef] [PubMed]
- Spanhel, L.; Anderson, M.A. Semiconductor clusters in the sol-gel process−quantized aggregation, gelation, and crystal-growth in concentrated ZnO colloids. J. Am. Chem. Soc. 1991, 113, 2826–2833. [Google Scholar] [CrossRef]
- Lee, J. D. Concise Inorganic Chemistry; Blackwell Science: Noida, India, 2005; p. 1032. [Google Scholar]
- Xiu, F.X.; Yang, Z.; Mandalapu, L.J.; Liu, J.L.; Beyermann, W.P. p-Type ZnO films with solid-source phosphorus doping by molecular-beam epitaxy. Appl. Phys. Lett. 2006, 88, 052106. [Google Scholar] [CrossRef]
- Viswanatha, R.; Chakraborty, S.; Basu, S.; Sarma, D.D. Blue-emitting copper-doped zinc oxide nanocrystals. J. Phys. Chem. B 2006, 110, 22310–22312. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.S.; Thomas, P.J.; O'Brien, P. Optical properties of ZnO nanocrystals doped with Cd, Mg, Mn, and Fe ions. J. Phys. Chem. B 2006, 110, 21412–21415. [Google Scholar] [CrossRef] [PubMed]
- Hirate, T.; Sasaki, S.; Li, W.C.; Miyashita, H.; Kimpara, T.; Satoh, T. Effects of laser-ablated impurity on aligned ZnO nanorods grown by chemical vapor deposition. Thin Solid Films 2005, 487, 35–39. [Google Scholar] [CrossRef]
- Liu, S.M.; Liu, F.Q.; Wang, Z.G. Relaxation of carriers in terbium-doped ZnO nanoparticles. Chem. Phys. Lett. 2001, 343, 489–492. [Google Scholar] [CrossRef]
- Cheng, B.C.; Xiao, Y.H.; Wu, G.S.; Zhang, L.D. Controlled growth and properties of one-dimensional ZnO nanostructures with Ce as activator/dopant. Adv. Func. Mater. 2004, 14, 913–919. [Google Scholar] [CrossRef]
- Ishizumi, A.; Kanemitsu, Y. Structural and luminescence properties of Eu-doped ZnO nanorods fabricated by a microemulsion method. Appl. Phys. Lett. 2005, 86, 253106. [Google Scholar] [CrossRef]
- Wu, G.S.; Zhuang, Y.L.; Lin, Z.Q.; Yuan, X.Y.; Xie, T.; Zhang, L.D. Synthesis and photoluminescence of dy-doped ZnO nanowires. Physica E 2006, 31, 5–8. [Google Scholar] [CrossRef]
- Zhang, X.T.; Liu, Y.C.; Zhang, J.Y.; Lu, Y.M.; Shen, D.Z.; Fan, X.W.; Kong, X.G. Structure and photoluminescence of Mn-passivated nanocrystalline ZnO thin films. J. Cryst. Growth 2003, 254, 80–85. [Google Scholar] [CrossRef]
- Xu, C.X.; Sun, X.W.; Dong, Z.L.; Tan, S.T.; Cui, Y.P.; Wang, B.P. Manganese-doped zinc oxide tetratubes and their photoluminescent properties. J. Appl. Phys. 2005, 98, 113513. [Google Scholar] [CrossRef]
- Yang, L.W.; Wu, X.L.; Huang, G.S.; Qiu, T.; Yang, Y.M. In situ synthesis of Mn-doped ZnO multileg nanostructures and Mn-related Raman vibration. J. Appl. Phys. 2005, 97, 014308. [Google Scholar] [CrossRef]
- Ronning, C.; Gao, P.X.; Ding, Y.; Wang, Z.L.; Schwen, D. Manganese-doped ZnO nanobelts for spintronics. Appl. Phys. Lett. 2004, 84, 783–785. [Google Scholar] [CrossRef]
- Shen, G.Z.; Cho, J.H.; Yoo, J.K.; Yi, G.C.; Lee, C.J. Synthesis and optical properties of S-doped ZnO nanostructures: Nanonails and nanowires. J. Phys. Chem. B 2005, 109, 5491–5496. [Google Scholar] [CrossRef] [PubMed]
- Foreman, J.V.; Li, J.Y.; Peng, H.Y.; Choi, S.J.; Everitt, H.O.; Liu, J. Time-resolved investigation of bright visible wavelength luminescence from sulfur-doped ZnO nanowires and micropowders. Nano Lett. 2006, 6, 1126–1130. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Lu, M.K.; Xu, D.; Yuan, D.L.; Zhou, G.J. ZnS nanocrystals co-activated by transition metals and rare-earth metals–a new class of luminescent materials. J. Lumin. 2001, 93, 101–105. [Google Scholar] [CrossRef]
- Qu, S.C.; Zhou, W.H.; Liu, F.Q.; Chen, N.F.; Wang, Z.G.; Pan, H.Y.; Yu, D.P. Photoluminescence properties of Eu3+-doped ZnS nanocrystals prepared in a water/methanol solution. Appl. Phys. Lett. 2002, 80, 3605–3607. [Google Scholar] [CrossRef]
- Suyver, J.F.; Wuister, S.F.; Kelly, J.J.; Meijerink, A. Synthesis and photoluminescence of nanocrystalline ZnS : Mn2+. Nano Lett. 2001, 1, 429–433. [Google Scholar] [CrossRef]
- Bhargava, R.N.; Gallagher, D.; Hong, X.; Nurmikko, A. Optical-Properties of Manganese-Doped Nanocrystals of Zns. Phys. Rev. Lett. 1994, 72, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Bol, A.A.; Meijerink, A. Long-lived Mn2+ emission in nanocrystalline ZnS : Mn2+. Phys. Rev. B 1998, 58, R15997–R16000. [Google Scholar] [CrossRef]
- Su, F.H.; Fang, Z.L.; Ma, B.S.; Ding, K.; Li, G.H.; Chen, W. Pressure dependence of Mn2+ luminescence in differently sized ZnS : Mn nanoparticles. J. Phys. Chem. B 2003, 107, 6991–6996. [Google Scholar] [CrossRef]
- Khosravi, A.A.; Kundu, M.; Kuruvilla, B.A.; Shekhawat, G.S.; Gupta, R.P.; Sharma, A.K.; Vyas, P.D.; Kulkarni, S.K. Manganese-doped zinc-sulfide nanoparticles by aqueous method. Appl. Phys. Lett. 1995, 67, 2506–2508. [Google Scholar] [CrossRef]
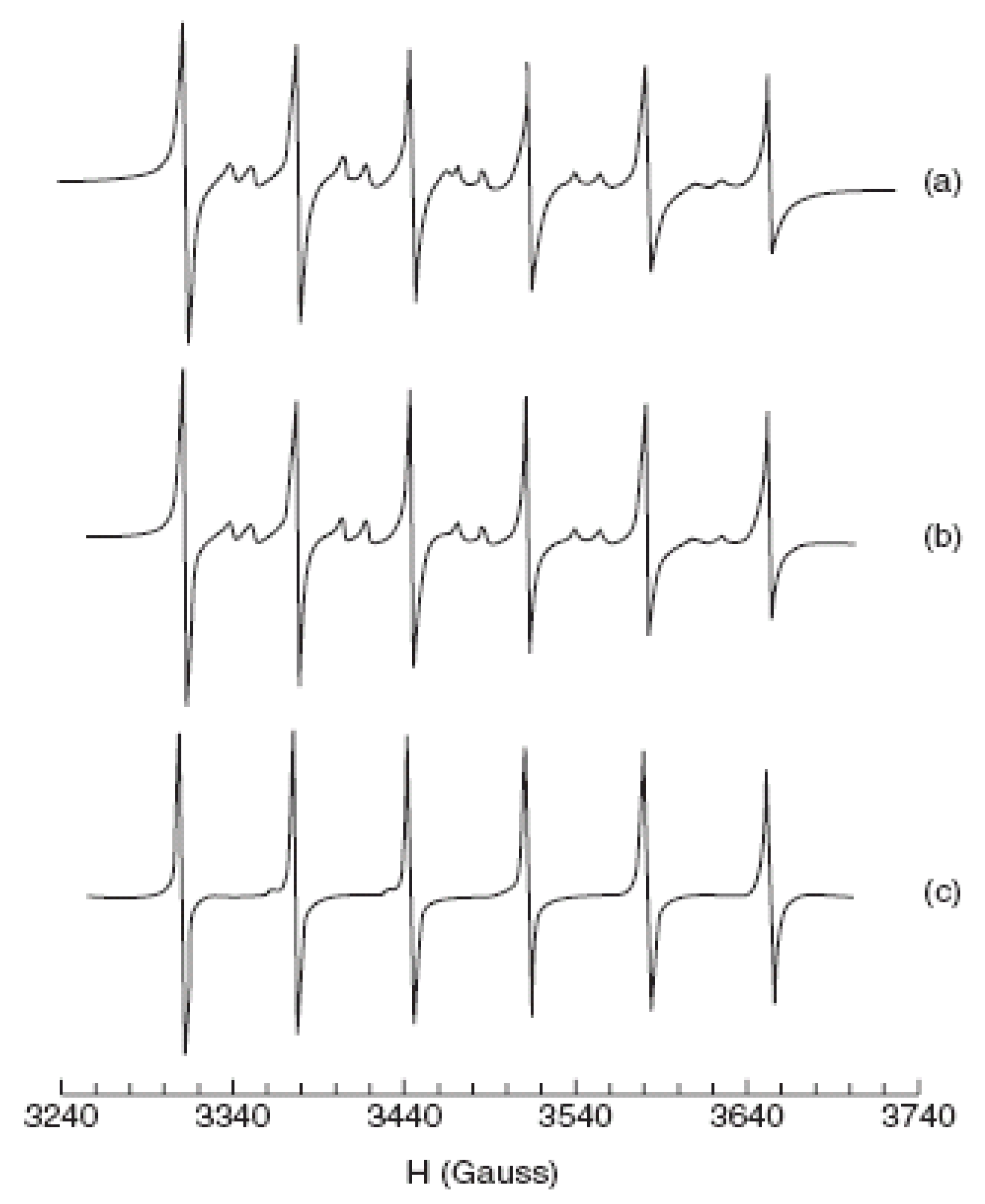
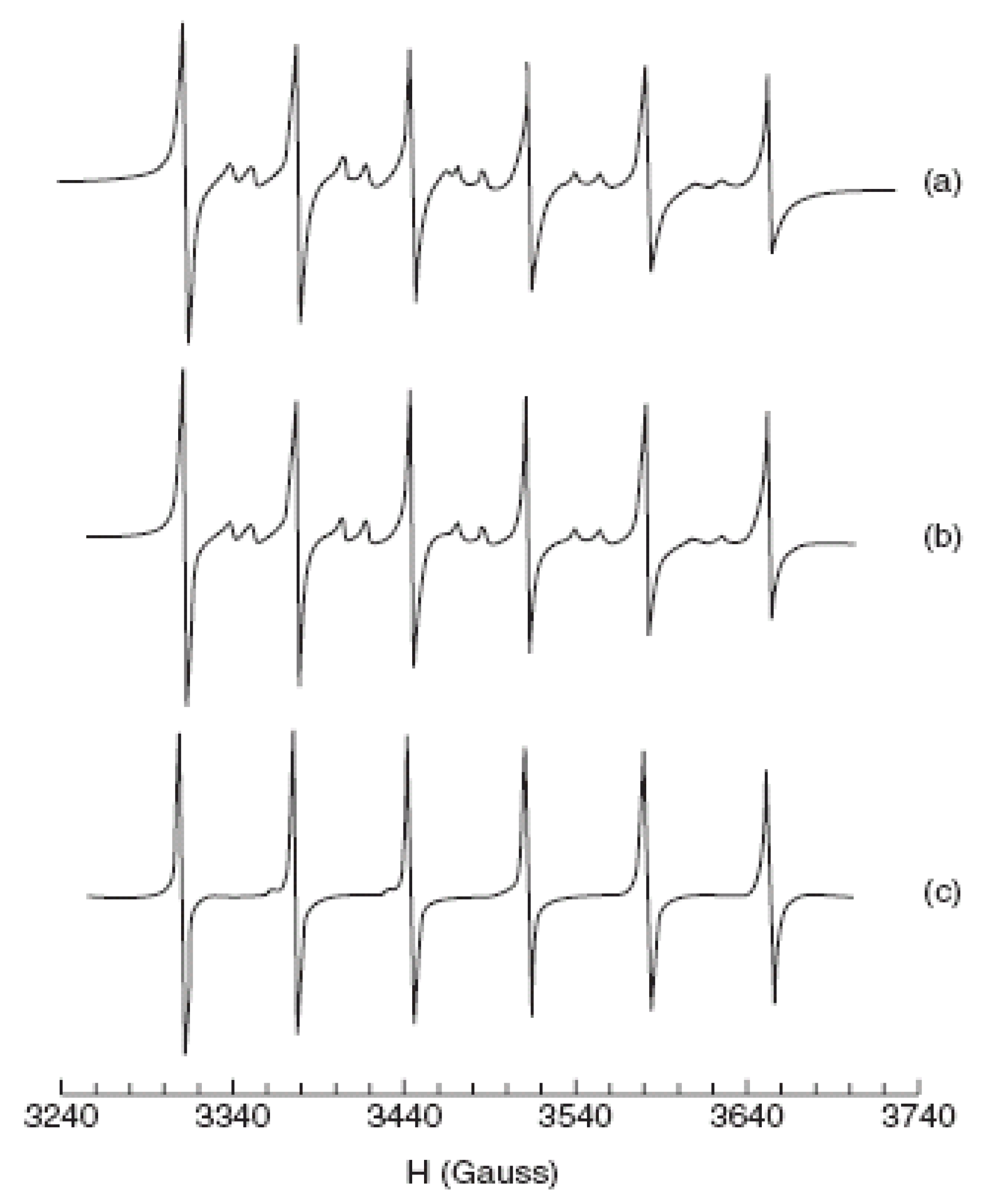
- Beermann, P.A.G.; McGarvey, B.R.; Muralidharan, S.; Sung, R.C.W. EPR spectra of Mn2+-doped ZnS quantum dots. Chem. Mater. 2004, 16, 915–918. [Google Scholar] [CrossRef]
- Eaton, D.F. Reference materials for fluorescence measurement. Pure Appl. Chem. 1988, 60, 1107–1114. [Google Scholar] [CrossRef]
- Qian, L.; Bera, D.; Holloway, P.H. Temporal evolution of white light emission from CdSe quantum dots. Nanotechnology 2008, 19, 285702. [Google Scholar] [CrossRef] [PubMed]
- Bera, D.; Qian, L.; Holloway, P.H. Time-evolution of photoluminescence properties of ZnO/MgO core/shell quantum dots. J. Phys. D 2008, 41, 182002. [Google Scholar] [CrossRef]
- Hines, M.A.; Guyot-Sionnest, P. Bright UV-blue luminescent colloidal ZnSe nanocrystals. J. Phys. Chem. B 1998, 102, 3655–3657. [Google Scholar] [CrossRef]
- Lee, J.; Sundar, V.C.; Heine, J.R.; Bawendi, M.G.; Jensen, K.F. Full color emission from II-VI semiconductor quantum dot-polymer composites. Adv. Mater. 2000, 12, 1102. [Google Scholar] [CrossRef]
- Talapin, D.V.; Rogach, A.L.; Kornowski, A.; Haase, M.; Weller, H. Highly luminescent monodisperse CdSe and CdSe/ZnS nanocrystals synthesized in a hexadecylamine-trioctylphosphine oxide-trioctylphospine mixture. Nano Lett. 2001, 1, 207–211. [Google Scholar] [CrossRef]
- Norris, D.J.; Yao, N.; Charnock, F.T.; Kennedy, T.A. High-quality manganese-doped ZnSe nanocrystals. Nano Lett. 2001, 1, 3–7. [Google Scholar] [CrossRef]
- Qu, L.H.; Peng, X.G. Control of photoluminescence properties of CdSe nanocrystals in growth. J. Am. Chem. Soc. 2002, 124, 2049–2055. [Google Scholar] [CrossRef] [PubMed]
- Steckel, J.S.; Coe-Sullivan, S.; Bulovic, V.; Bawendi, M.G. 1.3 mu m to 1.55 mu m tunable electroluminescence from PbSe quantum dots embedded within an organic device. Adv. Mater. 2003, 15, 1862–1866. [Google Scholar] [CrossRef]
- Donega, C.D.; Hickey, S.G.; Wuister, S.F.; Vanmaekelbergh, D.; Meijerink, A. Single-step synthesis to control the photoluminescence quantum yield and size dispersion of CdSe nanocrystals. J. Phys. Chem. B 2003, 107, 489–496. [Google Scholar] [CrossRef]
- Anikeeva, P.O.; Halpert, J.E.; Bawendi, M.G.; Bulovic, V. Electroluminescence from a mixed red-green-blue colloidal quantum dot monolayer. Nano Lett. 2007, 7, 2196–2200. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Jun, S.; Jang, E.; Baik, H.; Kim, H.; Cho, J. Preparation of highly luminescent nanocrystals and their application to light-emitting diodes. Adv. Mater. 2007, 19, 1927–1932. [Google Scholar] [CrossRef]
- van Dijken, A.; Makkinje, J.; Meijerink, A. The influence of particle size on the luminescence quantum efficiency of nanocrystalline ZnO particles. J. Lumin. 2001, 92, 323–328. [Google Scholar] [CrossRef]
- Chen, H.S.; Lo, B.; Hwang, J.Y.; Chang, G.Y.; Chen, C.M.; Tasi, S.J.; Wang, S.J.J. Colloidal ZnSe, ZnSe/ZnS, and ZnSe/ZnSeS quantum dots synthesized from ZnO. J. Phys. Chem. B 2004, 108, 17119–17123. [Google Scholar] [CrossRef]
- Bol, A.A.; Meijerink, A. Luminescence quantum efficiency of nanocrystalline ZnS : Mn2+. 1. Surface passivation and Mn2+ concentration. J. Phys. Chem. B 2001, 105, 10197–10202. [Google Scholar] [CrossRef]
- Hattori, Y.; Isobe, T.; Takahashi, H.; Itoh, S. Luminescent properties of ZnS:Mn2+ nanocrystals/SiO2 hybrid phosphor synthesized by in situ surface modification co-precipitation. J. Lumin. 2005, 113, 69–78. [Google Scholar] [CrossRef]
- Becker, W.G.; Bard, A.J. Photo-luminescence and photoinduced oxygen-adsorption of colloidal zinc-sulfide dispersions. J. Phys. Chem. 1983, 87, 4888–4893. [Google Scholar] [CrossRef]
- Baral, S.; Fojtik, A.; Weller, H.; Henglein, A. Photochemistry and radiation-chemistry of colloidal semiconductors .12. Intermediates of the oxidation of extremely small particles of CdS, ZnS, and Cd3P2 and size quantization effects (a pulse-radiolysis study). J. Am. Chem. Soc. 1986, 108, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Dunstan, D.E.; Hagfeldt, A.; Almgren, M.; Siegbahn, H.O.G.; Mukhtar, E. Importance of surface-reactions in the photochemistry of ZnS colloids. J. Phys. Chem. 1990, 94, 6797–6804. [Google Scholar] [CrossRef]
- Bol, A.A.; Meijerink, A. Luminescence quantum efficiency of nanocrystalline ZnS:Mn2+. 2. Enhancement by UV irradiation. J. Phys. Chem. B 2001, 105, 10203–10209. [Google Scholar] [CrossRef]
- Cao, L.X.; Zhang, J.H.; Ren, S.L.; Huang, S.H. Luminescence enhancement of core-shell ZnS : Mn/ZnS nanoparticles. Appl. Phys. Lett. 2002, 80, 4300–4302. [Google Scholar] [CrossRef]
- Jin, C.M.; Yu, J.Q.; Sun, L.D.; Dou, K.; Hou, S.G.; Zhao, J.L.; Chen, Y.M.; Huang, S.H. Luminescence of Mn2+ doped ZnS nanocrystallites. J. Lumin. 1995, 66–7, 315–318. [Google Scholar]
- Shavel, A.; Gaponik, N.; Eychmuller, A. Efficient UV-blue photoluminescing thiol-stabilized water-soluble alloyed ZnSe(S) nanocrystals. J. Phys. Chem. B 2004, 108, 5905–5908. [Google Scholar] [CrossRef]
- Bera, D.; Holloway, P.H.; Yang, H. Efficient and photostable ZnS-passivated CdS:Mn quantum dots. ECS Trans. 2006, 3, 5–17. [Google Scholar]
- Williams, E.W.; Hall, R. Luminescence and the light emitting diode, 1 Ed. ed; Pergomon Press: New York, NY, USA, 1977; p. 237. [Google Scholar]
- Klimov, V.I.; Mikhailovsky, A.A.; McBranch, D.W.; Leatherdale, C.A.; Bawendi, M.G. Quantization of multiparticle Auger rates in semiconductor quantum dots. Science 2000, 287, 1011–1013. [Google Scholar] [CrossRef] [PubMed]
- Scherer, A.; Craighead, H.G.; Beebe, E.D. Gallium-arsenide and aluminum gallium-arsenide reactive ion etching in boron-trichloride argon mixtures. J. Vac. Sci. Technol. B 1987, 5, 1599–1605. [Google Scholar] [CrossRef]
- Tsutsui, K.; Hu, E.L.; Wilkinson, C.D.W. Reactive ion etched II-VI quantum dots–dependence of etched profile on pattern geometry. Jpn. J. Appl. Phys. Part 1 1993, 32, 6233–6236. [Google Scholar] [CrossRef]
- Chason, E.; Picraux, S.T.; Poate, J.M.; Borland, J.O.; Current, M.I.; delaRubia, T.D.; Eaglesham, D.J.; Holland, O.W.; Law, M.E.; Magee, C.W.; Mayer, J.W.; Melngailis, J.; Tasch, A.F. Ion beams in silicon processing and characterization. J. Appl. Phys. 1997, 81, 6513–6561. [Google Scholar] [CrossRef]
- Burda, C.; Chen, X.B.; Narayanan, R.; El-Sayed, M.A. Chemistry and properties of nanocrystals of different shapes. Chem. Rev. 2005, 105, 1025–1102. [Google Scholar] [CrossRef] [PubMed]
- Sashchiuk, A.; Lifshitz, E.; Reisfeld, R.; Saraidarov, T.; Zelner, M.; Willenz, A. Optical and conductivity properties of PbS nanocrystals in amorphous zirconia sol-gel films. J. Sol-Gel Sci. Technol. 2002, 24, 31–38. [Google Scholar] [CrossRef]
- Hoener, C.F.; Allan, K.A.; Bard, A.J.; Campion, A.; Fox, M.A.; Mallouk, T.E.; Webber, S.E.; White, J.M. Demonstration of a shell core structure in layered CdSe-ZnSe small particles by x-ray photoelectron and Auger spectroscopies. J. Phys. Chem. 1992, 96, 3812–3817. [Google Scholar] [CrossRef]
- Yang, H. Syntheses and application of Mn-doped II-VI semiconductor nanocrystals. Dissertation, Univ. Florida, Gainesville, 2003. [Google Scholar]
- Yang, H.; Holloway, P.H. Enhanced photoluminescence from CdS:Mn/ZnS core/shell quantum dots. Appl. Phys. Lett. 2003, 82, 1965–1967. [Google Scholar] [CrossRef]
- Qu, L.H.; Peng, Z.A.; Peng, X.G. Alternative routes toward high quality CdSe nanocrystals. Nano Lett. 2001, 1, 333–337. [Google Scholar] [CrossRef]
- Yu, W.W.; Wang, Y.A.; Peng, X.G. Formation and stability of size-, shape-, and structure-controlled CdTe nanocrystals: Ligand effects on monomers and nanocrystals. Chem. Mater. 2003, 15, 4300–4308. [Google Scholar] [CrossRef]
- Talapin, D.V.; Haubold, S.; Rogach, A.L.; Kornowski, A.; Haase, M.; Weller, H. A novel organometallic synthesis of highly luminescent CdTe nanocrystals. J. Phys. Chem. B 2001, 105, 2260–2263. [Google Scholar] [CrossRef]
- Singh, S.; Rama, N.; Rao, M.S.R. Influence of d-d transition bands on electrical resistivity in Ni doped polycrystalline ZnO. Appl. Phys. Lett. 2006, 88, 222111 1–3. [Google Scholar]
- Yang, H.S.; Lee, H.; Holloway, P.H. Anisotropic growth of luminescent Eu3+- or Er3+-doped Gd2O3 nanocrystals. Nanotechnology 2005, 16, 2794–2798. [Google Scholar] [CrossRef]
- Chen, H.S.; Wang, S.J.J.; Lo, C.J.; Chi, J.Y. White-light emission from organics-capped ZnSe quantum dots and application in white-light-emitting diodes. Appl. Phys. Lett. 2005, 86, 131905. [Google Scholar] [CrossRef]
- Bakueva, L.; Musikhin, S.; Hines, M.A.; Chang, T.W.F.; Tzolov, M.; Scholes, G.D.; Sargent, E.H. Size-tunable infrared (1000–1600 nm) electroluminescence from PbS quantum-dot nanocrystals in a semiconducting polymer. Appl. Phys. Lett. 2003, 82, 2895–2897. [Google Scholar] [CrossRef]
- Battaglia, D.; Peng, X.G. Formation of high quality InP and InAs nanocrystals in a noncoordinating solvent. Nano Lett. 2002, 2, 1027–1030. [Google Scholar] [CrossRef]
- Bae, W.K.; Nam, M.K.; Char, K.; Lee, S. Gram-scale one-pot synthesis of highly luminescent blue emitting Cd1-xZnxS/ZnS nanocrystals. Chem. Mater. 2008, 20, 5307–5313. [Google Scholar] [CrossRef]
- Olshavsky, M.A.; Goldstein, A.N.; Alivisatos, A.P. Organometallic synthesis of GaAs crystallites exhibiting quantum confinement. J. Am. Chem. Soc. 1990, 112, 9438–9439. [Google Scholar] [CrossRef]
- Brennan, J.G.; Siegrist, T.; Carroll, P.J.; Stuczynski, S.M.; Reynders, P.; Brus, L.E.; Steigerwald, M.L. Bulk and nanostructure group-II-VI compounds from molecular organometallic precursors. Chem. Mater. 1990, 2, 403–409. [Google Scholar] [CrossRef]
- Peng, X.G.; Wickham, J.; Alivisatos, A.P. Kinetics of II-VI and III-V colloidal semiconductor nanocrystal growth: "Focusing" of size distributions. J. Am. Chem. Soc. 1998, 120, 5343–5344. [Google Scholar] [CrossRef]
- Kher, S.S.; Wells, R.L. A straightforward, new method for the synthesis of nanocrystalline GaAs and GaP. Chem. Mater. 1994, 6, 2056–2062. [Google Scholar] [CrossRef]
- Micic, O.I.; Sprague, J.R.; Curtis, C.J.; Jones, K.M.; Machol, J.L.; Nozik, A.J.; Giessen, H.; Fluegel, B.; Mohs, G.; Peyghambarian, N. Synthesis and characterization of InP, GaP, and GaInP2 quantum dots. J. Phys. Chem. 1995, 99, 7754–7759. [Google Scholar] [CrossRef]
- Guzelian, A.A.; Katari, J.E.B.; Kadavanich, A.V.; Banin, U.; Hamad, K.; Juban, E.; Alivisatos, A.P.; Wolters, R.H.; Arnold, C.C.; Heath, J.R. Synthesis of size-selected, surface-passivated InP nanocrystals. J. Phys. Chem. 1996, 100, 7212–7219. [Google Scholar] [CrossRef]
- Guzelian, A.A.; Banin, U.; Kadavanich, A.V.; Peng, X.; Alivisatos, A.P. Colloidal chemical synthesis and characterization of InAs nanocrystal quantum dots. Appl. Phys. Lett. 1996, 69, 1432–1434. [Google Scholar] [CrossRef]
- Cao, Y.W.; Banin, U. Synthesis and characterization of InAs/InP and InAs/CdSe core/shell nanocrystals. Angew. Chem. Int. Ed. 1999, 38, 3692–3694. [Google Scholar] [CrossRef]
- Peng, X.G.; Manna, L.; Yang, W.D.; Wickham, J.; Scher, E.; Kadavanich, A.; Alivisatos, A.P. Shape control of CdSe nanocrystals. Nature (London) 2000, 404, 59–61. [Google Scholar] [CrossRef]
- Manna, L.; Scher, E.C.; Alivisatos, A.P. Synthesis of soluble and processable rod-, arrow-, teardrop-, and tetrapod-shaped CdSe nanocrystals. J. Am. Chem. Soc. 2000, 122, 12700–12706. [Google Scholar] [CrossRef]
- Hikmet, R.A.M.; Talapin, D.V.; Weller, H. Study of conduction mechanism and electroluminescence in CdSe/ZnS quantum dot composites. J. Appl. Phys. 2003, 93, 3509–3514. [Google Scholar] [CrossRef]
- Peng, Z.A.; Peng, X.G. Formation of high-quality CdTe, CdSe, and CdS nanocrystals using CdO as precursor. J. Am. Chem. Soc. 2001, 123, 183–184. [Google Scholar] [CrossRef] [PubMed]
- Coe-Sullivan, S.; Steckel, J.S.; Woo, W.K.; Bawendi, M.G.; Bulovic, V. Large-area ordered quantum-dot monolayers via phase separation during spin-casting. Adv. Func. Mater. 2005, 15, 1117–1124. [Google Scholar] [CrossRef]
- Jun, S.; Jang, E.J.; Chung, Y.S. Alkyl thiols as a sulfur precursor for the preparation of monodisperse metal sulfide nanostructures. Nanotechnology 2006, 17, 4806–4810. [Google Scholar] [CrossRef]
- Zhu, J.J.; Koltypin, Y.; Gedanken, A. General sonochemical method far the preparation of nanophasic selenides: Synthesis of ZnSe nanoparticles. Chem. Mater. 2000, 12, 73–78. [Google Scholar] [CrossRef]
- Qian, H.F.; Li, L.; Ren, J.C. One-step and rapid synthesis of high quality alloyed quantum dots (CdSe-CdS) in aqueous phase by microwave irradiation with controllable temperature. Mater. Res. Bull. 2005, 40, 1726–1736. [Google Scholar] [CrossRef]
- Wang, L.C.; Chen, L.Y.; Luo, T.; Qian, Y.T. A hydrothermal method to prepare the spherical ZnS and flower-like CdS microcrystallites. Mater. Lett. 2006, 60, 3627–3630. [Google Scholar] [CrossRef]
- Yang, H.Q.; Yin, W.Y.; Zhao, H.; Yang, R.L.; Song, Y.Z. A complexant-assisted hydrothermal procedure for growing well-dispersed InP nanocrystals. J. Phys. Chem. Solids 2008, 69, 1017–1022. [Google Scholar] [CrossRef]
- Xie, Y.; Qian, Y.T.; Wang, W.Z.; Zhang, S.Y.; Zhang, Y.H. A benzene-thermal synthetic route to nanocrystalline GaN. Science 1996, 272, 1926–1927. [Google Scholar] [CrossRef] [PubMed]
- Rogach, A.L.; Katsikas, L.; Kornowski, A.; Su, D.S.; Eychmuller, A.; Weller, H. Synthesis and characterization of thiol-stabilized CdTe nanocrystals. Ber.Bunsen Ges. Phys. Chem. Chem. Phys. 1996, 100, 1772–1778. [Google Scholar] [CrossRef]
- O'Connor, E.; O'Riordan, A.; Doyle, H.; Moynihan, S.; Cuddihy, A.; Redmond, G. Near-infrared electroluminescent devices based on colloidal HgTe quantum dot arrays. Appl. Phys. Lett. 2005, 86, 201114. [Google Scholar] [CrossRef]
- Sankaran, V.; Cummins, C.C.; Schrock, R.R.; Cohen, R.E.; Silbey, R.J. Small PbS clusters prepared via romp block copolymer technology. J. Am. Chem. Soc. 1990, 112, 6858–6859. [Google Scholar] [CrossRef]
- Winiarz, J.G.; Zhang, L.M.; Park, J.; Prasad, P.N. Inorganic: Organic hybrid nanocomposites for photorefractivity at communication wavelengths. J. Phys. Chem. B 2002, 106, 967–970. [Google Scholar] [CrossRef]
- Leonard, D.; Krishnamurthy, M.; Reaves, C.M.; Denbaars, S.P.; Petroff, P.M. Direct formation of quantum-sized dots from uniform coherent islands of InGaAs on GaAs-surfaces. Appl. Phys. Lett. 1993, 63, 3203–3205. [Google Scholar] [CrossRef]
- Xin, S.H.; Wang, P.D.; Yin, A.; Kim, C.; Dobrowolska, M.; Merz, J.L.; Furdyna, J.K. Formation of self-assembling CdSe quantum dots on ZnSe by molecular beam epitaxy. Appl. Phys. Lett. 1996, 69, 3884–3886. [Google Scholar] [CrossRef]
- Leonardi, K.; Selke, H.; Heinke, H.; Ohkawa, K.; Hommel, D.; Gindele, F.; Woggon, U. Formation of self-assembling II-VI semiconductor nanostructures during migration enhanced epitaxy. J. Cryst. Growth 1998, 184, 259–263. [Google Scholar] [CrossRef]
- Kurtz, E.; Shen, J.; Schmidt, M.; Grun, M.; Hong, S.K.; Litvinov, D.; Gerthsen, D.; Oka, T.; Yao, T.; Klingshirn, C. Formation and properties of self-organized II-VI quantum islands. Thin Solid Films 2000, 367, 68–74. [Google Scholar] [CrossRef]
- Swihart, M.T. Vapor-phase synthesis of nanoparticles. Curr. Opin. Colloid Interface Sci. 2003, 8, 127–133. [Google Scholar] [CrossRef]
- Frank, F.C.; van der Merwe, J.H. One-dimentional dislocations. I. Static theory. Proc. Roy. Soc. London A 1949, 198, 205. [Google Scholar] [CrossRef]
- Volmer, M.; Weber, A. Keimbildung in ubersettigten gebilden. Z. Phys. Chem. 1926, 119, 277. [Google Scholar]
- Stranski, I.N.; Krastanow, V.L. Sitzungberichte der akademie der wissenschaften in wien. Akad. Wiss. Lit. Mainz Math.-Natur. KI. IIb 1939, 146, 797. [Google Scholar]
- Eaglesham, D.J.; Cerullo, M. Dislocation-free stranski-krastanow growth of Ge on Si(100). Phys. Rev. Lett. 1990, 64, 1943–1946. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Joh, Y.S.; Song, J.H.; Baek, K.S.; Chang, S.K.; Sim, E.D. Temperature-dependent photoluminescence of ZnSe/ZnS quantum dots fabricated under the Stranski-Krastanov mode. Appl. Phys. Lett. 2003, 83, 2656–2658. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Bell, G.R.; Arakawa, Y. Heteroepitaxial growth of InAs on GaAS(001) by in situ STM located inside MBE growth chamber. Microelectron. J. 2006, 37, 1498–1504. [Google Scholar] [CrossRef]
- Jiao, Y.H.; Wu, J.; Xu, B.; Jin, P.; Hu, L.J.; Liang, L.Y.; Wang, Z.G. MBE InAs quantum dots grown on metamorphic InGaAs for long wavelength emitting. Physica E 2006, 35, 194–198. [Google Scholar] [CrossRef]
- Nakamura, S.; Kitamura, K.; Umeya, H.; Jia, A.; Kobayashi, M.; Yoshikawa, A.; Shimotomai, M.; Nakamura, S.; Takahashi, K. Bright electroluminescence from CdS quantum dot LED structures. Electron. Lett. 1998, 34, 2435–2436. [Google Scholar] [CrossRef]
- Lobo, C.; Leon, R. InGaAs island shapes and adatom migration behavior on (100), (110), (111), and (311) GaAs surfaces. J. Appl. Phys. 1998, 83, 4168–4172. [Google Scholar] [CrossRef]
- Muller, C.D.; Falcou, A.; Reckefuss, N.; Rojahn, M.; Wiederhirn, V.; Rudati, P.; Frohne, H.; Nuyken, O.; Becker, H.; Meerholz, K. Multi-colour organic light-emitting displays by solution processing. Nature (London) 2003, 421, 829–833. [Google Scholar] [CrossRef]
- Meerheim, R.W.K.; He, G.; Pfeiffer, M.; Leo, K. Highly efficient organic light emitting diodes (OLED) for displays and lighting. Proc. SPIE Int. Soc. Opt. Eng. 2006, 6192, 1–16. [Google Scholar]
- Adachi, C.; Baldo, M.A.; Thompson, M.E.; Forrest, S.R. Nearly 100% internal phosphorescence efficiency in an organic light-emitting device. J. Appl. Phys. 2001, 90, 5048–5051. [Google Scholar] [CrossRef]
- Gu, G.; Garbuzov, D.Z.; Burrows, P.E.; Venkatesh, S.; Forrest, S.R.; Thompson, M.E. High-external-quantum-efficiency organic light-emitting devices. Opt. Lett. 1997, 22, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Cui, D.H.; Lewis, B.A.; Wang, A.Y.; Xu, S.Y.; Gerhold, M. Microcavity light-emitting devices based on colloidal semiconductor nanocrystal quantum dots. IEEE Photonics Technol. Lett. 2005, 17, 2008–2010. [Google Scholar] [CrossRef]
- Coe, S.; Woo, W.K.; Bawendi, M.; Bulovic, V. Electroluminescence from single monolayers of nanocrystals in molecular organic devices. Nature (London) 2002, 420, 800–803. [Google Scholar] [CrossRef]
- Caruge, J.M.; Halpert, J.E.; Bulovic, V.; Bawendi, M.G. NiO as an inorganic hole-transporting layer in quantum-dot light-emitting devices. Nano Lett. 2006, 6, 2991–2994. [Google Scholar] [CrossRef] [PubMed]
- Bae, W.K.; Kwak, J.; Lim, J.; Lee, D.; Nam, M.K.; Char, K.; Lee, C.; Lee, S. Deep blue light-emitting diodes based on Cd1-xZnxS@ZnS quantum dots. Nanotechnology 2009, 20, 075202. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, S.; Ozkan, M.; Chan, W.C.W. Trilayer hybrid polymer-quantum dot light-emitting diodes. Appl. Phys. Lett. 2004, 84, 2925–2927. [Google Scholar] [CrossRef]
- Irwin, M.D.; Buchholz, B.; Hains, A.W.; Chang, R.P.H.; Marks, T.J. p-Type semiconducting nickel oxide as an efficiency-enhancing anode interfacial layer in polymer bulk-heterojunction solar cells. Proc. Natl. Acad. Sci. USA 2008, 105, 2783–2787. [Google Scholar] [CrossRef]
- Mattoussi, H.; Radzilowski, L.H.; Dabbousi, B.O.; Thomas, E.L.; Bawendi, M.G.; Rubner, M.F. Electroluminescence from heterostructures of poly(phenylene vinylene) and inorganic CdSe nanocrystals. J. Appl. Phys. 1998, 83, 7965–7974. [Google Scholar] [CrossRef]
- Mattoussi, H.; Radzilowski, L.H.; Dabbousi, B.O.; Fogg, D.E.; Schrock, R.R.; Thomas, E.L.; Rubner, M.F.; Bawendi, M.G. Composite thin films of CdSe nanocrystals and a surface passivating/electron transporting block copolymer: Correlations between film microstructure by transmission electron microscopy and electroluminescence. J. Appl. Phys. 1999, 86, 4390–4399. [Google Scholar] [CrossRef]
- Coe-Sullivan, S.; Woo, W.K.; Steckel, J.S.; Bawendi, M.; Bulovic, V. Tuning the performance of hybrid organic/inorganic quantum dot light-emitting devices. Org. Electron. 2003, 4, 123–130. [Google Scholar] [CrossRef]
- Cho, K.S.; Lee, E.K.; Joo, W.J.; Jang, E.; Kim, T.H.; Lee, S.J.; Kwon, S.J.; Han, J.Y.; Kim, B.K.; Choi, B.L.; Kim, J.M. High-performance crosslinked colloidal quantum-dot light-emitting diodes. Nat. Photonics 2009, 3, 341–345. [Google Scholar] [CrossRef]
- Zhao, J.L.; Bardecker, J.A.; Munro, A.M.; Liu, M.S.; Niu, Y.H.; Ding, I.K.; Luo, J.D.; Chen, B.Q.; Jen, A.K.Y.; Ginger, D.S. Efficient CdSe/CdS quantum dot light-emitting diodes using a thermally polymerized hole transport layer. Nano Lett. 2006, 6, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Wang, Y.A.; Li, L.S.; Wang, D.Y.; Zhu, T.; Xu, J.; Yang, C.H.; Li, Y.F. Bright, multicoloured light-emitting diodes based on quantum dots. Nat. Photonics 2007, 1, 717–722. [Google Scholar] [CrossRef]
- Schlamp, M.C.; Peng, X.G.; Alivisatos, A.P. Improved efficiencies in light emitting diodes made with CdSe(CdS) core/shell type nanocrystals and a semiconducting polymer. J. Appl. Phys. 1997, 82, 5837–5842. [Google Scholar] [CrossRef]
- Mueller, A.H.; Petruska, M.A.; Achermann, M.; Werder, D.J.; Akhadov, E.A.; Koleske, D.D.; Hoffbauer, M.A.; Klimov, V.I. Multicolor light-emitting diodes based on semiconductor nanocrystals encapsulated in GaN charge injection layers. Nano Lett. 2005, 5, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.B.; Kagan, C.R.; Bawendi, M.G. Synthesis and characterization of monodisperse nanocrystals and close-packed nanocrystal assemblies. Annu. Rev. Mater. Sci. 2000, 30, 545–610. [Google Scholar] [CrossRef]
- Colvin, V.L.; Schlamp, M.C.; Alivisatos, A.P. Light-emitting-diodes made from cadmium selenide nanocrystals and a semiconducting polymer. Nature (London) 1994, 370, 354–357. [Google Scholar] [CrossRef]
- Caruge, J.M.; Halpert, J.E.; Wood, V.; Bulovic, V.; Bawendi, M.G. Colloidal quantum-dot light-emitting diodes with metal-oxide charge transport layers. Nat. Photonics 2008, 2, 247–250. [Google Scholar] [CrossRef]
- Zhu, T.; Shanmugasundaram, K.; Price, S.C.; Ruzyllo, J.; Zhang, F.; Xu, J.; Mohney, S.E.; Zhang, Q.; Wang, A.Y. Mist fabrication of light emitting diodes with colloidal nanocrystal quantum dots. Appl. Phys. Lett. 2008, 92, 023111 1–3. [Google Scholar]
- Kobayashi, S.; Tani, Y.; Kawazoe, H. Quantum dot activated all-inorganic electroluminescent device fabricated using solution-synthesized CdSe/ZnS nanocrystals. Jpn. J. Appl. Phys. Part 2 2007, 46, L966–L969. [Google Scholar] [CrossRef]
- Taylor, R.M.; Church, K.H.; Sluch, M.I. Red light emission from hybrid organic/inorganic quantum dot AC light emitting displays. Displays 2007, 28, 92–96. [Google Scholar] [CrossRef]
- Steckel, J.S.; Zimmer, J.P.; Coe-Sullivan, S.; Stott, N.E.; Bulovic, V.; Bawendi, M.G. Blue luminescence from (CdS)ZnS core-shell nanocrystals. Angew. Chem. Int. Ed. 2004, 43, 2154–2158. [Google Scholar] [CrossRef]
- Dabbousi, B.O.; Bawendi, M.G.; Onitsuka, O.; Rubner, M.F. Electroluminescence from CdSe quantum-dot polymer composites. Appl. Phys. Lett. 1995, 66, 1316–1318. [Google Scholar] [CrossRef]
- Yang, H.S.; Holloway, P.H. Electroluminescence from hybrid conjugated polymer− CdS:Mn/ZnS core/shell nanocrystals devices. J. Phys. Chem. B 2003, 107, 9705–9710. [Google Scholar] [CrossRef]
- Tessler, N.; Medvedev, V.; Kazes, M.; Kan, S.H.; Banin, U. Efficient near-infrared polymer nanocrystat light-emitting diodes. Science 2002, 295, 1506–1508. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.P.; Gerenser, L.J.; Jarman, C.M.; Fornalik, J.E. Thin-film encapsulation of organic light-emitting devices. Appl. Phys. Lett. 2005, 86. [Google Scholar]
- McElvain, J.; Antoniadis, H.; Hueschen, M.R.; Miller, J.N.; Roitman, D.M.; Sheats, J.R.; Moon, R.L. Formation and growth of black spots in organic light-emitting diodes. J. Appl. Phys. 1996, 80, 6002–6007. [Google Scholar] [CrossRef]
- Tan, Z.N.; Zhang, F.; Zhu, T.; Xu, J.; Wang, A.Y.; Dixon, D.; Li, L.S.; Zhang, Q.; Mohney, S.E.; Ruzyllo, J. Bright and color-saturated emission from blue light-emitting diodes based on solution-processed colloidal nanocrystal quantum dots. Nano Lett. 2007, 7, 3803–3807. [Google Scholar] [CrossRef] [PubMed]
- Bae, W.K.; Kwak, J.; Park, J.W.; Char, K.; Lee, C.; Lee, S. Highly efficient green-light-emitting diodes based on CdSe@ZnS quantum dots with a chemical-composition gradient. Adv. Mater. 2009, 21, 1690–1694. [Google Scholar] [CrossRef]
- Gao, M.Y.; Lesser, C.; Kirstein, S.; Mohwald, H.; Rogach, A.L.; Weller, H. Electroluminescence of different colors from polycation/CdTe nanocrystal self-assembled films. J. Appl. Phys. 2000, 87, 2297–2302. [Google Scholar] [CrossRef]
- Chen, H.S.; Hsu, C.K.; Hong, H.Y. InGaN-CdSe-ZnSe quantum dots white LEDs. IEEE Photonics Technol. Lett. 2006, 18, 193–195. [Google Scholar] [CrossRef]
- Nizamoglu, S.; Ozel, T.; Sari, E.; Demir, H.V. White light generation using CdSe/ZnS core-shell nanocrystals hybridized with InGaN/GaN light emitting diodes. Nanotechnology 2007, 18, 065709. [Google Scholar] [CrossRef]
- Yeh, D.M.; Huang, C.F.; Lu, Y.C.; Yang, C.C. White-light light-emitting device based on surface plasmon-enhanced CdSe/ZnS nanocrystal wavelength conversion on a blue/green two-color light-emitting diode. Appl. Phys. Lett. 2008, 92, 09112 1–3. [Google Scholar]
- Gratzel, M. From space to earth: The story of solar electricity. Nature (London) 2000, 403, 363. [Google Scholar] [CrossRef]
- Shockley, W.; Queisser, H.J. Detailed balance limit of efficiency of p-n junction solar cells. J. Appl. Phys. 1961, 32, 510–519. [Google Scholar] [CrossRef]
- King, R.R.; Law, D.C.; Edmondson, K.M.; Fetzer, C.M.; Kinsey, G.S.; Yoon, H.; Sherif, R.A.; Karam, N.H. 40% efficient metamorphic GaInP/GaInAs/Ge multijunction solar cells. Appl. Phys. Lett. 2007, 90, 183516. [Google Scholar] [CrossRef]
- Hoppe, H.; Sariciftci, N.S. Organic solar cells: An overview. J. Mater. Res. 2004, 19, 1924–1945. [Google Scholar] [CrossRef]
- Guldi, D.M.; Rahman, G.M.A.; Sgobba, V.; Kotov, N.A.; Bonifazi, D.; Prato, M. CNT-CdTe versatile donor-acceptor nanohybrids. J. Am. Chem. Soc. 2006, 128, 2315–2323. [Google Scholar] [CrossRef] [PubMed]
- Han, L.L.; Qin, D.H.; Jiang, X.; Liu, Y.S.; Wang, L.; Chen, J.W.; Cao, Y. Synthesis of high quality zinc-blende CdSe nanocrystals and their application in hybrid solar cells. Nanotechnology 2006, 17, 4736–4742. [Google Scholar] [CrossRef] [PubMed]
- Oey, C.C.; Djurisic, A.B.; Wang, H.; Man, K.K. Y.; Chan, W.K.; Xie, M.H.; Leung, Y.H.; Pandey, A.; Nunzi, J.M.; Chui, P.C. Polymer-TiO2 solar cells: TiO2\ interconnected network for improved cell performance. Nanotechnology 2006, 17, 706–713. [Google Scholar] [CrossRef]
- Olson, D.C.; Piris, J.; Collins, R.T.; Shaheen, S.E.; Ginley, D.S. Hybrid photovoltaic devices of polymer and ZnO nanofiber composites. Thin Solid Films 2006, 496, 26–29. [Google Scholar] [CrossRef]
- Michler, P.; Imamoglu, A.; Mason, M.D.; Carson, P.J.; Strouse, G.F.; Buratto, S.K. Quantum correlation among photons from a single quantum dot at room temperature. Nature (London) 2000, 406, 968–970. [Google Scholar] [CrossRef]
- Cui, D.H.; Xu, J.; Zhu, T.; Paradee, G.; Ashok, S.; Gerhold, M. Harvest of near infrared light in PbSe nanocrystal-polymer hybrid photovoltaic cells. Appl. Phys. Lett. 2006, 88, 183111. [Google Scholar] [CrossRef]
- O'Regan, B.; Schwartz, D.T.; Zakeeruddin, S.M.; Gratzel, M. Electrodeposited nanocomposite n-p heterojunctions for solid-state dye-sensitized photovoltaics. Adv. Mater. 2000, 12, 1263–1267. [Google Scholar] [CrossRef]
- Bereznev, S.; Konovalov, I.; Opik, A.; Kois, J. Hybrid CuInS2/polypyrrole and CuInS2 /poly(3,4-ethylenedioxythiophene) photovoltaic structures. Synth. Met. 2005, 152, 81–84. [Google Scholar] [CrossRef]
- Nozik, A.J. Exciton multiplication and relaxation dynamics in quantum dots: Applications to ultrahigh-efficiency solar photon conversion. Inorg. Chem. 2005, 44, 6893–6899. [Google Scholar] [CrossRef] [PubMed]
- Pijpers, J.J.H.; Ulbricht, R.; Tielrooij, K.J.; Osherov, A.; Golan, Y.; Delerue, C.; Allan, G.; Bonn, M. Assessment of carrier-multiplication efficiency in bulk PbSe and PbS. Nat. Physics 2009, 5, 811–814. [Google Scholar] [CrossRef]
- Beard, M.C.; Ellingson, R.J. Multiple exciton generation in semiconductor nanocrystals: Toward efficient solar energy conversion. Laser Photonics Rev. 2008, 2, 377–399. [Google Scholar] [CrossRef]
- Allan, G.; Delerue, C. Influence of electronic structure and multiexciton spectral density on multiple-exciton generation in semiconductor nanocrystals: Tight-binding calculations. Phys. Rev. B 2008, 77, 125340. [Google Scholar] [CrossRef]
- Beard, M.C.; Knutsen, K.P.; Yu, P.R.; Luther, J.M.; Song, Q.; Metzger, W.K.; Ellingson, R.J.; Nozik, A.J. Multiple exciton generation in colloidal silicon nanocrystals. Nano Lett. 2007, 7, 2506–2512. [Google Scholar] [CrossRef] [PubMed]
- Nozik, A.J. Quantum dot solar cells. Physica E 2002, 14, 115–120. [Google Scholar] [CrossRef]
- Oregan, B.; Gratzel, M. A low-cost, high-efficiency solar-cell based on dye-sensitized colloidal TiO2 films. Nature (London) 1991, 353, 737–740. [Google Scholar] [CrossRef]
- Gratzel, M. Photoelectrochemical cells. Nature (London) 2001, 414, 338–344. [Google Scholar] [CrossRef]
- Vogel, R.; Hoyer, P.; Weller, H. Quantum-sized PbS, CdS, Ag2S, Sb2S3, and Bi2S3 particles as sensitizers for various nanoporous wide-bandgap semiconductors. J. Phys. Chem. 1994, 98, 3183–3188. [Google Scholar] [CrossRef]
- Vogel, R.; Pohl, K.; Weller, H. Sensitization of highly porous, polycrystalline TiO2 electrodes by quantum sized CdS. Chem. Phys. Lett. 1990, 174, 241–246. [Google Scholar] [CrossRef]
- Gopidas, K.R.; Bohorquez, M.; Kamat, P.V. Photoelectrochemistry in semiconductor particulate systems .16. Photophysical and photochemical aspects of coupled semiconductors−Charge-transfer processes in colloidal CdS-TiO2 and CdS-AgI systems. J. Phys. Chem. 1990, 94, 6435–6440. [Google Scholar] [CrossRef]
- Plass, R.; Pelet, S.; Krueger, J.; Gratzel, M.; Bach, U. Quantum dot sensitization of organic-inorganic hybrid solar cells. J. Phys. Chem. B 2002, 106, 7578–7580. [Google Scholar] [CrossRef]
- Peter, L.M.; Riley, D.J.; Tull, E.J.; Wijayantha, K.G.U. Photosensitization of nanocrystalline TiO2 by self-assembled layers of CdS quantum dots. Chem. Commun. 2002, 1030–1031. [Google Scholar] [CrossRef]
- Lin, S.C.; Lee, Y.L.; Chang, C.H.; Shen, Y.J.; Yang, Y.M. Quantum-dot-sensitized solar cells: Assembly of CdS-quantum-dots coupling techniques of self-assembled monolayer and chemical bath deposition. Appl. Phys. Lett. 2007, 90, 143517. [Google Scholar] [CrossRef]
- Robel, I.; Subramanian, V.; Kuno, M.; Kamat, P.V. Quantum dot solar cells. Harvesting light energy with CdSe nanocrystals molecularly linked to mesoscopic TiO2 films. J. Am. Chem. Soc. 2006, 128, 2385–2393. [Google Scholar] [CrossRef] [PubMed]
- Niitsoo, O.; Sarkar, S.K.; Pejoux, C.; Ruhle, S.; Cahen, D.; Hodes, G. Chemical bath deposited CdS/CdSe-sensitized porous TiO2 solar cells. J. Photochem. Photobiol. A 2006, 181, 306–313. [Google Scholar] [CrossRef]
- Diguna, L.J.; Shen, Q.; Kobayashi, J.; Toyoda, T. High efficiency of CdSe quantum-dot-sensitized TiO2 inverse opal solar cells. Appl. Phys. Lett. 2007, 91, 023116 1–3. [Google Scholar]
- Sun, W.T.; Yu, Y.; Pan, H.Y.; Gao, X.F.; Chen, Q.; Peng, L.M. CdS quantum dots sensitized TiO2 nanotube-array photoelectrodes. J. Am. Chem. Soc. 2008, 130, 1124–1125. [Google Scholar] [CrossRef] [PubMed]
- Karakoti, A.S.; Filmalter, R.; Bera, D.; Kuchibhatla, S.; Vincent, A.; Seal, S. Spiral growth of one dimensional titania nanostructures using anodic oxidation. J. Nanosci. Nanotech. 2006, 6, 2084–2089. [Google Scholar] [CrossRef]
- Lee, Y.L.; Lo, Y.S. Highly efficient quantum-dot-sensitized solar cell based on co-sensitization of CdS/CdSe. Adv. Func. Mater. 2009, 19, 604–609. [Google Scholar] [CrossRef]
- Zaban, A.; Micic, O.I.; Gregg, B.A.; Nozik, A.J. Photosensitization of nanoporous TiO2 electrodes with InP quantum dots. Langmuir 1998, 14, 3153–3156. [Google Scholar] [CrossRef]
- Wang, Y.; Herron, N. Photoconductivity of CdS nanocluster-doped polymers. Chem. Phys. Lett. 1992, 200, 71–75. [Google Scholar] [CrossRef]
- Greenham, N.C.; Peng, X.G.; Alivisatos, A.P. Charge separation and transport in conjugated polymer cadmium selenide nanocrystal composites studied by photoluminescence quenching and photoconductivity. Synth. Met. 1997, 84, 545–546. [Google Scholar] [CrossRef]
- Arango, A.C.; Carter, S.A.; Brock, P.J. Charge transfer in photovoltaics consisting of interpenetrating networks of conjugated polymer and TiO2 nanoparticles. Appl. Phys. Lett. 1999, 74, 1698–1700. [Google Scholar] [CrossRef]
- Huynh, W.U.; Dittmer, J.J.; Alivisatos, A.P. Hybrid nanorod-polymer solar cells. Science 2002, 295, 2425–2427. [Google Scholar] [CrossRef] [PubMed]
- Beek, W.J.E.; Wienk, M.M.; Janssen, R.A.J. Efficient hybrid solar cells from zinc oxide nanoparticles and a conjugated polymer. Adv. Mater. 2004, 16, 1009–1013. [Google Scholar] [CrossRef]
- Sun, B.Q.; Greenham, N.C. Improved effciency of photovoltaics based on CdSe nanorods and poly(3-hexylthiophene) nanofibers. Phys. Chem. Chem. Phys. 2006, 8, 3557–3560. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Abrusci, A.; Wong, H.M.P.; Svensson, M.; Andersson, M.R.; Greenham, N.C. Photoinduced charge transfer and efficient solar energy conversion in a blend of a red polyfluorene copolymer with CdSe nanoparticles. Nano Lett. 2006, 6, 1789–1793. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.Q.; Snaith, H.J.; Dhoot, A.S.; Westenhoff, S.; Greenham, N.C. Vertically segregated hybrid blends for photovoltaic devices with improved efficiency. J. Appl. Phys. 2005, 97, 014914. [Google Scholar] [CrossRef]
- Sun, B.Q.; Marx, E.; Greenham, N.C. Photovoltaic devices using blends of branched CdSe nanoparticles and conjugated polymers. Nano Lett. 2003, 3, 961–963. [Google Scholar] [CrossRef]
- Dayal, S.; Kopidakis, N.; Olson, D.C.; Ginley, D.S.; Rumbles, G. Photovoltaic devices with a low band gap polymer and CdSe nanostructures exceeding 3% efficiency. Nano Lett. 2010, 10, 239–242. [Google Scholar] [CrossRef]
- Gur, I.; Fromer, N.A.; Chen, C.P.; Kanaras, A.G.; Alivisatos, A.P. Hybrid solar cells with prescribed nanoscale morphologies based on hyperbranched semiconductor nanocrystals. Nano Lett. 2007, 7, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.S.; Tanaka, T.; Sivula, K.; Alivisatos, A.P.; Frechet, J.M.J. Employing end-functional polythiophene to control the morphology of nanocrystal-polymer composites in hybrid solar cells. J. Am. Chem. Soc. 2004, 126, 6550–6551. [Google Scholar] [CrossRef] [PubMed]
- Watt, A.A.R.; Blake, D.; Warner, J.H.; Thomsen, E.A.; Tavenner, E.L.; Rubinsztein-Dunlop, H.; Meredith, P. Lead sulfide nanocrystal: conducting polymer solar cells. J. Phys. D 2005, 38, 2006–2012. [Google Scholar] [CrossRef]
- Klimov, V.I.; Ivanov, S.A.; Nanda, J.; Achermann, M.; Bezel, I.; McGuire, J.A.; Piryatinski, A. Single-exciton optical gain in semiconductor nanocrystals. Nature (London) 2007, 447, 441–446. [Google Scholar] [CrossRef]
- Wang, C.J.; Wehrenberg, B.L.; Woo, C.Y.; Guyot-Sionnest, P. Light emission and amplification in charged CdSe quantum dots. J. Phys. Chem. B 2004, 108, 9027–9031. [Google Scholar] [CrossRef]
- Roither, J.; Pichler, S.; Kovalenko, M.V.; Heiss, W.; Feychuk, P.; Panchuk, O.; Allam, J.; Murdin, B.N. Two- and one-dimensional light propagations and gain in layer-by-layer-deposited colloidal nanocrystal waveguides. Appl. Phys. Lett. 2006, 89, 111120. [Google Scholar] [CrossRef]
- Liu, B.; Li, H.P.; Chew, C.H.; Que, W.X.; Lam, Y.L.; Kam, C.H.; Gan, L.M.; Xu, G.Q. PbS-polymer nanocomposite with third-order nonlinear optical response in femtosecond regime. Mater. Lett. 2001, 51, 461–469. [Google Scholar] [CrossRef]
- Choudhury, K.R.; Sahoo, Y.; Prasad, P.N. Hybrid quantum-dot-polymer nanocomposites for infrared photorefractivity at an optical communication wavelength. Adv. Mater. 2005, 17, 2877–2881. [Google Scholar] [CrossRef]
- Williams, D.P.; Andreev, A.D.; O'Reilly, E.P. Dependence of exciton energy on dot size in GaN/AlN quantum dots. Phys. Rev. B 2006, 73, 241301. [Google Scholar] [CrossRef]
- Pan, G.Q.; Kordesch, M.E.; Van Patten, P.G. New pyrolysis route to GaN quantum dots. Chem. Mater. 2006, 18, 3915–3917. [Google Scholar] [CrossRef]
- Barik, S.; Tan, H.H.; Jagadish, C.; Vukmirovic, N.; Harrison, P. Selective wavelength tuning of self-assembled InAs quantum dots grown on InP. Appl. Phys. Lett. 2006, 88. [Google Scholar]
- Cassidy, P.J.; Radda, G.K. Molecular imaging perspectives. J. R. Soc. Interface 2005, 2, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Schillaci, O.; Danieli, R.; Padovano, F.; Testa, A.; Simonetti, G. Molecular imaging of atheroslerotic plaque with nuclear medicine techniques (Review). Int. J. Mol. Med. 2008, 22, 3–7. [Google Scholar] [PubMed]
- Lecchi, M.; Ottobrini, L.; Martelli, C.; Del Sole, A.; Lucignani, G. Instrumentation and probes for molecular and cellular imaging. Q. J. Nucl. Med. Mol. Imaging 2007, 51, 111–126. [Google Scholar] [PubMed]
- Pichler, B.J.; Wehrl, H.F.; Judenhofer, M.S. Latest advances in molecular imaging instrumentation. J. Nucl. Med. 2008, 49, S5–S23. [Google Scholar]
- Levenson, R.M.; Lynch, D.T.; Kobayashi, H.; Backer, J.M.; Backer, M.V. Multiplexing with multispectral imaging: From mice to microscopy. Ilar J. 2008, 49, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Brown, S.; Walter, G.; Santra, S.; Moudgil, B. Nanoparticles for bioimaging. Adv. Colloid Interface Sci. 2006, 123, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Deng, Y.L.; Qing, H.; Xie, H.Y. Advance in real-time and dynamic biotracking and bioimaging based on quantum dots. Chem. J. Chin. Univer.-Chin. 2008, 29, 661–668. [Google Scholar]
- Parak, W.J.; Pellegrino, T.; Plank, C. Labelling of cells with quantum dots. Nanotechnology 2005, 16, R9–R25. [Google Scholar] [PubMed]
- Li, Z.B.; Cai, W.; Chen, X.Y. Semiconductor quantum dots for in vivo imaging. J. Nanosci. Nanotech. 2007, 7, 2567–2581. [Google Scholar] [CrossRef]
- Kumar, S.; Richards-Kortum, R. Optical molecular imaging agents for cancer diagnostics and therapeutics. Nanomedicine 2006, 1, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Dave, S.; Nie, S.M.; True, L.; Gao, X.H. Multicolor quantum dots for molecular diagnostics of cancer. Expert Rev. Mol. Diagn. 2006, 6, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.H.; Yang, L.L.; Petros, J.A.; Marshal, F.F.; Simons, J.W.; Nie, S.M. In vivo molecular and cellular imaging with quantum dots. Curr. Opin. Biotechnol. 2005, 16, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, J.K.; Goldman, E.R.; Mattoussi, H.; Simon, S.M. Use of quantum dots for live cell imaging. Nat. Methods 2004, 1, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Duan, H.W.; Mohs, A.M.; Nie, S.M. Bioconjugated quantum dots for in vivo molecular and cellular imaging. Adv. Drug Delivery Rev. 2008, 60, 1226–1240. [Google Scholar] [CrossRef]
- Gao, X.H.; Dave, S.R. Quantum dots for cancer molecular imaging. In Bio-Applications of Nanoparticles; 2007; Volume 620, pp. 57–73. [Google Scholar]
- Zrazhevskiy, P.; Gao, X. Quantum dots for cancer molecular imaging. Minerva Biotecnologica 2009, 21, 37–52. [Google Scholar]
- Hillman, E.M.C. Optical brain imaging in vivo: Techniques and applications from animal to man. J. Biomed. Optics 2007, 12, 051402. [Google Scholar] [CrossRef]
- Luker, G.D.; Luker, K.E. Optical imaging: Current applications and future directions. J. Nucl. Med. 2008, 49, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.W.; Li, A.D. Optically switchable nanoparticles for biological imaging. Nanomedicine 2007, 2, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Weissleder, R. A clearer vision for in vivo imaging. Nat. Biotechnol. 2001, 19, 316–317. [Google Scholar] [CrossRef] [PubMed]
- Bruchez, M.; Moronne, M.; Gin, P.; Weiss, S.; Alivisatos, A.P. Semiconductor nanocrystals as fluorescent biological labels. Science 1998, 281, 2013–2016. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.C.W.; Nie, S.M. Quantum dot bioconjugates for ultrasensitive nonisotopic detection. Science 1998, 281, 2016–2018. [Google Scholar] [CrossRef] [PubMed]
- Parak, W.J.; Boudreau, R.; Le Gros, M.; Gerion, D.; Zanchet, D.; Micheel, C.M.; Williams, S.C.; Alivisatos, A.P.; Larabell, C. Cell motility and metastatic potential studies based on quantum dot imaging of phagokinetic tracks. Adv. Mater. 2002, 14, 882–885. [Google Scholar] [CrossRef]
- Akerman, M.E.; Chan, W.C.W.; Laakkonen, P.; Bhatia, S.N.; Ruoslahti, E. Nanocrystal targeting in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 12617–12621. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lim, Y.T.; Soltesz, E.G.; De Grand, A.M.; Lee, J.; Nakayama, A.; Parker, J.A.; Mihaljevic, T.; Laurence, R.G.; Dor, D.M.; Cohn, L.H.; Bawendi, M.G.; Frangioni, J.V. Near-infrared fluorescent type II quantum dots for sentinel lymph node mapping. Nat. Biotechnol. 2004, 22, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Medintz, I.L.; Clapp, A.R.; Mattoussi, H.; Goldman, E.R.; Fisher, B.; Mauro, J.M. Self-assembled nanoscale biosensors based on quantum dot FRET donors. Nat. Mater. 2003, 2, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Sapsford, K.E.; Pons, T.; Medintz, I.L.; Mattoussi, H. Biosensing with luminescent semiconductor quantum dots. Sensors 2006, 6, 925–953. [Google Scholar] [CrossRef]
- Sukhanova, A.; Devy, M.; Venteo, L.; Kaplan, H.; Artemyev, M.; Oleinikov, V.; Klinov, D.; Pluot, M.; Cohen, J.H.M.; Nabiev, I. Biocompatible fluorescent nanocrystals for immunolabeling of membrane proteins and cells. Anal. Biochem. 2004, 324, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Dahan, M.; Levi, S.; Luccardini, C.; Rostaing, P.; Riveau, B.; Triller, A. Diffusion dynamics of glycine receptors revealed by single-quantum dot tracking. Science 2003, 302, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Michalet, X.; Pinaud, F.F.; Bentolila, L.A.; Tsay, J.M.; Doose, S.; Li, J.J.; Sundaresan, G.; Wu, A.M.; Gambhir, S.S.; Weiss, S. Quantum dots for live cells, in vivo imaging, and diagnostics. Science 2005, 307, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Klaunberg, B.A. Biomedical applications of fluorescence imaging in vivo. Comp. Med. 2004, 54, 635–644. [Google Scholar] [PubMed]
- Medintz, I.L.; Uyeda, H.T.; Goldman, E.R.; Mattoussi, H. Quantum dot bioconjugates for imaging, labelling and sensing. Nat. Mater. 2005, 4, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, J.; Wang, P.N.; Chen, J.Y.; Lu, Z.J.; Lu, D.R.; Guo, J.; Wang, C.C.; Yang, W.L. Time-dependent photoluminescence blue shift of the quantum dots in living cells: Effect of oxidation by singlet oxygen. J. Am. Chem. Soc. 2006, 128, 13396–13401. [Google Scholar] [CrossRef] [PubMed]
- Dahan, M.; Laurence, T.; Pinaud, F.; Chemla, D.S.; Alivisatos, A.P.; Sauer, M.; Weiss, S. Time-gated biological imaging by use of colloidal quantum dots. Opt. Lett. 2001, 26, 825–827. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Hong, J.Q.; Guo, X.Q.; Huang, C.B.; Lai, J.P.; Zheng, J.S.; Chen, J.B.; Mu, X.; Zhao, Y.B. Fluorescent core-shell silica nanoparticles as tunable precursors: Towards encoding and multifunctional nano-probes. Chem. Comm. 2008, 750–752. [Google Scholar] [CrossRef]
- Han, M.Y.; Gao, X.H.; Su, J.Z.; Nie, S. Quantum-dot-tagged microbeads for multiplexed optical coding of biomolecules. Nat. Biotechnol. 2001, 19, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Dubertret, B.; Skourides, P.; Norris, D.J.; Noireaux, V.; Brivanlou, A.H.; Libchaber, A. In vivo imaging of quantum dots encapsulated in phospholipid micelles. Science 2002, 298, 1759–1762. [Google Scholar] [CrossRef] [PubMed]
- Larson, D.R.; Zipfel, W.R.; Williams, R.M.; Clark, S.W.; Bruchez, M.P.; Wise, F.W.; Webb, W.W. Water-soluble quantum dots for multiphoton fluorescence imaging in vivo. Science 2003, 300, 1434–1436. [Google Scholar] [CrossRef] [PubMed]
- Holloway, P.H.; Yang, H.; Lee, H.; Seo, S.; Santra, S.; Qian, L.; Bera, D. Nanophosphor: PL, EL, and Biological Markers Proc. Int. Symp. Display lighting Phosphor Mater, ISDLPMW’06. 2006.
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy,, 2nd ed.; Academic Publishers: New York, NY, USA, 1999. [Google Scholar]
- Tran, P.T.; Goldman, E.R.; Anderson, G.P.; Mauro, J.M.; Mattoussi, H. Use of luminescent CdSe-ZnS nanocrystal bioconjugates in quantum dot-based nanosensors. Phys. Status Solidi B 2002, 229, 427–432. [Google Scholar] [CrossRef]
- Clapp, A.R.; Medintz, I.L.; Mauro, J.M.; Fisher, B.R.; Bawendi, M.G.; Mattoussi, H. Fluorescence resonance energy transfer between quantum dot donors and dye-labeled protein acceptors. J. Am. Chem. Soc. 2004, 126, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Clapp, A.R.; Medintz, I.L.; Fisher, B.R.; Anderson, G.P.; Mattoussi, H. Can luminescent quantum dots be efficient energy acceptors with organic dye donors? J. Am. Chem. Soc. 2005, 127, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Patolsky, F.; Gill, R.; Weizmann, Y.; Mokari, T.; Banin, U.; Willner, I. Lighting-up the dynamics of telomerization and DNA replication by CdSe-ZnS quantum dots. J. Am. Chem. Soc. 2003, 125, 13918–13919. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Shin, D.M. Monoclonal antibodies to target epidermal growth factor receptor-positive tumors–A new paradigm for cancer therapy. Cancer 2002, 94, 1593–1611. [Google Scholar] [CrossRef] [PubMed]
- Reuter, C.W.M.; Morgan, M.A.; Eckardt, A. Targeting EGF-receptor-signalling in squamous cell carcinomas of the head and neck. Br. J. Cancer 2007, 96, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Peng, X.-H.; Ansari, D.O.; Yin-Goen, Q.; Chen, G.Z.; Shin, D.M.; Yang, L.; Young, A.N.; Wang, M.D.; Nie, S. In vivo tumor targeting and spectroscopic detection with surface-enhanced Raman nanoparticle tags. Nat. Biotech. 2008, 26, 83–90. [Google Scholar] [CrossRef]
- Martin, C.R.; Mitchell, D.T. Nanomaterials in analytical chemistry. Anal. Chem. 1998, 70, A322–A327. [Google Scholar]
- Tan, W.B.; Zhang, Y. Multi-functional chitosan nanoparticles encapsulating quantum dots and Gd-DTPA as imaging probes for bio-applications. J. Nanosci. Nanotech. 2007, 7, 2389–2393. [Google Scholar] [CrossRef]
- Mulder, W.J.M.; Koole, R.; Brandwijk, R.J.; Storm, G.; Chin, P.T.K.; Strijkers, G.J.; Donega, C.D.; Nicolay, K.; Griffioen, A.W. Quantum dots with a paramagnetic coating as a bimodal molecular imaging probe. Nano Lett. 2006, 6, 1–6. [Google Scholar] [CrossRef] [PubMed]
- van Tilborg, G.A.F.; Mulder, W.J.M.; Chin, P.T.K.; Storm, G.; Reutelingsperger, C.P.; Nicolay, K.; Strijkers, G.J. Annexin A5-conjugated quantum dots with a paramagnetic lipidic coating for the multimodal detection of apoptotic cells. Bioconjug. Chem. 2006, 17, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Bakalova, R.; Zhelev, Z.; Aoki, I.; Ohba, H.; Imai, Y.; Kanno, I. Silica-shelled single quantum dot micelles as imaging probes with dual or multimodality. Anal. Chem. 2006, 78, 5925–5932. [Google Scholar] [CrossRef] [PubMed]
- Toth, E.; Bolskar, R.D.; Borel, A.; Gonzalez, G.; Helm, L.; Merbach, A.E.; Sitharaman, B.; Wilson, L.J. Water-soluble gadofullerenes: Toward high-relaxivity, pH-responsive MRI contrast agents. J. Am. Chem. Soc. 2005, 127, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, M.; Bauer, H.; Mintorovitch, J.; Requardt, M.; Weinmann, H.J. Comparison of magnetic properties of MRI contrast media solutions at different magnetic field strengths. Invest. Radiol. 2005, 40, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Pierre, V.C.; Botta, M.; Raymond, K.N. Dendrimeric gadolinium chelate with fast water exchange and high relaxivity at high magnetic field strength. J. Am. Chem. Soc. 2005, 127, 504–505. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.B.; Zhang, Y. Multifunctional quantum-dot-based magnetic chitosan nanobeads. Adv. Mater. 2005, 17, 2375–2380. [Google Scholar] [CrossRef]
- Son, D.H.; Hughes, S.M.; Yin, Y.D.; Alivisatos, A.P. Cation exchange reactions-in ionic nanocrystals. Science 2004, 306, 1009–1012. [Google Scholar] [CrossRef] [PubMed]
- Green, M. Semiconductor quantum dots as biological imaging agents. Angew. Chem., Int. Ed. 2004, 43, 4129–4131. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bera, D.; Qian, L.; Tseng, T.-K.; Holloway, P.H. Quantum Dots and Their Multimodal Applications: A Review. Materials 2010, 3, 2260-2345. https://doi.org/10.3390/ma3042260
Bera D, Qian L, Tseng T-K, Holloway PH. Quantum Dots and Their Multimodal Applications: A Review. Materials. 2010; 3(4):2260-2345. https://doi.org/10.3390/ma3042260
Chicago/Turabian StyleBera, Debasis, Lei Qian, Teng-Kuan Tseng, and Paul H. Holloway. 2010. "Quantum Dots and Their Multimodal Applications: A Review" Materials 3, no. 4: 2260-2345. https://doi.org/10.3390/ma3042260
APA StyleBera, D., Qian, L., Tseng, T.-K., & Holloway, P. H. (2010). Quantum Dots and Their Multimodal Applications: A Review. Materials, 3(4), 2260-2345. https://doi.org/10.3390/ma3042260