The One-Step Pickering Emulsion Polymerization Route for Synthesizing Organic-Inorganic Nanocomposite Particles

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
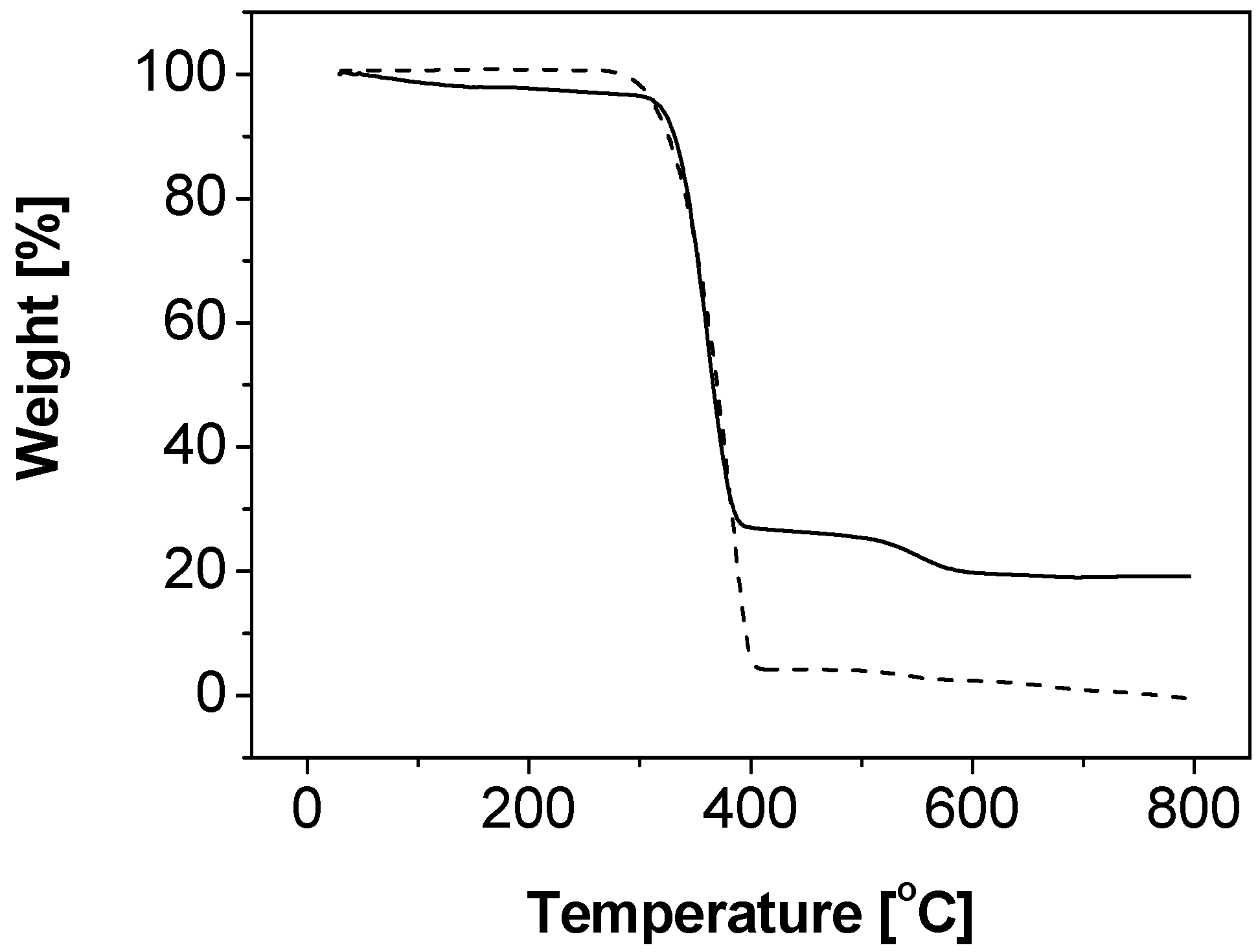
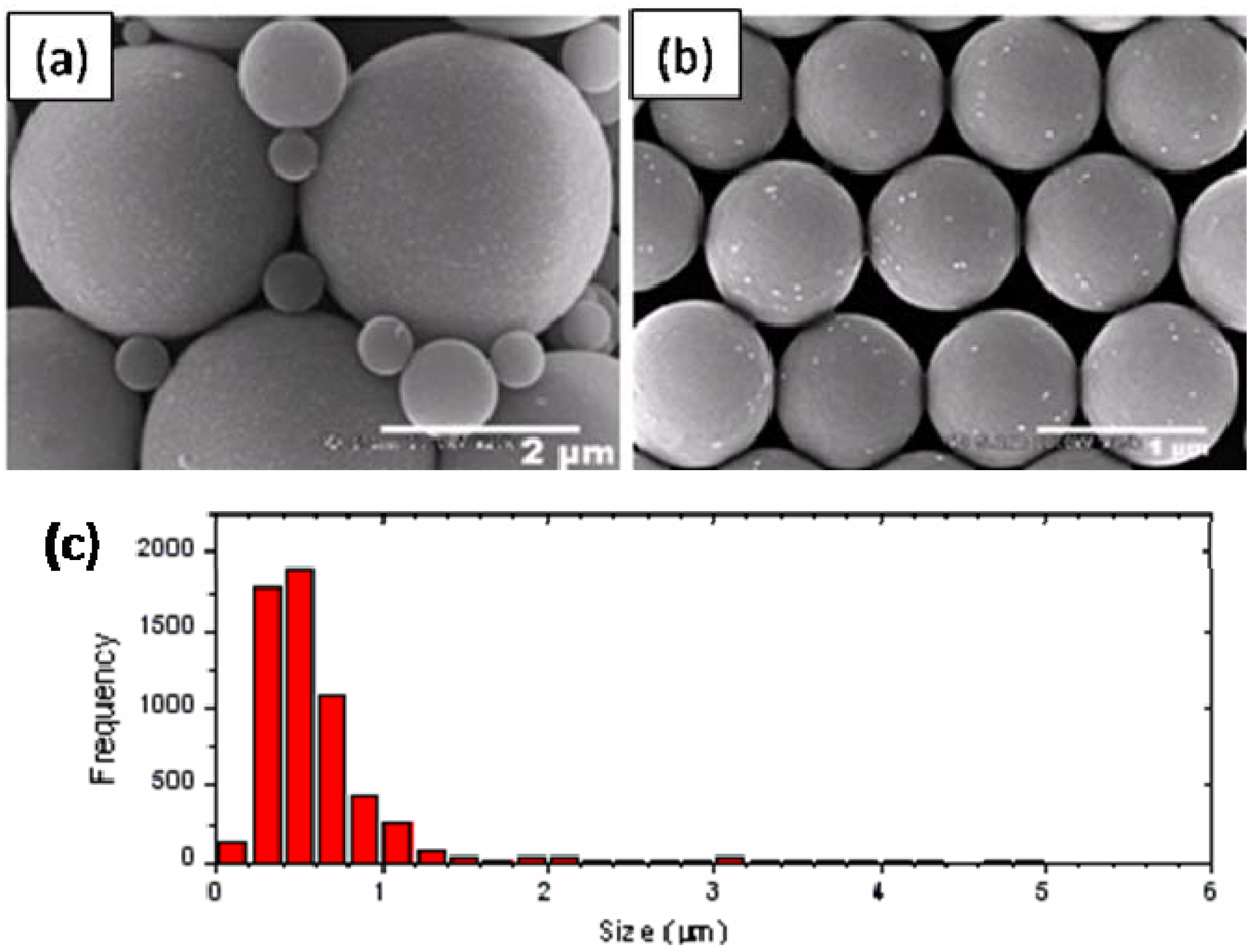
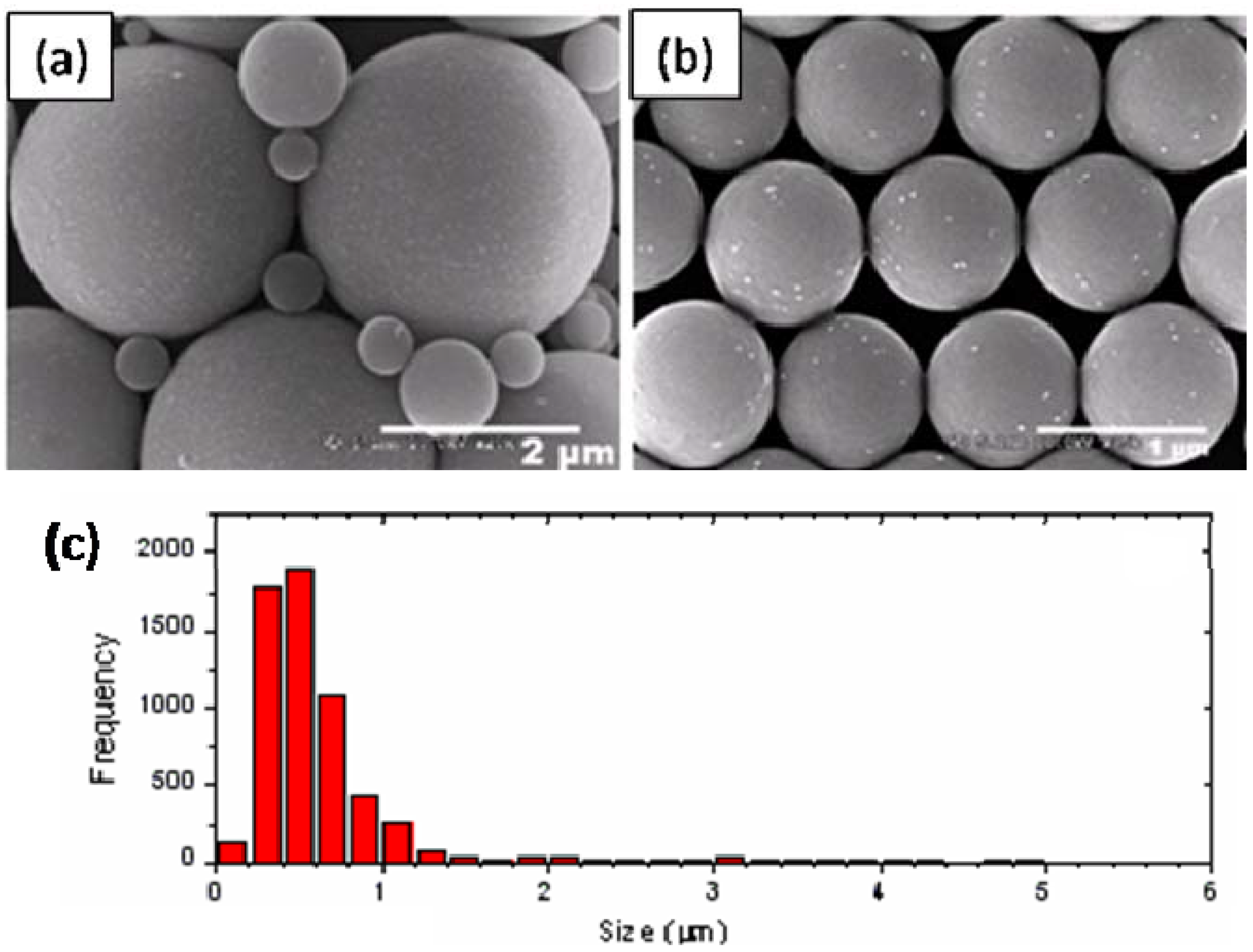
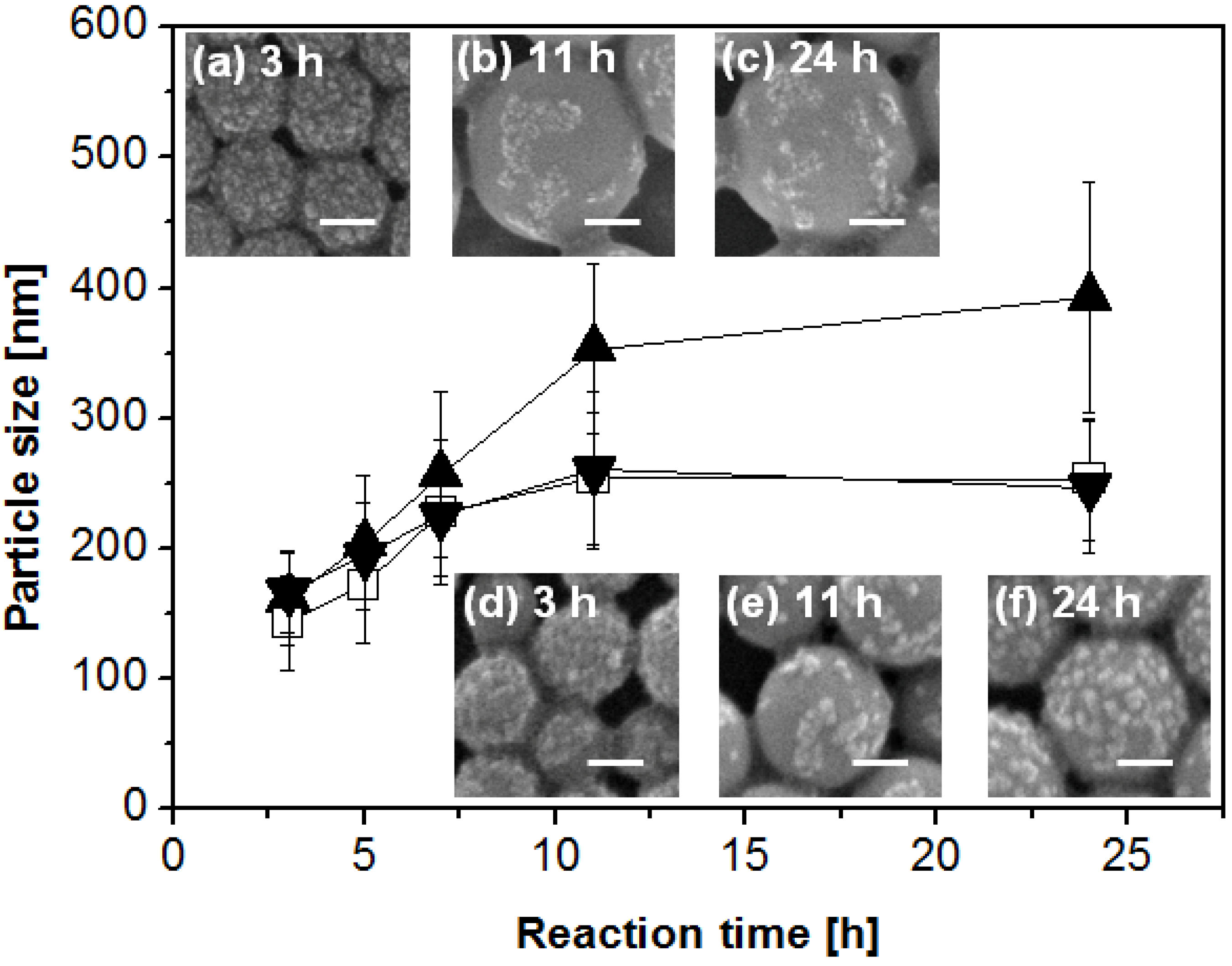
2.1. Synthesis and Characterizations of Nanocomposite Particles



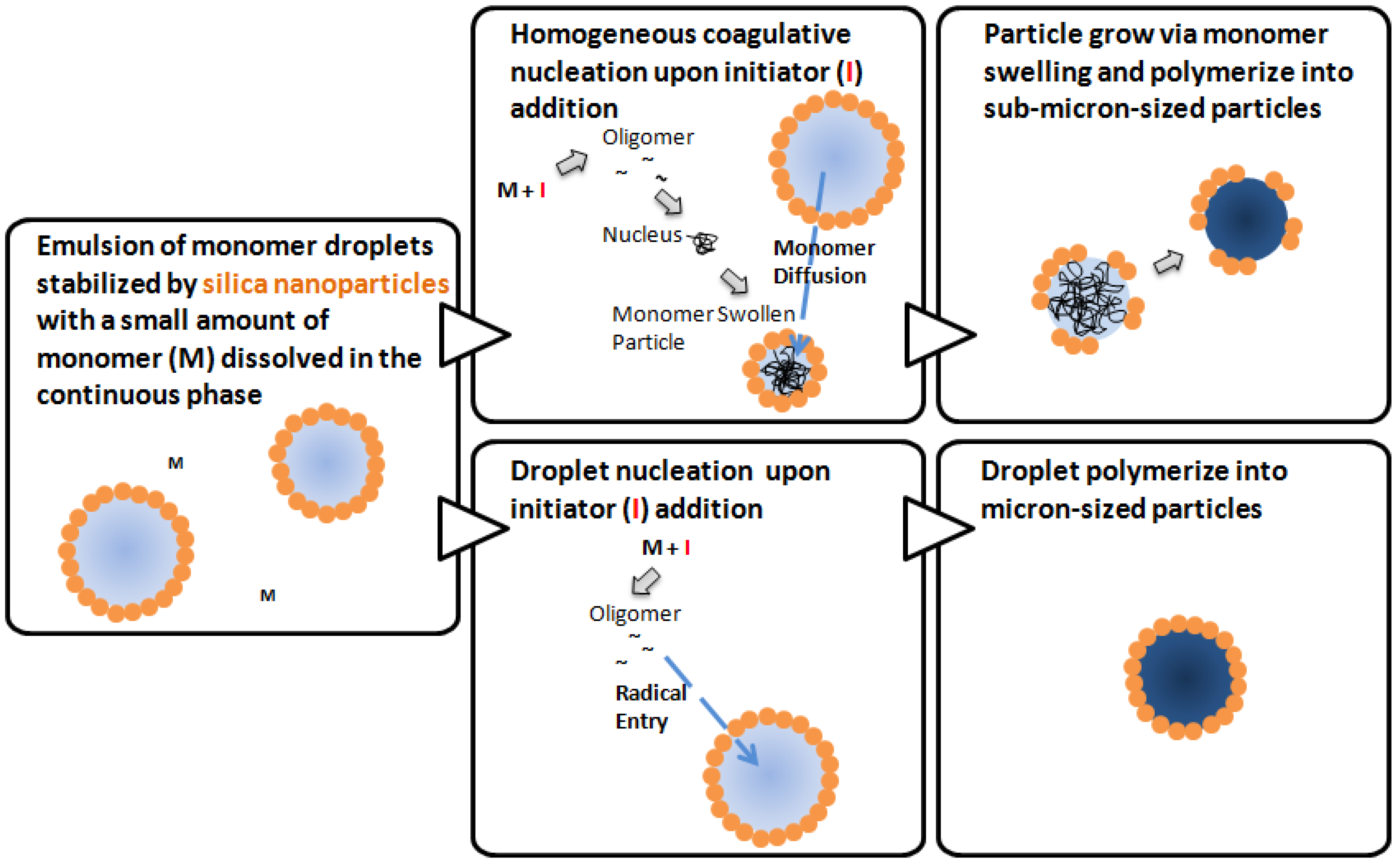
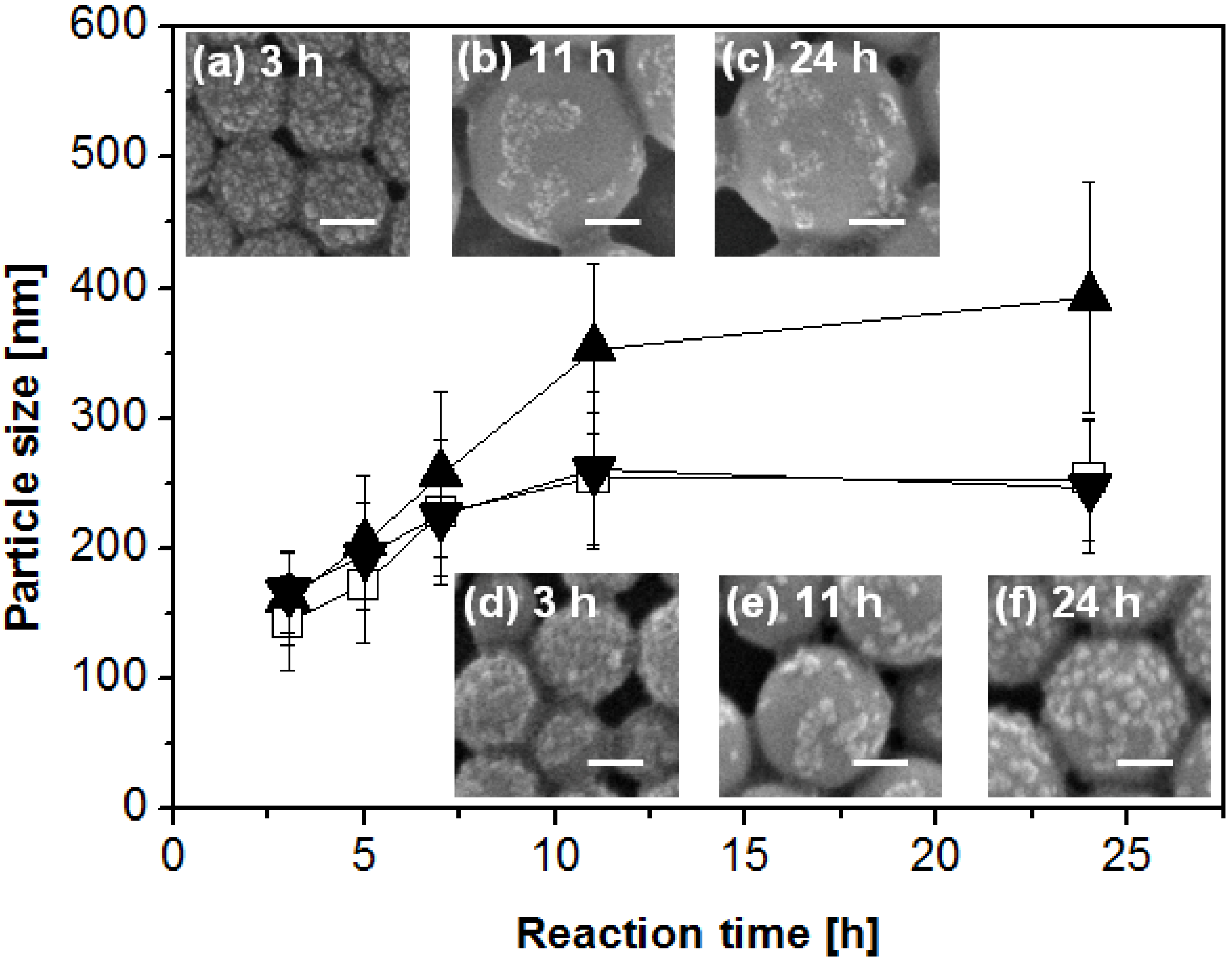
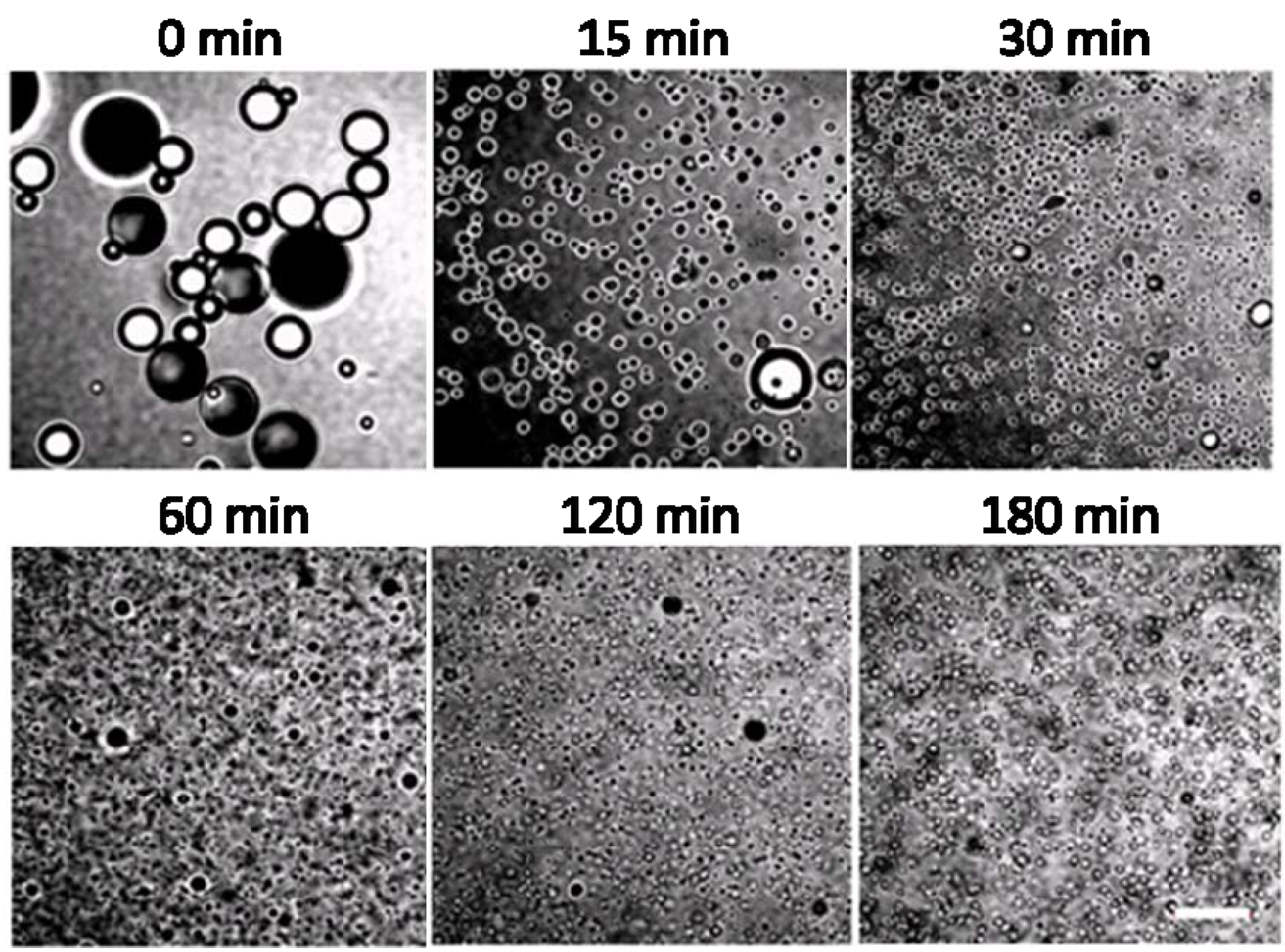
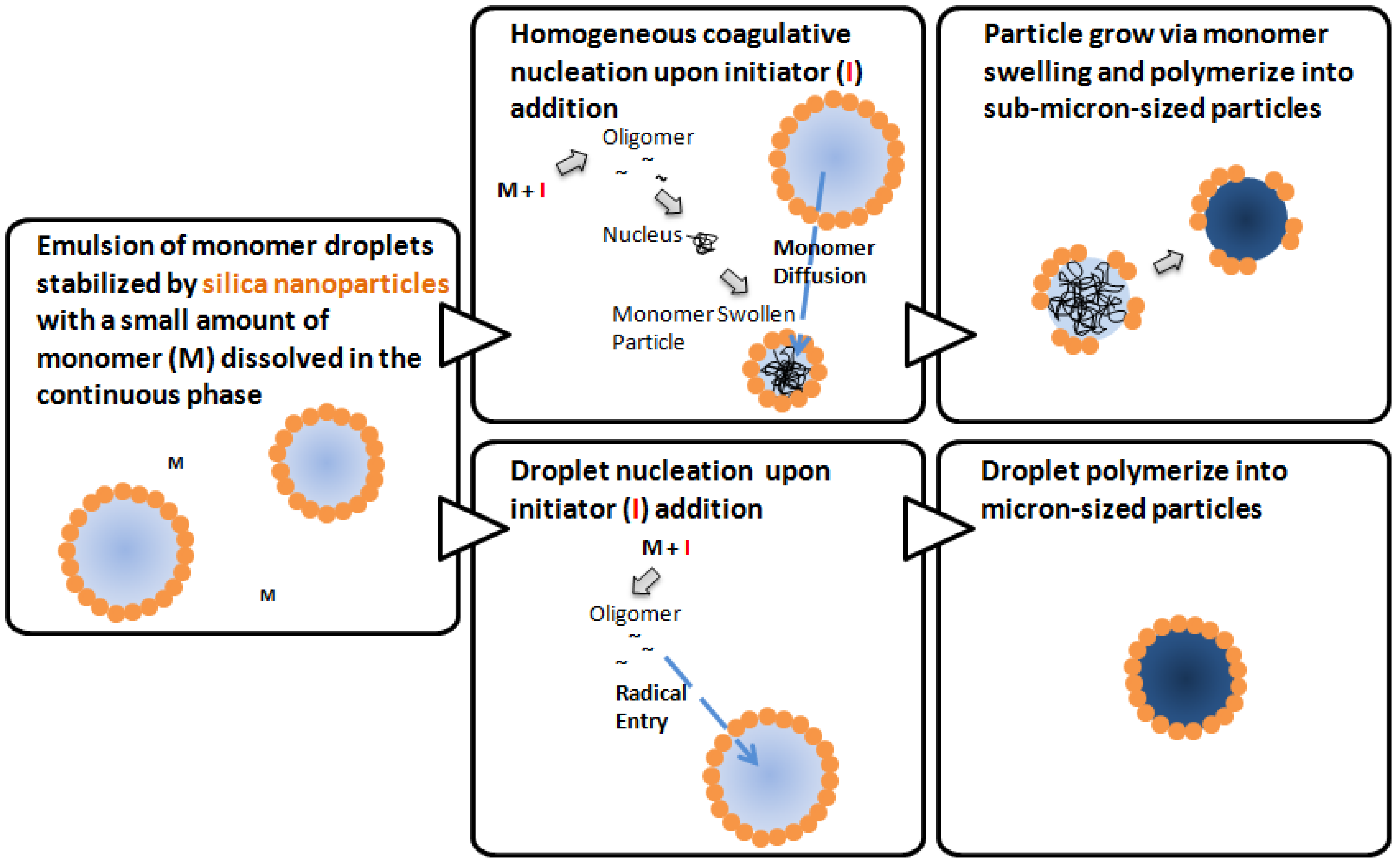
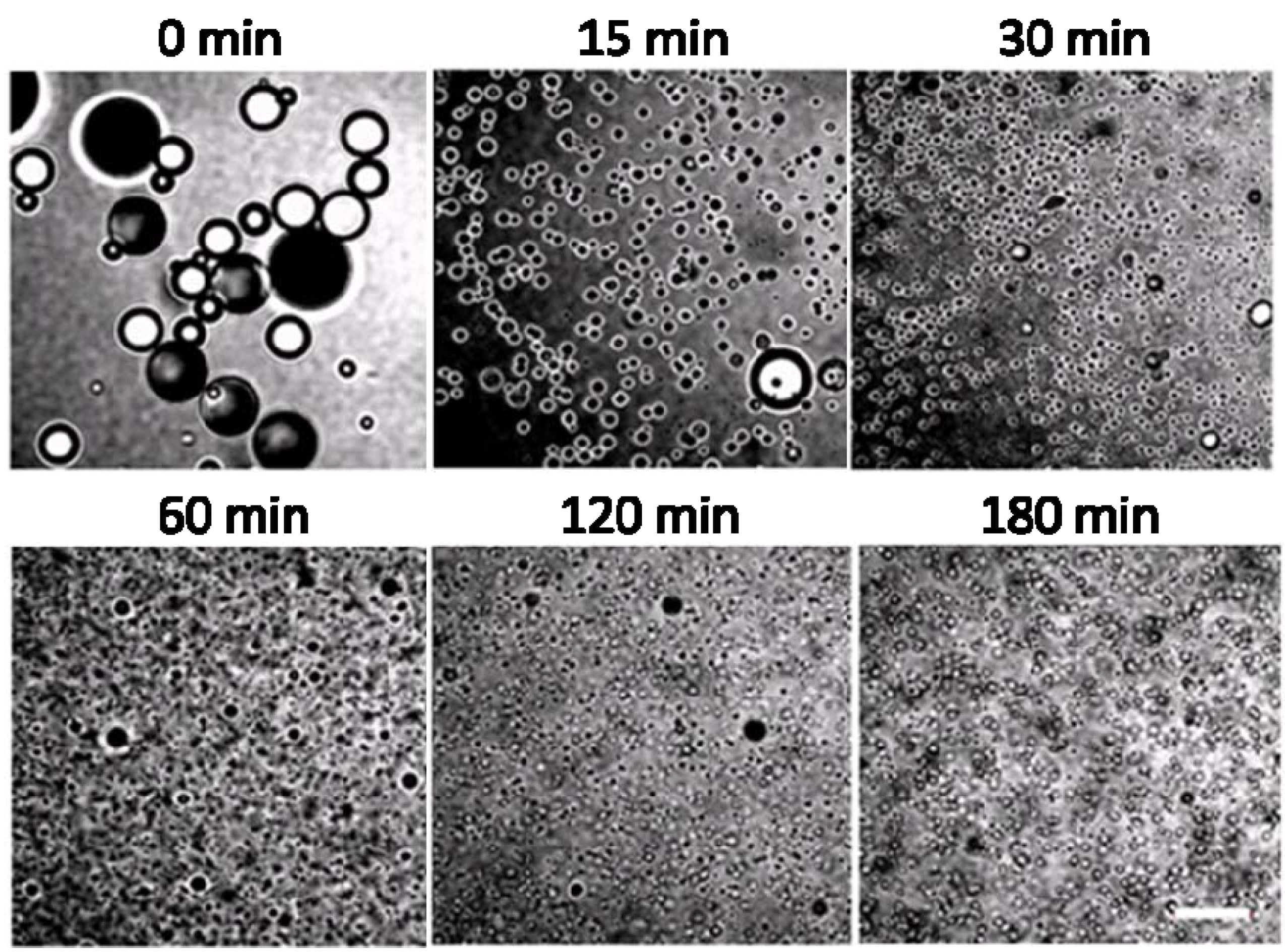
2.2. Mechanisms of Pickering Emulsion Polymerization
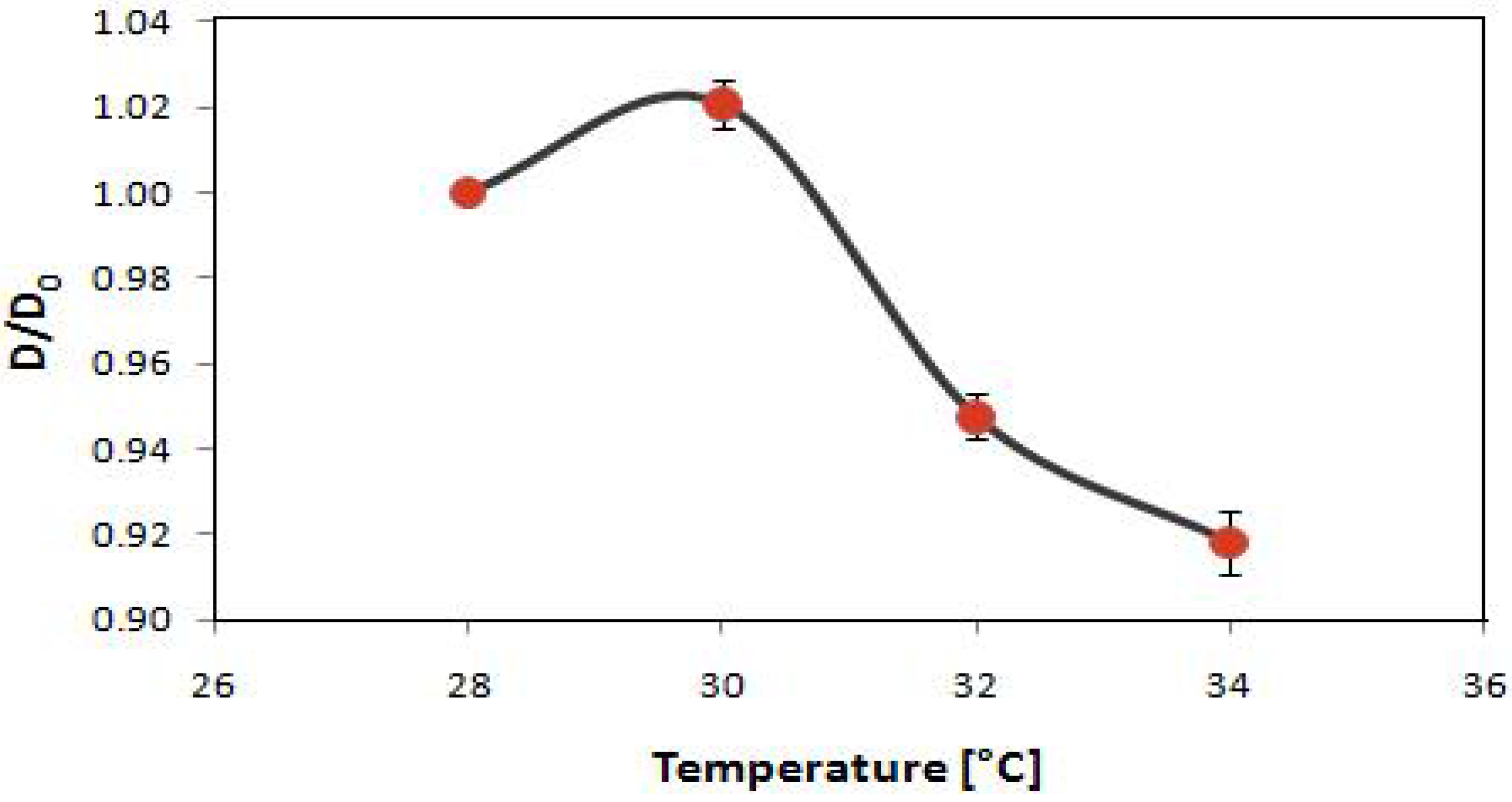
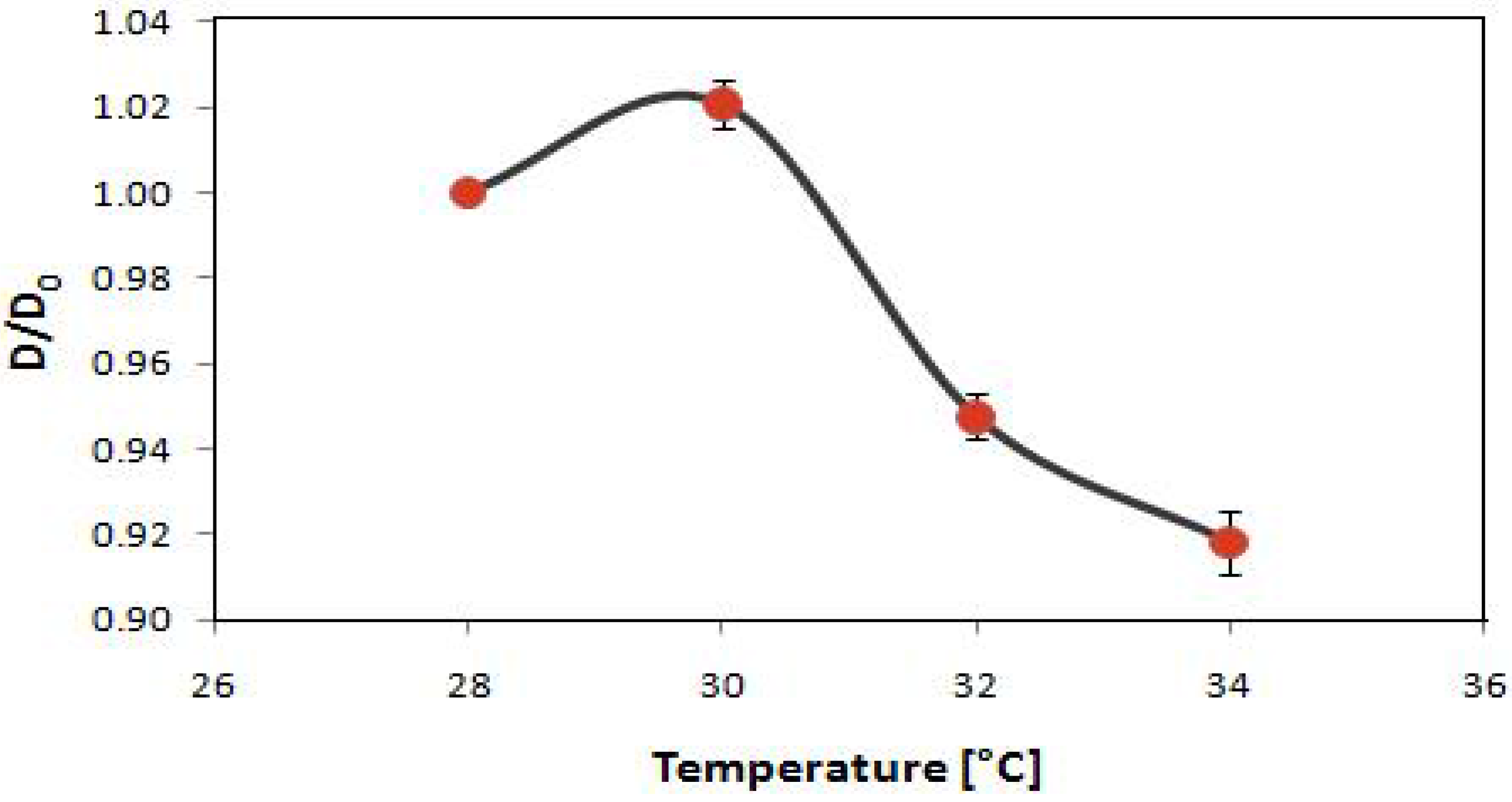
2.3. Potential Applications of Composite Nanoparticles in Controlled Drug Delivery

3. Experimental Section
3.1. Materials
3.2. Composite Particle/Nanoparticle Synthesis
3.3. Hydrofluoric Acid Etching
3.4. Determination of Particle Size
3.5. Particle Morphology and Composition Characterization
3.6. Confocal Microscope Observations of the Sampled Mixtures during Polymerization
3.7. Cellular Uptake Experiments
4. Conclusions
Acknowledgements
References and Notes
- Ramsden, W. Separation of solids in the surface-layers of solutions and 'suspensions' (observations on surface-membranes, bubbles, emulsions, and mechanical coagulation). preliminary account. Proc. R. Soc. London 1903, 72, 156–164. [Google Scholar] [CrossRef]
- Pickering, S.U. Emulsions. J. Chem. Soc. 1907, 91, 2001–2021. [Google Scholar] [CrossRef]
- Dai, L.L.; Sharma, R.; Wu, C.Y. Self-assembled structure of nanoparticles at a liquid-liquid interface. Langmuir 2005, 21, 2641–2643. [Google Scholar] [CrossRef] [PubMed]
- Tarimala, S.; Dai, L.L. Structure of microparticles in solid-stabilized emulsions. Langmuir 2004, 20, 3492–3494. [Google Scholar] [CrossRef] [PubMed]
- Tarimala, S.; Ranabothu, S.R.; Vernetti, J.P.; Dai, L.L. Mobility and in situ aggregation of charged microparticles at oil-water interfaces. Langmuir 2004, 20, 5171–5173. [Google Scholar] [CrossRef] [PubMed]
- Tarimala, S.; Wu, C.; Dai, L.L. Dynamics and collapse of two-dimensional colloidal lattices. Langmuir 2006, 22, 7458–7461. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Dai, L.L. One-particle microrheology at liquid-liquid interfaces. Appl. Phys. Lett. 2006, 89, 094107. [Google Scholar] [CrossRef]
- Wu, J.; Dai, L.L. Apparent microrheology of oil-water interfaces by single-particle tracking. Langmuir 2007, 23, 4324–4331. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Song, Y.; Dai, L.L. Two-particle microrheology at oil-water interfaces. Appl. Phys. Lett. 2009, 95, 144104. [Google Scholar] [CrossRef]
- Bourgeat-Lami, E.; Lang, J. Encapsulation of inorganic particles by dispersion polymerization in polar media: 1. silica nanoparticles encapsulated by polystyrene. J. Colloid Interface Sci. 1998, 197, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Caruso, F. Nanoengineering of particle surfaces. Adv. Mater. 2001, 13, 11–22. [Google Scholar] [CrossRef]
- Gu, S.; Kondo, T.; Konno, M. Preparation of silica–polystyrene core–shell particles up to micron sizes. J. Colloid Interface Sci. 2004, 272, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.K.; Wang, M.Y.; Zhang, H.Q.; Liu, G.D.; Zhang, L.Q.; Qu, X.W. The surface modification of nanosilica, preparation of nanosilica. J. Appl. Polym. Sci. 2008, 107, 2671–2680. [Google Scholar] [CrossRef]
- Nagao, D.; Yokoyama, M.; Saeki, S.; Kobayashi, Y.; Konno, M. Preparation of composite particles with magnetic silica core and fluorescent polymer shell. Colloid Polym. Sci. 2008, 286, 959–964. [Google Scholar] [CrossRef]
- Tianbin, W.; Yangchuan, K. Preparation of silica-PS composite particles and their application in PET. Eur. Polym. J. 2006, 42, 274–285. [Google Scholar] [CrossRef]
- Barthet, C.; Hickey, A.J.; Cairns, D.B.; Armes, S.P. Synthesis of novel polymer-silica colloidal nanocomposites via free-radical polymerization of vinyl monomers. Adv. Mater. 1999, 11, 408–410. [Google Scholar] [CrossRef]
- Ding, X.F.; Jiang, Y.Q.; Yu, K.F.; Hari-Bala; Tao, N.N.; Zhao, J.Z.; Wang, Z.C. Silicon dioxide as coating on polystyrene nanoparticles in situ emulsion polymerization. Mater. Lett. 2004, 58, 1722–1725. [Google Scholar] [CrossRef]
- Lynch, D.E.; Nawaz, Y.; Bostrom, T. Preparation of sub-micrometer silica shells using poly(1-methylpyrrol-2-ylsquaraine). Langmuir 2005, 21, 6572–6575. [Google Scholar] [CrossRef] [PubMed]
- Dokoutchaev, A.; James, J.T.; Koene, S.C.; Pathak, S.; Prakash, G.K.S.; Thompson, M.E. Colloidal metal deposition onto functionalized polystyrene microspheres. Chem. Mater. 1999, 11, 2389–2399. [Google Scholar] [CrossRef]
- Caruso, F.; Susha, A.S.; Giersig, M.; Möhwald, H. Magnetic core-shell particles: Preparation of magnetite multilayers on polymer latex microspheres. Adv. Mater. 1999, 11, 950–953. [Google Scholar] [CrossRef]
- Caruso, R.A.; Susha, A.; Caruso, F. Multilayered titania, silica, and laponite nanoparticle coatings on polystyrene colloidal templates and resulting inorganic hollow spheres. Chem. Mater. 2001, 13, 400–409. [Google Scholar] [CrossRef]
- Bon, S.A.F.; Colver, P.J. Pickering miniemulsion polymerization using laponite clay as a stabilizer. Langmuir 2007, 23, 8316–8322. [Google Scholar] [CrossRef] [PubMed]
- Cauvin, S.; Colver, P.J.; Bon, S.A.F. Pickering stabilized miniemulsion polymerization: Preparation of clay armored latexes. Macromolecules 2005, 38, 7887–7889. [Google Scholar] [CrossRef]
- Schmid, A.; Fujii, S.; Armes, S.P.; Leite, C.A.P.; Galembeck, F.; Minami, H.; Saito, N.; Okubo, M. Polystyrene-silica colloidal nanocomposite particles prepared by alcoholic dispersion polymerization. Chem. Mater. 2007, 19, 2435–2445. [Google Scholar] [CrossRef]
- Schmid, A.; Fujii, S.; Armes, S.P. Polystyrene-silica nanocomposite particles via alcoholic dispersion polymerization using a cationic azo initiator. Langmuir 2006, 22, 4923–4927. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hasell, T.; Wang, W.X.; Li, J.; Brown, P.D.; Poliakoff, M.; Lester, E.; Howdle, S.M. Preparation of hybrid polymer nanocomposite microparticles by a nanoparticle stabilised dispersion polymerisation. J. Mater. Chem. 2008, 18, 998–1001. [Google Scholar] [CrossRef]
- Duan, L.; Chen, M.; Zhou, S.; Wu, L. Synthesis and characterization of poly(N-isopropylacrylamide)/Silica composite microspheres via inverse pickering suspension polymerization. Langmuir 2009, 25, 3467–3472. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Wang, C.; Liu, H.; Wang, C.; Liu, X.; Tong, Z. Suspension polymerization based on inverse pickering emulsion droplets for thermo-sensitive hybrid microcapsules with tunable supracolloidal structures. Polymer 2009, 50, 2587–2594. [Google Scholar] [CrossRef]
- Voorn, D.J.; Ming, W.; van Herk, A.M. Polymer-clay nanocomposite latex particles by inverse pickering emulsion polymerization stabilized with hydrophobic montmorillonite platelets. Macromolecules 2006, 39, 2137–2143. [Google Scholar] [CrossRef]
- Lee, J.; Hong, C.K.; Choe, S.; Shim, S.E. Synthesis of polystyrene/silica composite particles by soap-free emulsion polymerization using positively charged colloidal silica. J. Colloid Interface Sci. 2007, 310, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Vignati, E.; Piazza, R.; Lockhart, T.P. Pickering emulsions: Interfacial tension, colloidal layer morphology, and trapped-particle motion. Langmuir 2003, 19, 6650–6656. [Google Scholar] [CrossRef]
- Xu, H.; Melle, S.; Golemanov, K.; Fuller, G. Shape and buckling transitions in solid-stabilized drops. Langmuir 2005, 21, 10016–10020. [Google Scholar] [CrossRef] [PubMed]
- Torii, H.; Fujimoto, K.; Kawaguchi, H. Chemical properties of water-soluble, nonionic azo compounds as initiators for emulsion polymerization. J. Polym. Sci. A Polym. Chem. 1996, 34, 1237–1243. [Google Scholar] [CrossRef]
- Ou, J.; Yang, J.; Chen, H. Styrene/potassium persulfate/water systems: Effects of hydrophilic comonomers and solvent additives on the nucleation mechanism and the particle size. Eur. Polym. J. 2001, 37, 789–799. [Google Scholar] [CrossRef]
- Tauer, K.; Deckwer, R.; Kuhn, I.; Schellenberg, C. A comprehensive experimental study of surfactant-free emulsion polymerization of styrene. Colloid Polym. Sci. 1999, 277, 607–626. [Google Scholar] [CrossRef]
- Harkins, W.D. A general theory of the mechanism of emulsion polymerization II. J. Am. Chem. Soc. 1947, 69, 1428–1444. [Google Scholar] [CrossRef] [PubMed]
- Chern, C.S. Emulsion polymerization mechanisms and kinetics. Prog. Polym. Sci. 2006, 31, 443–486. [Google Scholar] [CrossRef]
- Tauer, K.; Hernandez, H.; Kozempel, S.; Lazareva, O.; Nazaran, P. Towards a consistent mechanism of emulsion polymerization—new experimental details. Colloid Polym. Sci. 2008, 286, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Kanda, Y.; Higashitani, K. Initial growth process of polystyrene particle investigated by AFM. J. Colloid Interface Sci. 2006, 299, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Kanda, Y.; Higashitani, K. Molecular-scale observation of formation of nuclei in soap-free polymerization of styrene. Langmuir 2004, 20, 4400–4405. [Google Scholar] [CrossRef] [PubMed]
- Feeney, P.J.; Napper, D.H.; Gilbert, R.G. Surfactant-Free Emulsion Polymerizations: Predictions of the coagulative nucleation theory. Macromolecules 1987, 20, 2922–2930. [Google Scholar] [CrossRef]
- Feeney, P.J.; Napper, D.H.; Gilbert, R.G. Coagulative nucleation and particle size distributions in emulsion polymerization. Macromolecules 1984, 17, 2520–2529. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nakayama, M.; Kanda, Y.; Higashitani, K. Growth mechanism of soap-free polymerization of styrene investigated by AFM. J. Colloid Interface Sci. 2006, 297, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Dai, L.L. Synthesis of polystyrene-silica composite particles via one-step nanoparticle-stabilized emulsion polymerization. J. Colloid Interface Sci. 2009, 333, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Julian, B.; Belleville, P.; Popall, M. Applications of hybrid organic-inorganic nanocomposites. J. Mater. Chem. 2005, 15, 3559–3592. [Google Scholar] [CrossRef]
- Schild, H.G. Poly(N-isopropylacrylamide): Experiment, theory and application. Prog. Polym. Sci. 1992, 17, 163–249. [Google Scholar] [CrossRef]
- Qiu, Y.; Park, K. Environment-sensitive hydrogels for drug delivery. Adv. Drug Deliv. Rev. 2001, 53, 321–339. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Fraylich, M.; Saunders, B.R. Thermoresponsive copolymers: From fundamental studies to applications. Colloid Polym. Sci. 2009, 287, 627–643. [Google Scholar] [CrossRef]
- Duracher, D.; Sauzedde, F.; Elaissari, A.; Perrin, A.; Pichot, C. Cationic amino-containing N-isopropylacrylamide-styrene copolymer latex particles: 1 - particle size and morphology vs. polymerization process. Colloid Polym. Sci. 1998, 276, 219–231. [Google Scholar] [CrossRef]
- Huang, D.M.; Hung, Y.; Ko, B.S.; Hsu, S.C.; Chen, W.H.; Chien, C.L.; Tsai, C.P.; Kuo, C.T.; Kang, J.C.; Yang, C.S.; Mou, C.Y.; Chen, Y.C. Highly efficient cellular labeling of mesoporous nanoparticles in human mesenchymal stem cells: Implication for stem cell tracking. FASEB J. 2005, 19, 2014–2016. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.C.; Latouche, J.; Krause, A.; Heston, W.D.W.; Bander, N.H.; Sadelain, M. Cancer patient T cells genetically targeted to prostate-specific membrane antigen specifically lyse prostate cancer cells and release cytokines in response to prostate-specific membrane antigen. Neoplasia 1999, 1, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Han, M.G.; Armes, S.P. Preparation and characterization of polypyrrole-silica colloidal nanocomposites in water-methanol mixtures. J. Colloid Interface Sci. 2003, 262, 418–427. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ma, H.; Luo, M.; Sanyal, S.; Rege, K.; Dai, L.L. The One-Step Pickering Emulsion Polymerization Route for Synthesizing Organic-Inorganic Nanocomposite Particles. Materials 2010, 3, 1186-1202. https://doi.org/10.3390/ma3021186
Ma H, Luo M, Sanyal S, Rege K, Dai LL. The One-Step Pickering Emulsion Polymerization Route for Synthesizing Organic-Inorganic Nanocomposite Particles. Materials. 2010; 3(2):1186-1202. https://doi.org/10.3390/ma3021186
Chicago/Turabian StyleMa, Huan, Mingxiang Luo, Sriya Sanyal, Kaushal Rege, and Lenore L. Dai. 2010. "The One-Step Pickering Emulsion Polymerization Route for Synthesizing Organic-Inorganic Nanocomposite Particles" Materials 3, no. 2: 1186-1202. https://doi.org/10.3390/ma3021186
APA StyleMa, H., Luo, M., Sanyal, S., Rege, K., & Dai, L. L. (2010). The One-Step Pickering Emulsion Polymerization Route for Synthesizing Organic-Inorganic Nanocomposite Particles. Materials, 3(2), 1186-1202. https://doi.org/10.3390/ma3021186