Insight into the Broad Field of Polymer Nanocomposites: From Carbon Nanotubes to Clay Nanoplatelets, via Metal Nanoparticles

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction to Polymer Nanocomposites
2. Polymer/Carbon Nanotubes Nanocomposites
2.1. Polystyrene/Carbon Nanotubes Composites
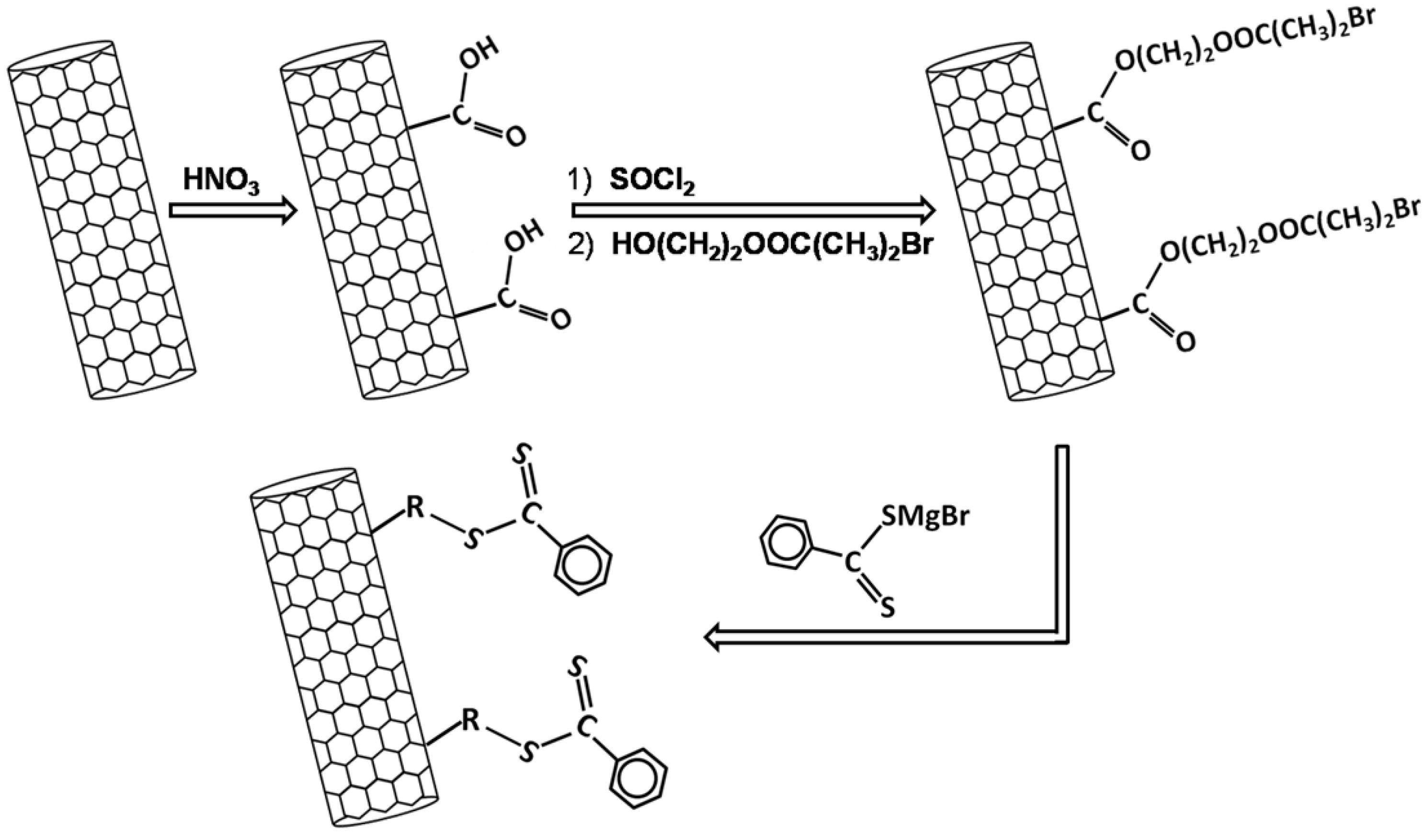
2.2. Poly(Ethylene Oxide)/Carbon Nanotubes Composites
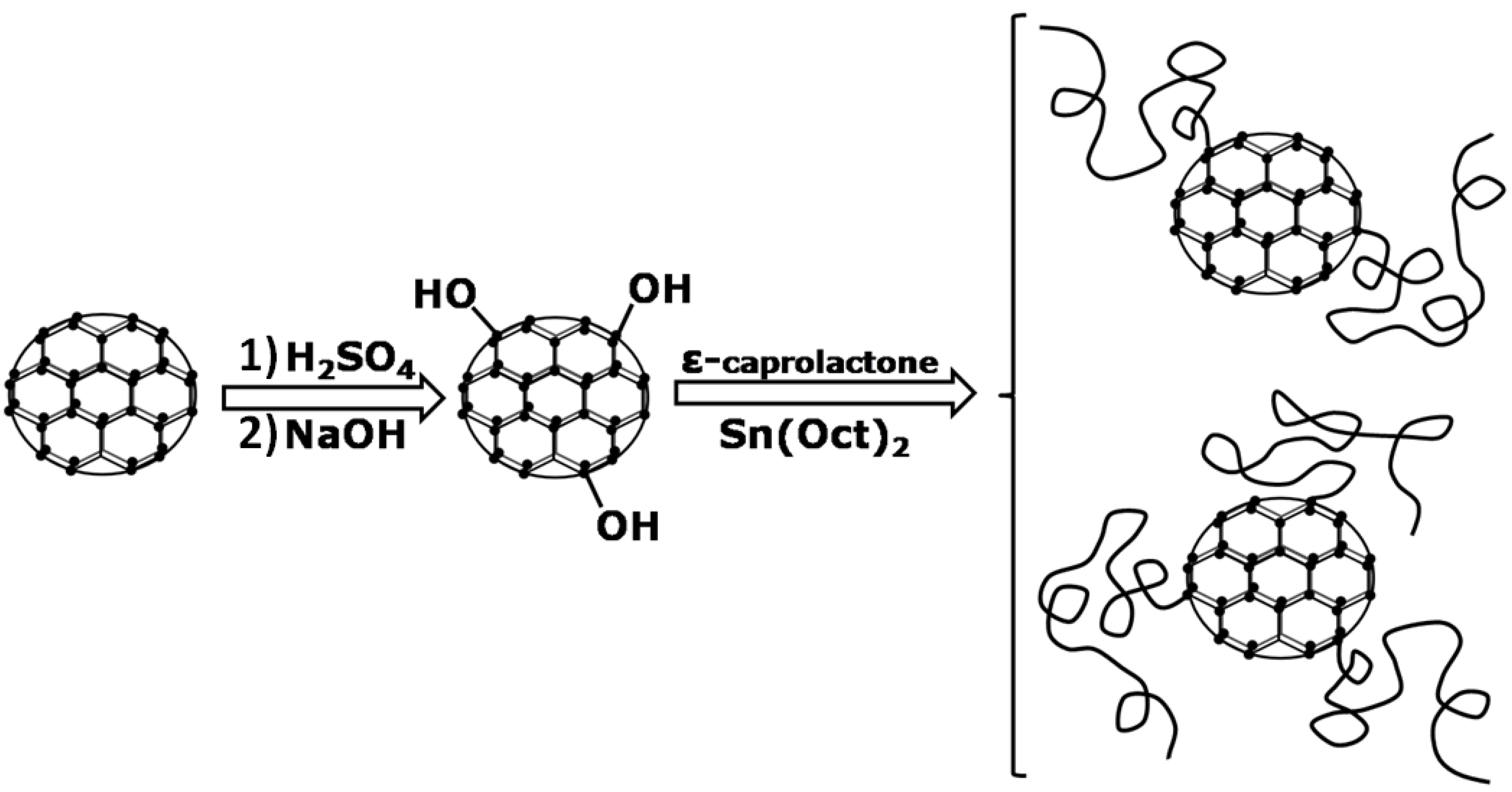
2.3. Poly(ε-Caprolactone)/Carbon Nanotubes Composites
2.4. Polypropylene/Carbon Nanotubes Composites
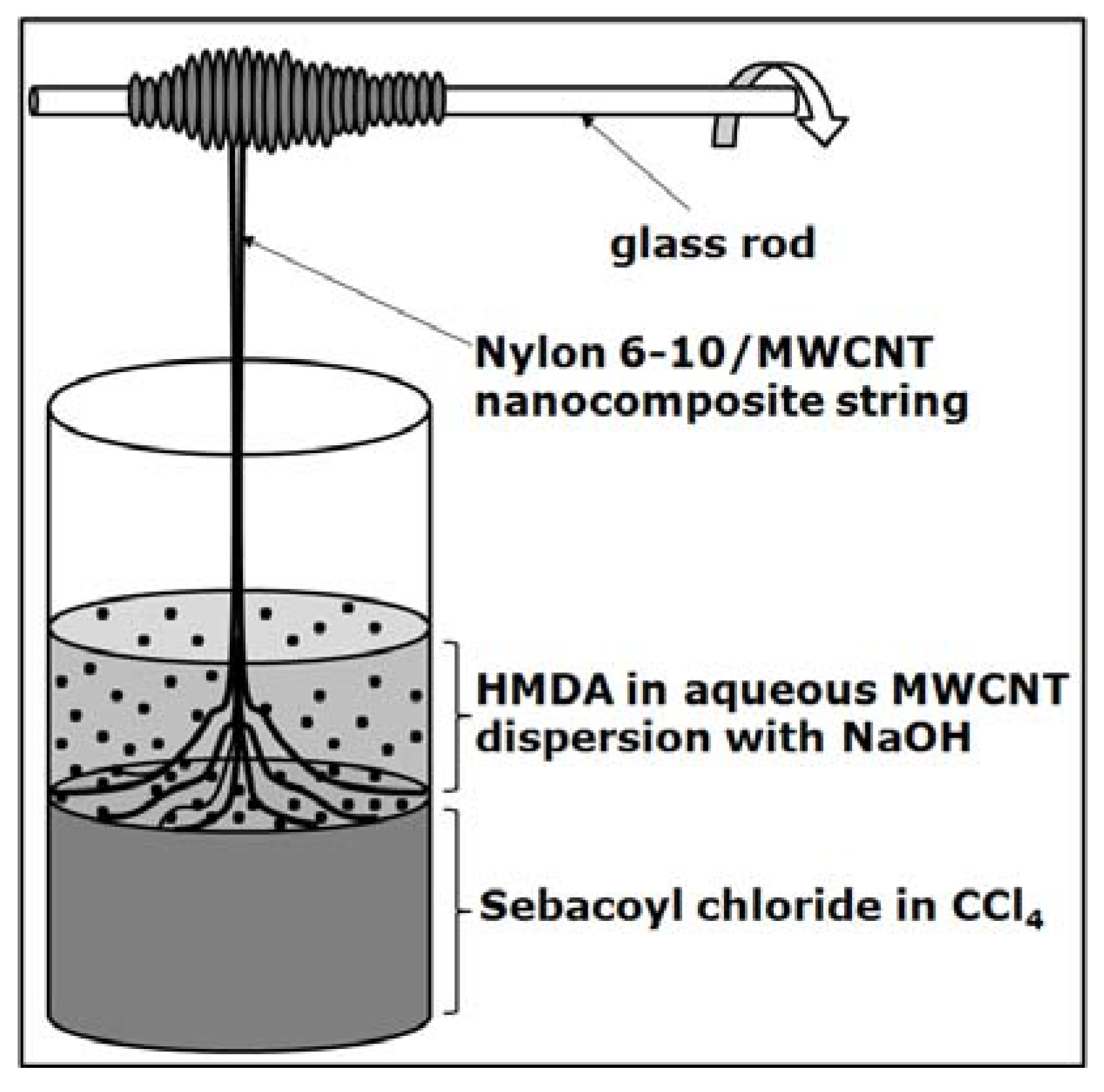
2.5. Nylon/Carbon Nanotubes Composites
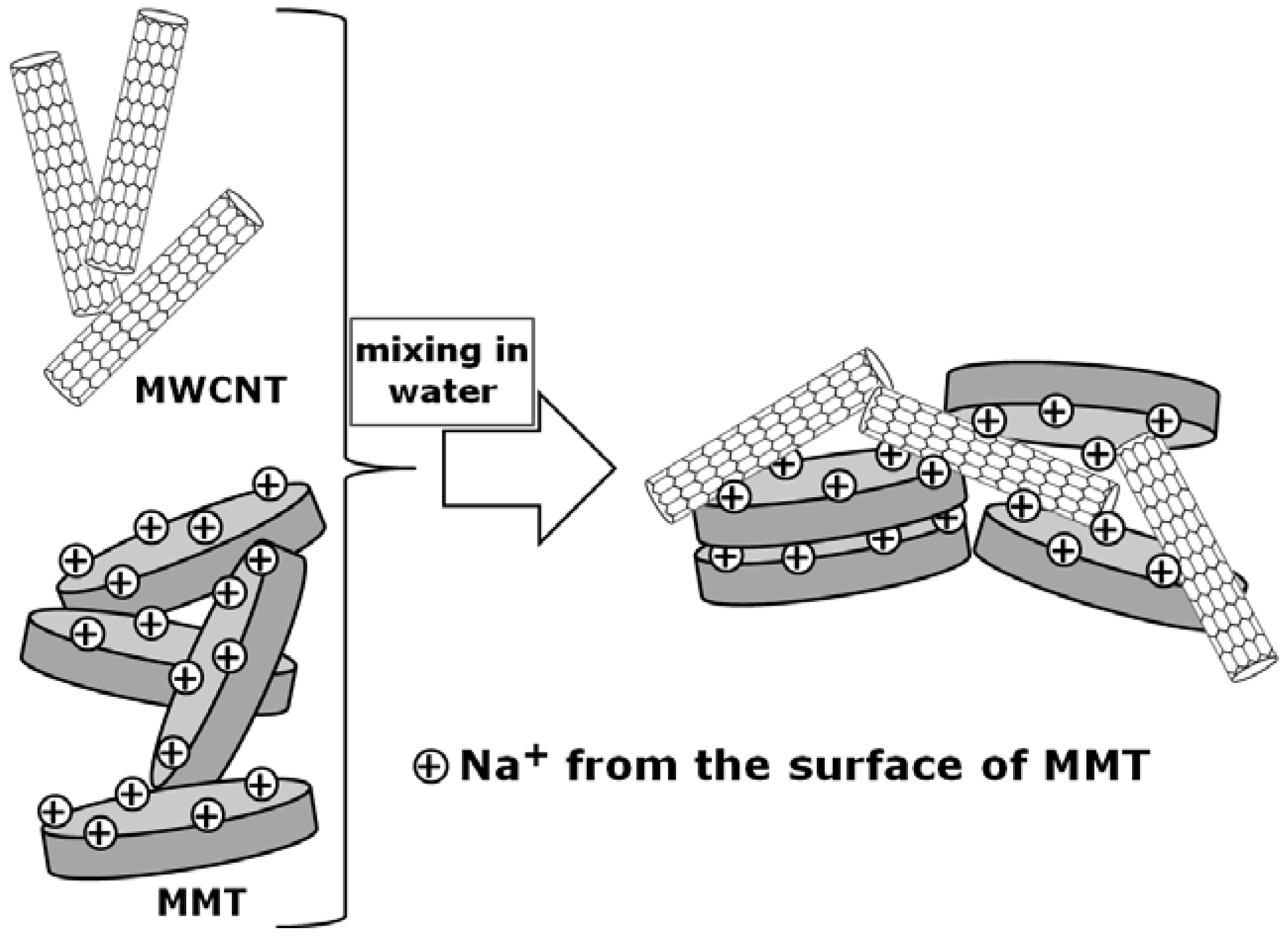
2.6. Poly(Ethylene Terephthalate)/Carbon Nanotubes Composites
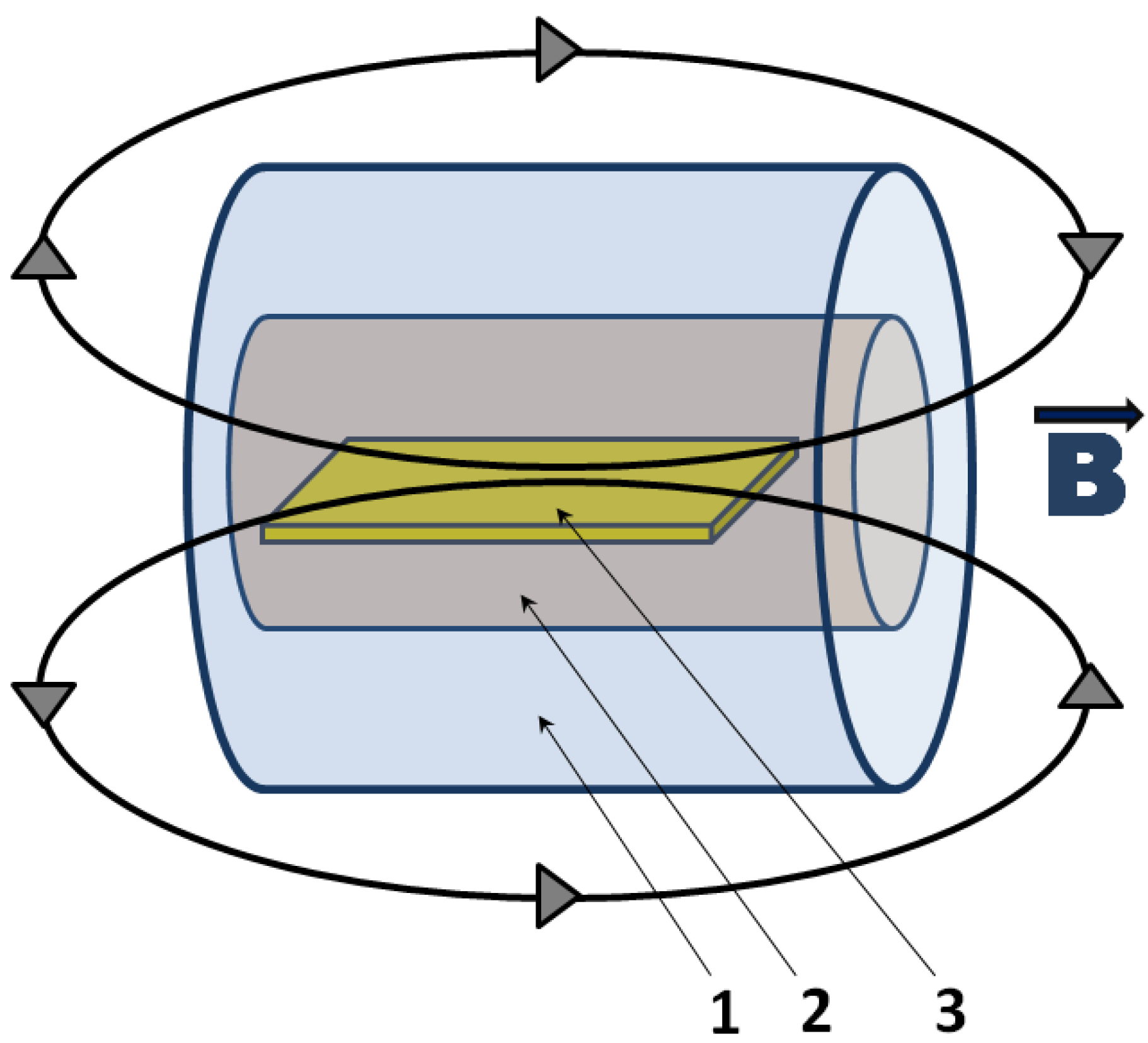
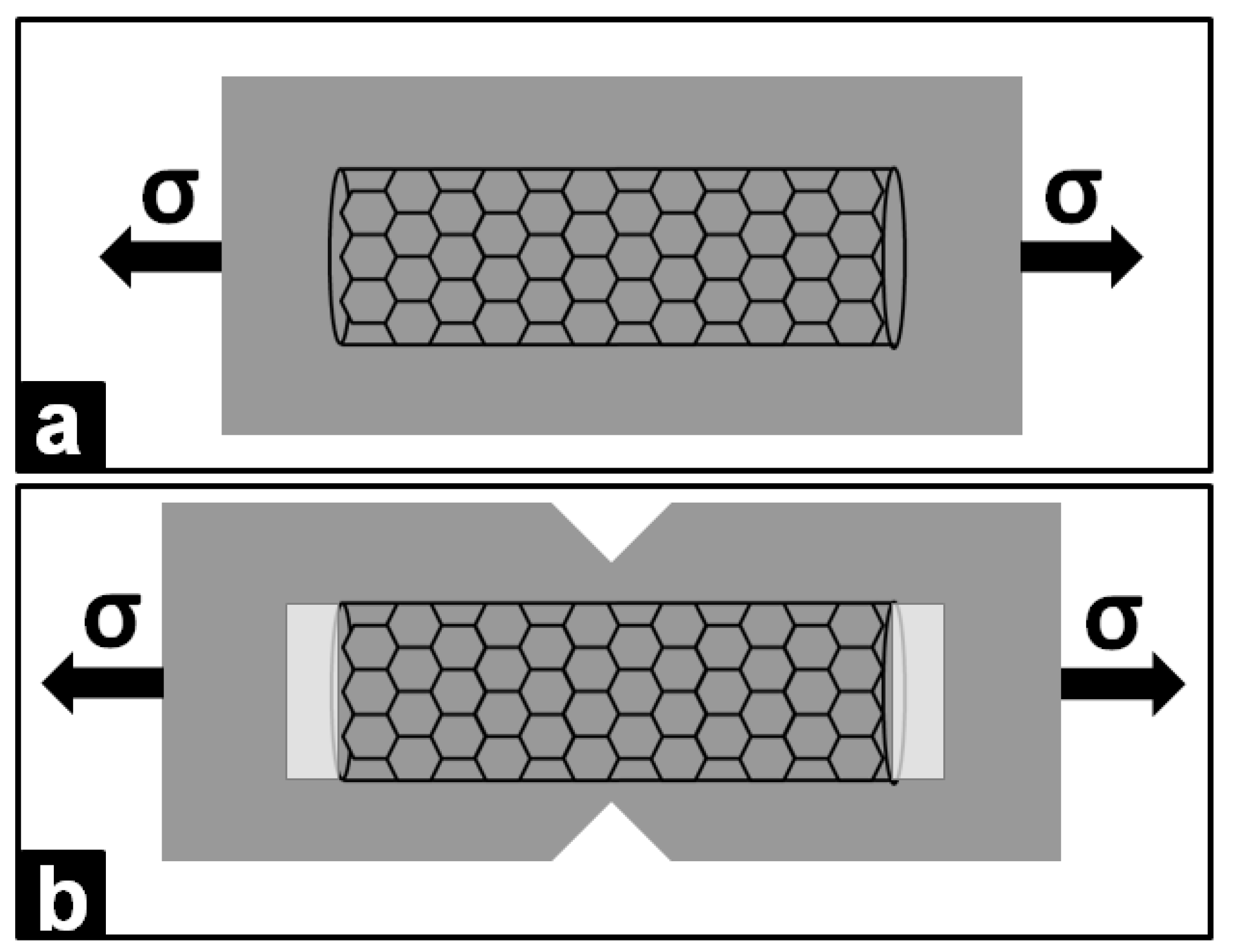
3. Polymer/Metal Nanocomposites
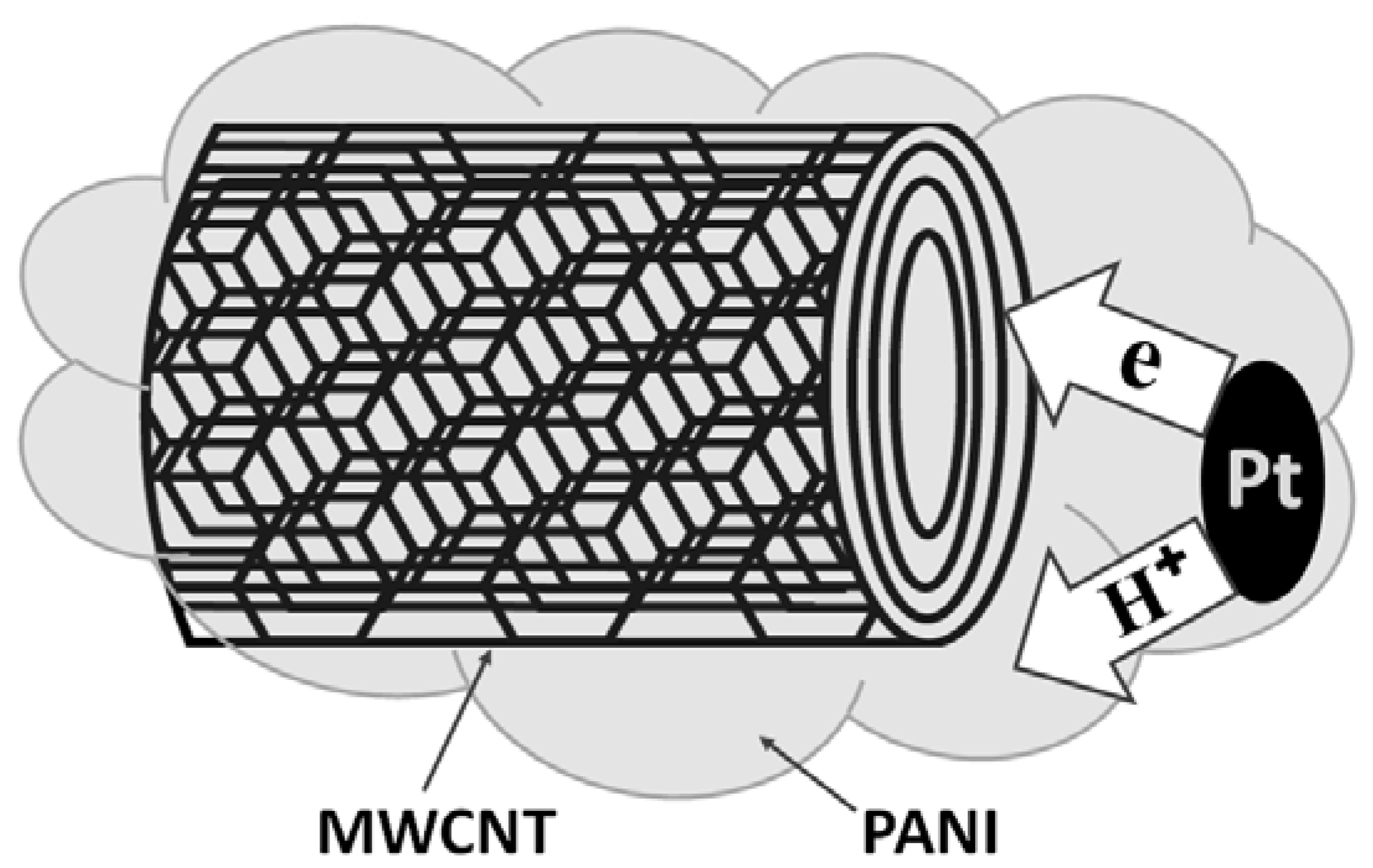
3.1. Polymer/Platinum Nanocomposites
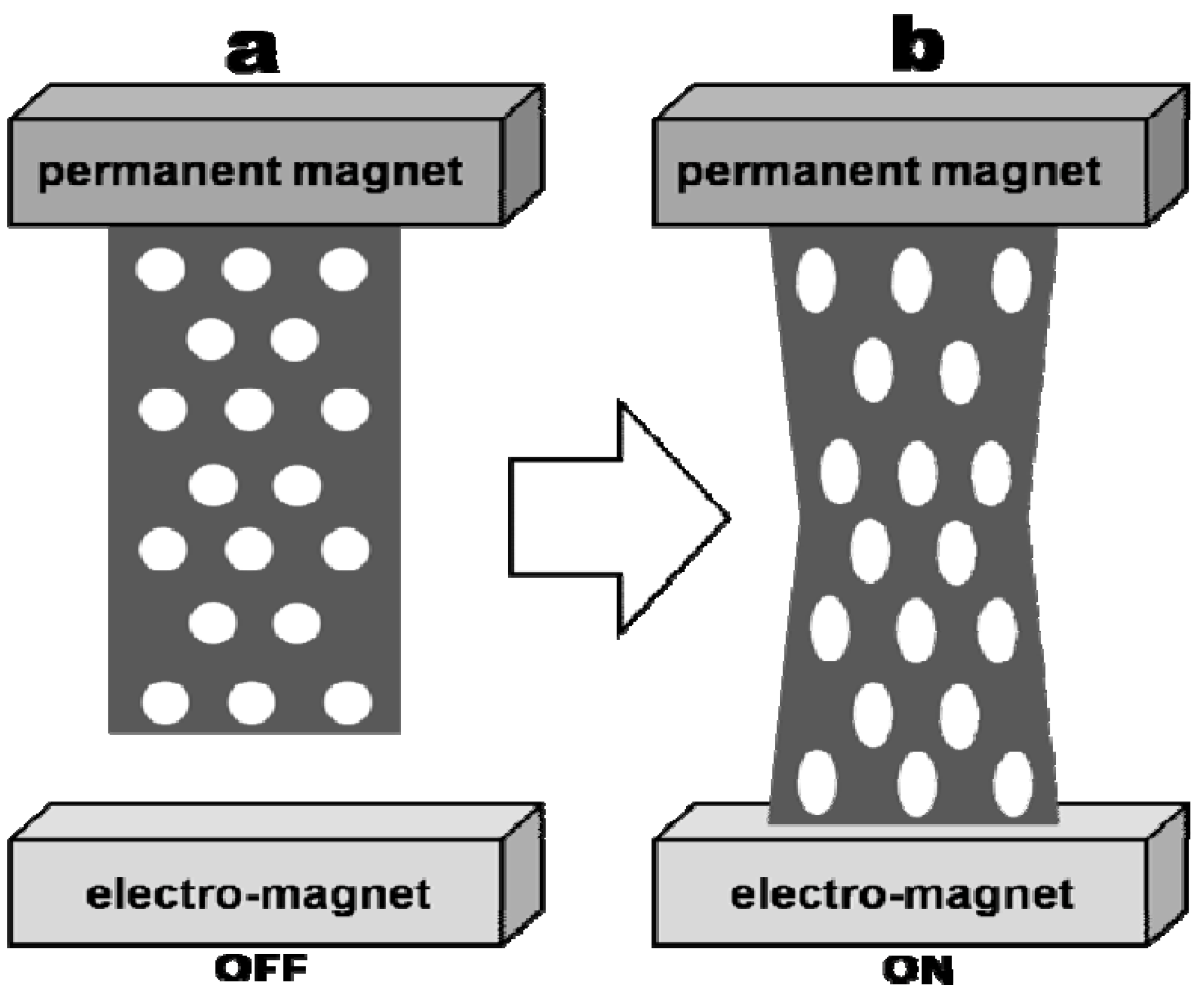
3.2. Polymer/Cobalt Nanocomposites
3.3. Polymer/Nickel Nanocomposites
3.4. Polymer/Silver Nanocomposites
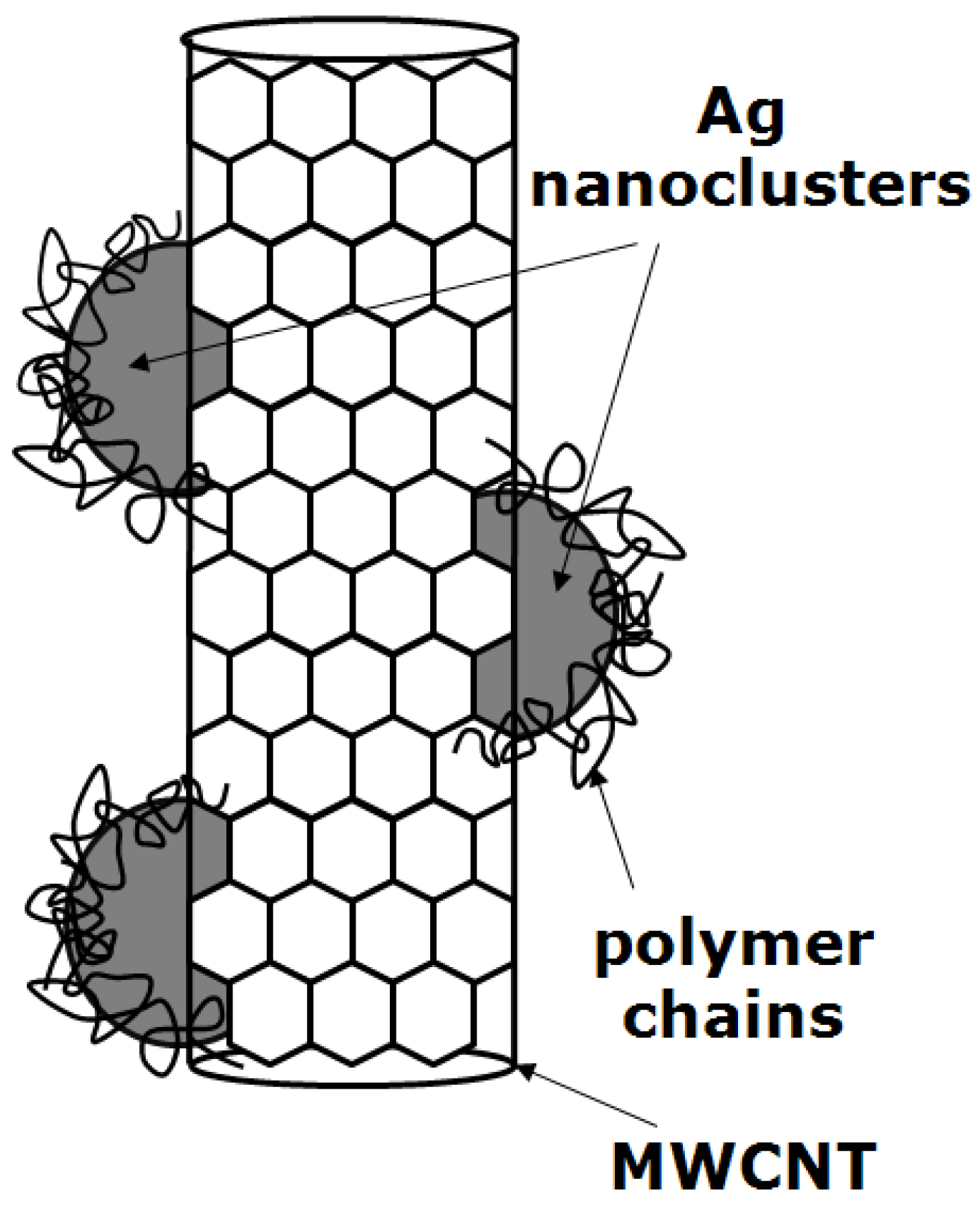
3.5. Polymer/Palladium Nanocomposites
3.6. Polymer/Gold Nanocomposites

3.7. Other Metals/Polymer Nanocomposites
4. Polymer/Clay Nanocomposites
4.1. Poly(Urethane Urea) Composites
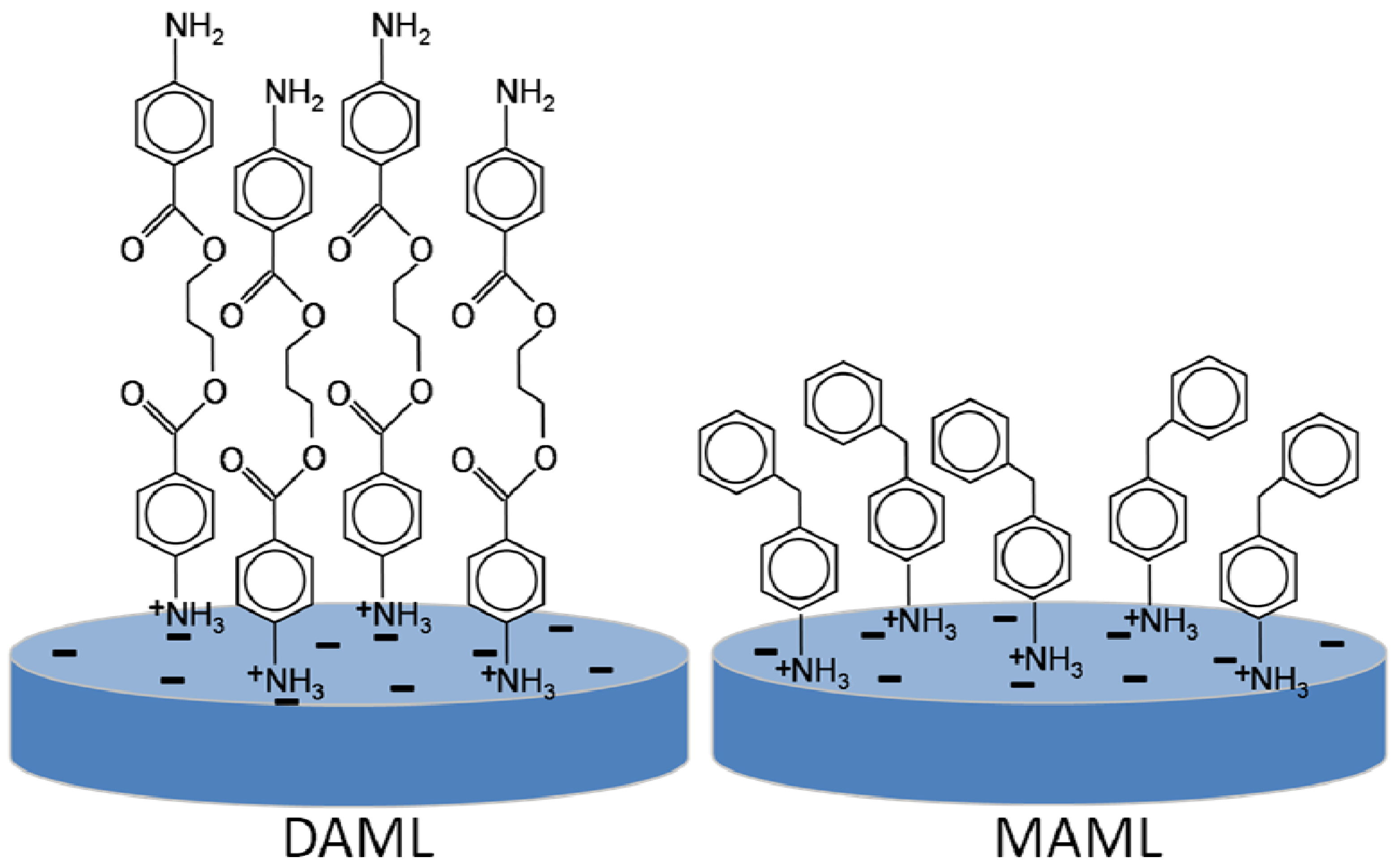
4.2. Poly(Methyl Methacrylate)/Laponite® Composites
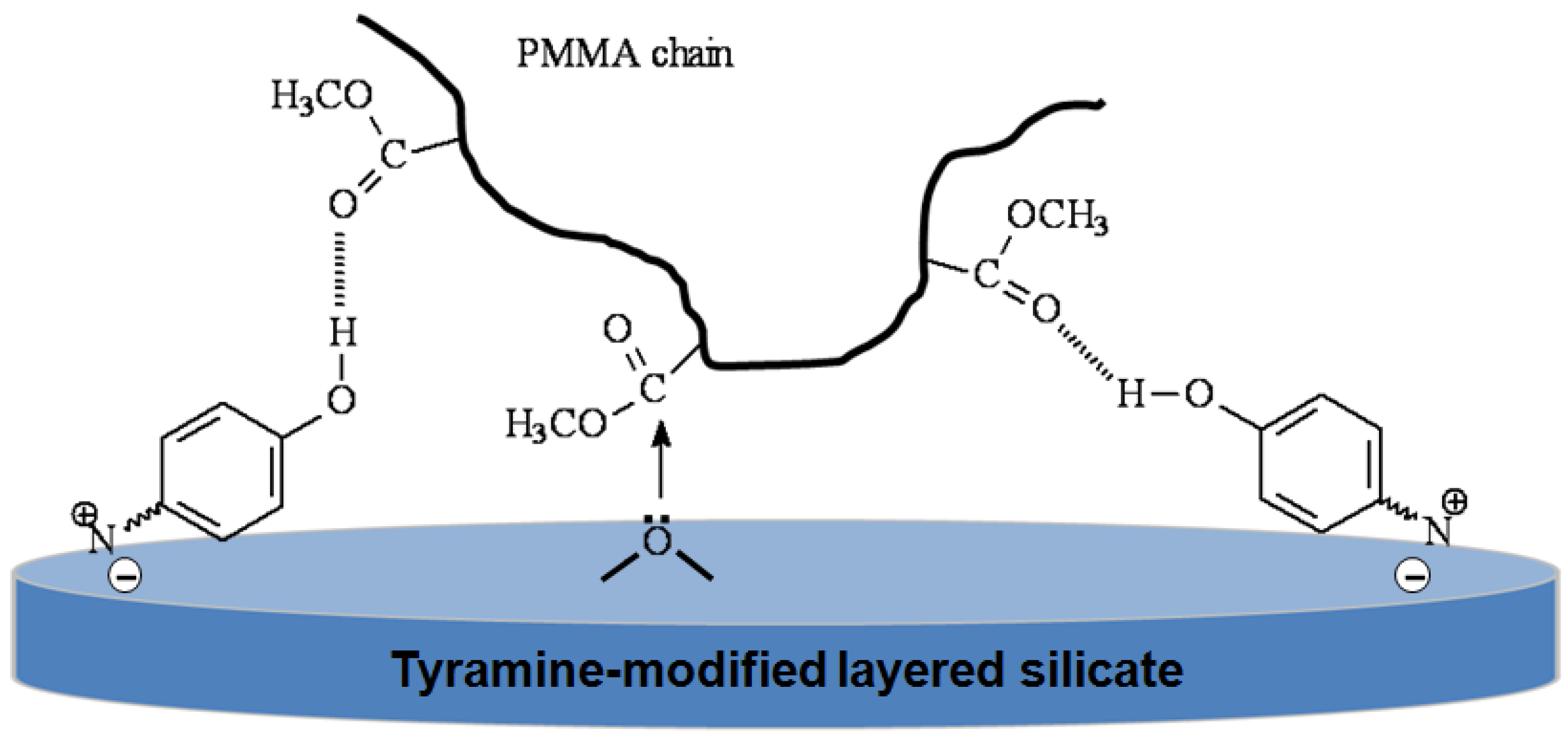
4.3. Polystyrene/Laponite® Composites
4.4. Poly(Ethylene Oxide)/Laponite® Composites
4.5. Diblock and Triblock Copolymers/Laponite® Composites
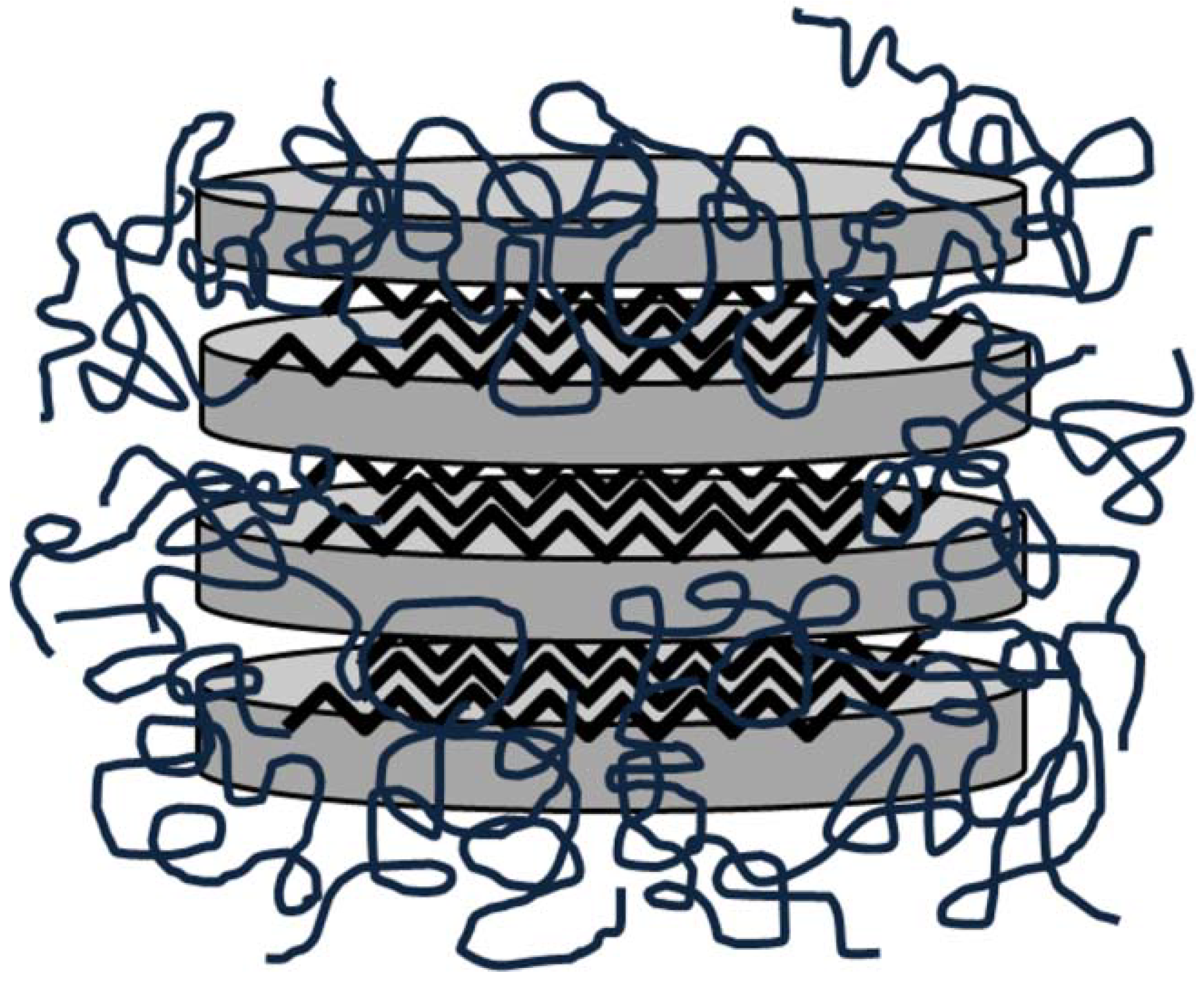
4.6. Polyurethane/Montmorillonite Composites
4.7. Poly(ε-Caprolactone)/Montmorillonite Composites
4.8. Polylactic Acid and/or Poly(Lactic-co-Glycolic Acid)/Montmorillonite Composites
4.9. Poly(Ethylene Oxide)/Montmorillonite Composites
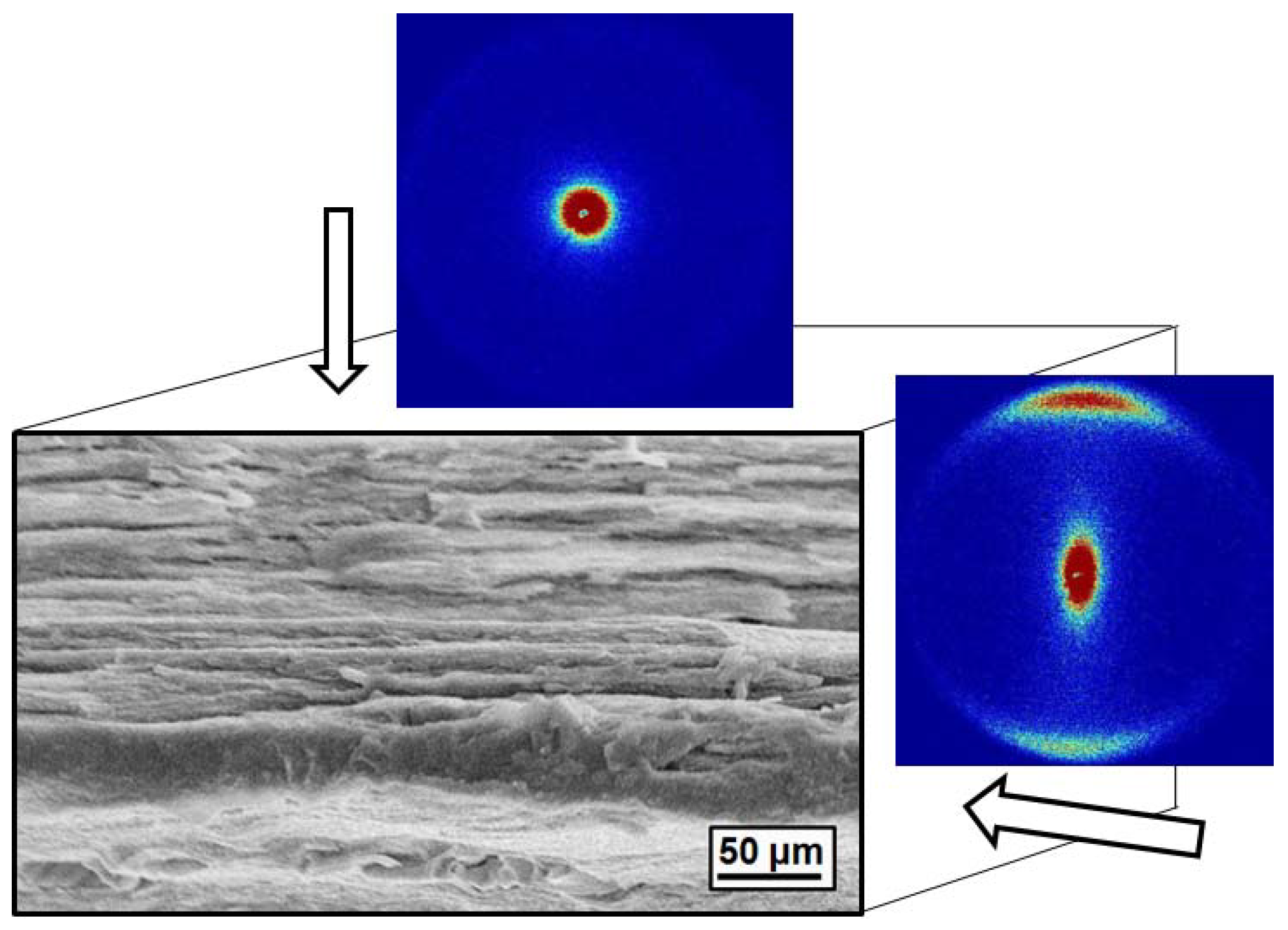
4.10. Polypropylene/Montmorillonite Composites
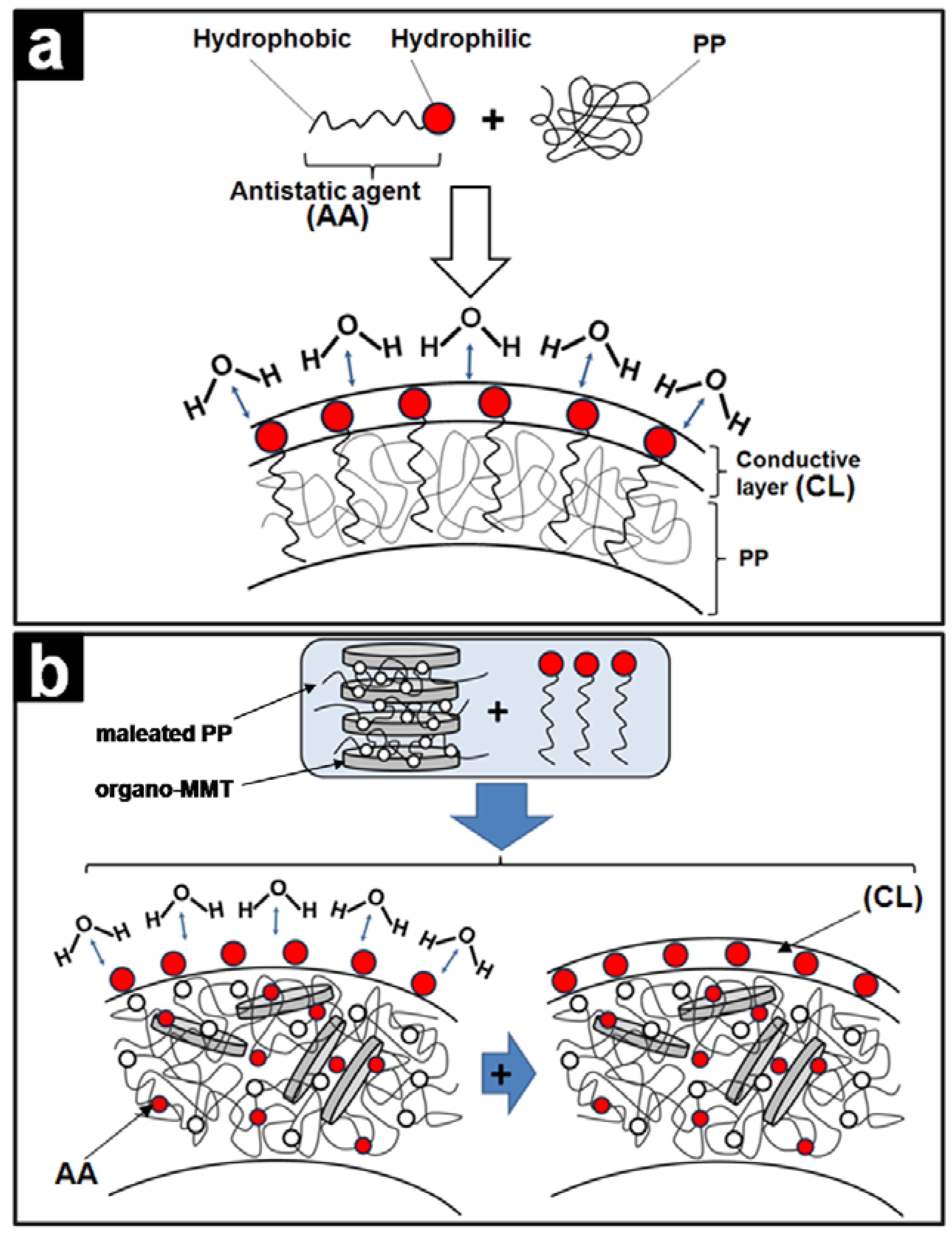
5. Conclusions
Acknowledgements
References
- Loizou, E.; Butler, P.; Porcar, L.; Kesselman, E.; Talmon, Y.; Dundigalla, A.; Schmidt, G. Large scale structures in nanocomposite hydrogels. Macromolecules 2005, 38, 2047–2049. [Google Scholar] [CrossRef]
- Loizou, E.; Butler, P.; Porcar, L.; Schmidt, G. Dynamic responses in nanocomposite hydrogels. Macromolecules 2006, 39, 1614–1619. [Google Scholar] [CrossRef]
- Schmidt, G.; Nakatani, A.I.; Butler, P.D.; Karim, A.; Han, C.C. Shear orientation of viscoelastic polymer-clay solutions probed by flow birefringence and SANS. Macromolecules 2000, 33, 7219–7222. [Google Scholar] [CrossRef]
- Schmidt, G.; Nakatani, A.I.; Han, C.C. Rheology and flow-birefringence from viscoelastic polymer-clay solutions. Rheol. Acta 2002, 41, 45–54. [Google Scholar] [CrossRef]
- Stefanescu, E.A.; Dundigalla, A.; Ferreiro, V.; Loizou, E.; Porcar, L.; Negulescu, I.; Garno, J.; Schmidt, G. Supramolecular structures in nanocomposite multilayered films. Phys. Chem. Chem. Phys. 2006, 8, 1739–1746. [Google Scholar] [CrossRef] [PubMed]
- Stefanescu, E.A.; Daly, W.H.; Negulescu, I.I. Hybrid polymer/clay nanocomposites : Effect of clay size on the structure of multilayered films. Macromol. Mater. Eng. 2008, 293, 651–656. [Google Scholar] [CrossRef]
- Dai, X.; Xu, J.; Guo, X.; Lu, Y.; Shen, D.; Zhao, N.; Luo, X.; Zhang, X. Study on structure and orientation action of polyurethane nanocomposites. Macromolecules 2004, 37, 5615–5623. [Google Scholar] [CrossRef]
- Chatterjee, T.; Mitchell, C.A.; Hadjiev, V.G.; Krishnamoorti, R. Hierarchical polymer-nanotube composites. Adv. Mater. 2007, 19, 3850–3853. [Google Scholar] [CrossRef]
- Jiang, H.; Moon, K.; Li, Y.; Wong, C.P. Surface functionalized silver nanoparticles for ultrahigh conductive polymer composites. Chem. Mater. 2006, 18, 2969–2973. [Google Scholar] [CrossRef]
- Keng, P.Y.; Shim, I.; Korth, B.D.; Douglas, J.F.; Pyun, J. Synthesis and self-assembly of polymer-coated ferromagnetic nanoparticles. ACS Nano 2007, 1, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Kwong, H.Y.; Wong, Y.W.; Wong, K.H. Temperature dependence of magnetoresistivity of cobalt-polytetrafluoroethylene granular composite films. J. Appl. Phys. 2007, 102, 114303. [Google Scholar] [CrossRef]
- Pirkkalainen, K.; Leppänen, K.; Vainio, U.; Webb, M.A.; Elbra, T.; Kohout, T.; Nykänen, A.; Ruokolainen, J.; Kotelnikova, N.; Serimaa, R. Nanocomposites of magnetic cobalt nanoparticles and cellulose. Eur. Phys. J. D 2008, 49, 333–342. [Google Scholar] [CrossRef]
- Mohanraj, G.T.; Dey, P.K.; Chaki, T.K.; Chakraborty, A.; Khastgir, D. Effect of temperature, pressure, and composition on DC resistivity and AC conductivity of conductive styrene-butadiene rubber-particulate metal alloy nanocomposites. Polym. Compos. 2007, 28, 696–704. [Google Scholar] [CrossRef]
- Panda, M.; Srinivas, V.; Thakur, A.K. Surface and interfacial effect of filler particle on electrical properties of polyvinyledene fluoride/nickel composites. Appl. Phys. Lett. 2008, 93, 242908. [Google Scholar] [CrossRef]
- Valmikanathan, O.P.; Ostroverkhova, O.; Mulla, I.S.; Vijayamohanan, K.; Atre, S.V. The effect of synthesis procedure on the structure and properties of palladium/polycarbonate nanocomposites. Polymer 2008, 49, 3413–3418. [Google Scholar] [CrossRef]
- Tang, Z.Y.; Kotov, N.A. One-dimensional assemblies of nanoparticles: Preparation, properties, and promise. Adv. Mater. 2005, 17, 951–962. [Google Scholar] [CrossRef]
- Tripp, S.L.; Dunin-Borkowski, R.E.; Wei, A. Flux closure in self-assembled cobalt nanoparticle rings. Angew. Chem., Int. Ed. 2003, 42, 5591–5593. [Google Scholar] [CrossRef]
- Farrell, D.; Ding, Y.; Majetich, S.A.; Sanchez-Hanke, C.; Kao, C.C. Structural ordering effects in Fe nanoparticle two- and three-dimensional arrays. J. Appl. Phys. 2004, 95, 6636–6638. [Google Scholar] [CrossRef]
- Hilgendorff, M.; Tesche, B.; Giersig, M. Creation of 3-D crystals from single cobalt nanoparticles in external magnetic fields. Aust. J. Chem. 2001, 54, 497–501. [Google Scholar] [CrossRef]
- Carotenuto, G.; Martorana, B.; Perlo, P.; Nicolais, L. A universal method for the synthesis of metal and metal sulfide clusters embedded in polymer matrices. J. Mater. Chem. 2003, 13, 2927–2930. [Google Scholar] [CrossRef]
- Jose, M.V.; Steinert, B.W.; Thomas, V.; Dean, D.R.; Abdalla, M.A.; Price, G.; Janowski, G.M. Morphology and mechanical properties of Nylon 6/MWNT nanofibers. Polymer 2007, 48, 1096–1104. [Google Scholar] [CrossRef]
- Chrissafis, K.; Antoniadis, G.; Paraskevopoulos, K.M.; Vassiliou, A.; Bikiaris, D.N. Comparative study of the effect of different nanoparticles on the mechanical properties and thermal degradation mechanism of in situ prepared poly(E-caprolactone) nanocomposites. Compos. Sci. Technol. 2007, 67, 2165–2174. [Google Scholar] [CrossRef]
- Thomassin, J.M.; Lou, X.; Pagnoulle, C.; Saib, A.; Bednarz, L.; Huynen, I.; Jerome, R.; Detrembleur, c. multiwalled carbon nanotube/poly(epsilon-caprolactone) nanocomposites with exceptional electromagnetic interference shielding properties. J. Phys. Chem. C 2007, 111, 11186–11192. [Google Scholar] [CrossRef]
- Abraham, T.N.; Debdatta, R.; Siengchin, S.; Karger-Kocsis, J. Rheological and thermal properties of poly(ethylene oxide)/multiwall carbon nanotube composites. J. Appl. Polym. Sci. 2008, 110, 2094–2101. [Google Scholar] [CrossRef]
- Narh, A.K.; Jallo, L.; Rhee, K., Y. The effect of carbon nanotube agglomeration on the thermal and mechanical properties of polyethylene oxide. Polym. Compos. 2008, 29, 809–817. [Google Scholar] [CrossRef]
- Song, Y.S. Effect of surface treatment for carbon nanotubes on morphological and rheological properties of poly(ethylene oxide) nanocomposites. Polym. Eng. Sci. 2006, 46, 1350–1357. [Google Scholar] [CrossRef]
- Averett, R.D.; Realff, M.L.; Jacob, K.I. The effects of fatigue and residual strain on the mechanical behavior of poly(ethylene terephthalate) unreinforced and nanocomposite fibers. Compos. Part A Appl. Sci. Manuf. 2009, 40, 709–723. [Google Scholar] [CrossRef]
- Steinert, B.W.; Dean, D.R. Magnetic field alignment and electrical properties of solution cast PET-carbon nanotube composite films. Polymer 2009, 50, 898–904. [Google Scholar] [CrossRef]
- Brosse, A.C.; Tence-Girault, S.; Piccione, P.M.; Leibler, L. Effect of multi-walled carbon nanotubes on the lamellae morphology of polyamide-6. Polymer 2008, 49, 4680–4686. [Google Scholar] [CrossRef]
- Li, Y.; Shimizu, H. Conductive PVDF/PA6/CNTs nanocomposites fabricated by dual formation of cocontinuous and nanodispersion structures. Macromolecules 2008, 41, 5339–5344. [Google Scholar] [CrossRef]
- Mitchell, C.A.; Krishnamoorti, R. Dispersion of single-walled carbon nanotubes in poly(epsilon-caprolactone). Macromolecules 2007, 40, 1538–1545. [Google Scholar] [CrossRef]
- Chatterjee, T.; Krishnamoorti, R. Steady Shear response of carbon nanotube networks dispersed in poly(ethylene oxide). Macromolecules 2008, 41, 5333–5338. [Google Scholar] [CrossRef]
- Chatterjee, T.; Yurekli, K.; Hadjiev, V.G.; Krishnamoorti, R. Single-Walled carbon nanotube dispersions in poly(ethylene oxide). Adv. Funct. Mater. 2005, 15, 1832–1838. [Google Scholar] [CrossRef]
- Yoo, H.J.; Jung, Y.C.; Cho, J.W. Effect of interaction between poly(ethylene terephthalate) and carbon nanotubes on the morphology and properties of their nanocomposites. J. Polym. Sci., Part B: Polym. Phys. 2008, 46, 900–910. [Google Scholar] [CrossRef]
- Ahn, B.W.; Chi, Y.S.; Kang, T.J. Preparation and characterization of multi-walled carbon nanotube/poly(ethylene terephthalate) nanoweb. J. Appl. Polym. Sci. 2008, 110, 4055–4063. [Google Scholar] [CrossRef]
- Wang, K.; Li, W.W.; Gao, C. Poly(epsilon-caprolactone)-functionalized carbon nanofibers by surface-initiated ring-opening polymerization. J. Appl. Polym. Sci. 2007, 105, 629–640. [Google Scholar] [CrossRef]
- Wakamatsu, N.; Takamori, H.; Fujigaya, T.; Nakashima, N. Self-Organized single-walled carbon nanotube conducting thin films with honeycomb structures on flexible plastic films. Adv. Funct. Mater. 2009, 19, 311–316. [Google Scholar] [CrossRef]
- Chen, H.; Liu, Z.; Cebe, P. Chain confinement in electrospun nanofibers of PET with carbon nanotubes. Polymer 2009, 50, 872–880. [Google Scholar] [CrossRef]
- Hu, G.J.; Feng, X.Y.; Zhang, S.M.; Yang, M.S. Crystallization behavior of poly(ethylene terephthalate)/multiwalled carbon nanotubes composites. J. Appl. Polym. Sci. 2008, 108, 4080–4089. [Google Scholar] [CrossRef]
- Nyczyk, A.; Hasik, M.; Turek, W.; Sniechota, A. Nanocomposites of polyaniline, its derivatives and platinum prepared using aqueous Pt sol. Synth. Met. 2009, 159, 561–567. [Google Scholar] [CrossRef]
- Zotti, G.; Vercelli, B.; Berlin, A. Gold nanoparticle linking to polypyrrole and polythiophene: monolayers and multilayers. Chem. Mater. 2008, 20, 6509–6516. [Google Scholar] [CrossRef]
- Huang, S.W.; Neoh, K.G.; Kang, E.T.; Han, H.S.; Tan, K.L. Palladium-containing polyaniline and polypyrrole microparticles. J. Mater. Chem. 1998, 8, 1743–1748. [Google Scholar] [CrossRef]
- Wilson, J.L.; Poddar, P.; Frey, N.A.; Srikanth, H.; Mohomed, K.; Harmon, J.P.; Kotha, S.; Wachsmuth, J. Synthesis and magnetic properties of polymer nanocomposites with embedded iron nanoparticles. J. Appl. Phys. 2004, 95, 1439. [Google Scholar] [CrossRef]
- Yurkov, G.; Fionov, A.; Koksharov, Y.; Koleso, V.; Gubin, S. Electrical and magnetic properties of nanomaterials containing iron or cobalt nanoparticles. Inorg. Mater. 2007, 43, 834–844. [Google Scholar] [CrossRef]
- Sarkar, A.; Kapoor, S.; Yashwant, G.; Salunke, H.G.; Mukherjee, T. Preparation and characterization of ultrafine Co and Ni particles in a polymer matrix. J. Phys. Chem. B 2005, 109, 7203–7207. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Sun, J.; Liu, M.; Chen, Q. Mechanical strength of carbon nanotube-nickel nanocomposites. Nanotechnology 2007, 18, 505704–505704. [Google Scholar] [CrossRef]
- Balan, L.; Jin, M.; Malval, J.P.; Chaumeil, H.; Defoin, A.; Vidal, L. Fabrication of silver nanoparticle-embedded polymer promoted by combined photochemical properties of a 2,7-diaminofluorene derivative dye. Macromolecules 2008, 41, 9359–9365. [Google Scholar] [CrossRef]
- Dundigalla, A.; Lin Gibson, S.; Ferreiro, V.; Malwitz, M.M.; Schmidt, G. Unusual multilayered structures in PEO/Laponite® nanocomposite films. Macromol. Rapid Commun. 2005, 26, 143–149. [Google Scholar] [CrossRef]
- Elmahdy, M.M.; Chrissopoulou, K.; Afratis, A.; Floudas, G.; Anastasiadis, S.H. Effect of confinement on polymer segmental motion and ion mobility in PEO/layered silicate nanocomposites. Macromolecules 2006, 39, 5170–5173. [Google Scholar] [CrossRef]
- Inyang, H.I.; Bae, S.; Mbamalu, G.; Park, S.-W. Aqueous polymer effects on volumetric swelling of Na-montmorillonite. J. Mater. Civil Eng. 2007, 19:1, 84–90. [Google Scholar] [CrossRef]
- Loiseau, A.; Tassin, J.F. Model nanocomposites based on Laponite® and poly(elhylene oxide): preparation and rheology. Macromolecules 2006, 39, 9185–9191. [Google Scholar] [CrossRef]
- Loyens, W.; Maurer, F.H.J.; Jannasch, P. Melt-compounded salt-containing poly(ethylene oxide)/clay nanocomposites for polymer electrolyte membranes. Polymer 2005, 46, 7334–7345. [Google Scholar] [CrossRef]
- Qiu, W.L.; Pyda, M.; Nowak-Pyda, E.; Habenschuss, A.; Wunderlich, B. Reversibility between glass and melting transitions of poly(oxyethylene). Macromolecules 2005, 38, 8454–8467. [Google Scholar] [CrossRef]
- Stefanescu, E.A.; Schexnailder, P.J.; Dundigalla, A.; Negulescu, I.I.; Schmidt, G. Structure and thermal properties of multilayered Laponite®/PEO nanocomposite films. Polymer 2006, 47, 7339–7348. [Google Scholar] [CrossRef]
- Stefanescu, E.A.; Stefanescu, C.; Daly, W.H.; Schmidt, G.; Negulescu, I.I. Hybrid polymer-clay nanocomposites: A mechanical study on gels and multilayered films. Polymer 2008, 49, 3785–3794. [Google Scholar] [CrossRef]
- Arias, C.B.; Zaman, A.A.; Talton, J. Rheological behavior and wear abrasion resistanceof polyethylene oxide/laponie nanocomposites. J. Dispersion Sci. Technol. 2007, 28, 247–254. [Google Scholar] [CrossRef]
- Xu, Y.; Higgins, B.; Brittain, W.J. Bottom-up synthesis of PS–CNF nanocomposites. Polymer 2005, 46, 799–810. [Google Scholar] [CrossRef]
- Wang, C.; Huang, C.-L.; Chen, Y.-C.; Hwang, G.-L.; Tsai, S.-J. Carbon nanocapsules-reinforced syndiotactic polystyrene nanocomposites: Crystallization and morphological features. Polymer 2008, 49, 5564–5574. [Google Scholar] [CrossRef]
- Shen, J.; Zeng, C.; Lee, L.J. Synthesis of polystyrene–carbon nanofibers nanocomposite foams. Polymer 2005, 46, 5218–5224. [Google Scholar] [CrossRef]
- Mu, M.; Walker, A.M.; Torkelson, J.M.; Winey, K.I. Cellular structures of carbon nanotubes in a polymer matrix improve properties relative to composites with dispersed nanotubes. Polymer 2008, 49, 1332–1337. [Google Scholar] [CrossRef]
- Chang; Kisliuk, A.; Rhodes, S.M.; Brittain, W.J.; Sokolov, A.P. Conductivity and mechanical properties of well-dispersed single-wall carbon nanotube/polystyrene composite. Polymer 2006, 47, 7740–7746. [Google Scholar] [CrossRef]
- Nyden, M.R.; Stoliarov, S.I. Calculations of the energy of mixing carbon nanotubes with polymers. Polymer 2007, 49, 635–641. [Google Scholar] [CrossRef]
- Xie, L.; Xu, F.; Qiu, F.; Lu, H.; Yang, Y. Single-walled carbon nanotubes functionalized with high bonding density of polymer layers and enhanced mechanical properties of composites. Macromolecules 2007, 40, 3296–3305. [Google Scholar] [CrossRef]
- Putz, K.; Krishnamoorti, R.; Green, P.F. The role of interfacial interactions in the dynamic mechanical response of functionalized SWNTePS nanocomposites. Polymer 2007, 48, 3540–3545. [Google Scholar] [CrossRef]
- Cipriano, B.H.; Kota, A.K.; Gershon, A.L.; Laskowski, C.J.; Kashiwagi, T.; Bruck, H.A.; Raghavan, S.R. Conductivity enhancement of carbon nanotube and nanofiber-based polymer nanocomposites by melt annealing. Polymer 2008, 49, 4846–4851. [Google Scholar] [CrossRef]
- Kota, A.K.; Cipriano, B.H.; Duesterberg, M.K.; Gershon, A.L.; Powell, D.; Raghavan, S.R.; Bruck, H.A. Electrical and rheological percolation in polystyrene/MWCNT nanocomposites. Macromolecules 2007, 40, 7400–7406. [Google Scholar] [CrossRef]
- Cipiriano, B.H.; Kashiwagi, T.; Raghavan, S.R.; Yang, Y.; Grulke, E.A.; Yamamoto, K.; Shields, J.R.; Douglas, J.F. Effects of aspect ratio of MWNT on the flammability properties of polymer nanocomposites. Polymer 2007, 48, 6086–6096. [Google Scholar] [CrossRef]
- Cui, J.; Wang, W.P.; You, Y.; Liu, C.; Wang, P. Functionalization of multiwalled carbon nanotubes by reversible addition fragmentation chain-transfer polymerization. Polymer 2004, 45, 8717–8721. [Google Scholar] [CrossRef]
- Xu, G.; Wu, W.-T.; Wang, Y.; Pang, W.; Zhu, Q.; Wang, P.; You, Y. Constructing polymer brushes on multiwalled carbon nanotubes by in situ reversible addition fragmentation chain transfer polymerization. Polymer 2006, 47, 5909–5918. [Google Scholar] [CrossRef]
- Jinqi, X.; Jeremy, W.B.; David, A.B.; Tzu-Chia, T.; Michael, E.M.; Karen, L.W. Hierarchical inorganic-organic nanocomposites possessing amphiphilic and morphological complexities: influence of nanofiller dispersion on mechanical performance. Adv. Funct. Mater. 2008, 18, 2733–2744. [Google Scholar] [CrossRef]
- Yang, B.-X.; Shi, J.-H.; Pramoda, K.P.; Goh, S.H. Enhancement of stiffness, strength, ductility and toughness of poly(ethylene oxide) using phenoxy-grafted multiwalled carbon nanotubes. Nanotechnology 2007, 18, 125606. [Google Scholar] [CrossRef]
- Jin, J.; Song, M.; Pan, F. A DSC study of effect of carbon nanotubes on crystallisation behaviour of poly(ethylene oxide). Thermochim. Acta 2007, 456, 25–31. [Google Scholar] [CrossRef]
- Wang, Z.; Meng, X.; Li, J.; Du, X.; Li, S.; Jiang, Z.; Tang, T. A simple method for preparing carbon nanotubes/clay hybrids in water. J. Phys. Chem. C 2009, 113, 8058–8064. [Google Scholar] [CrossRef]
- Song, Y.S. Rheological characterization of carbon nanotubes/poly(ethylene oxide) composites. Rheol. Acta 2006, 46, 231–238. [Google Scholar] [CrossRef]
- Priftis, D.; Petzetakis, N.; Sakelllariou, G.; Pitsikalis, M.; Baskaran, D.; Mays, J.W.; Hadjichristidis, N. Surface-Initiated titanium-mediated coordination polymerization from catalyst-functionalized single and multiwalled carbon nanotubes. Macromolecules 2009, 42, 3340–3346. [Google Scholar] [CrossRef]
- Hua, C.W.; Chen, Z.M.; Xu, Q.; He, L.H. Ring-Banded spherulites in PCL and PCL/MWCNT solution-casting films and effect of compressed CO2 on them. J. Polym. Sci. Pol. Phys. 2009, 47, 784–792. [Google Scholar] [CrossRef]
- Ruelle, B.; Peeterbroeck, S.; Gouttebaron, R.; Godfroid, T.; Monteverde, F.; Dauchot, J.P.; Alexandre, M.; Hecq, M.; Dubois, P. Functionalization of carbon nanotubes by atomic nitrogen formed in a microwave plasma Ar+N-2 and subsequent poly(epsilon-caprolactone) grafting. J. Mater. Chem. 2007, 17, 157–159. [Google Scholar] [CrossRef]
- Wu, D.F.; Zhang, Y.S.; Zhang, M.; Yu, W. Selective localization of multiwalled carbon nanotubes in Poly(epsilon-caprolactone)/Polylactide Blend. Biomacromolecules 2009, 10, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.M.; Chen, E.C. Isothermal and nonisothermal crystallization kinetics of poly(epsilon-caprolactone)/multi-walled carbon nanotube composites. Polym. Eng. Sci. 2006, 46, 1309–1317. [Google Scholar] [CrossRef]
- Xu, G.Y.; Du, L.C.; Wang, H.; Xia, R.; Meng, X.C.; Zhu, Q.R. Nonisothermal crystallization kinetics and thermomechanical properties of multiwalled carbon nanotube-reinforced poly(epsilon-caprolactone) composites. Polym. Int. 2008, 57, 1052–1066. [Google Scholar] [CrossRef]
- Castro, M.; Lu, J.B.; Bruzaud, S.; Kumar, B.; Feller, J.F. Carbon nanotubes/poly(epsilon-caprolactone) composite vapour sensors. Carbon 2009, 47, 1930–1942. [Google Scholar] [CrossRef]
- Kai, W.H.; Hua, L.; Dong, T.L.; Pan, P.J.; Zhu, B.; Inoue, Y. Synthesis and characterization of fullerene grafted poly(epsilon-caprolactone). J. Appl. Polym. Sci. 2008, 107, 4029–4035. [Google Scholar] [CrossRef]
- Kai, W.H.; Hua, L.; Dong, T.L.; Pan, P.J.; Zhu, B.; Inoue, Y. Fullerene end-capped biodegradable poly(epsilon-caprolactone). Macromol. Chem. Phys. 2008, 209, 104–111. [Google Scholar] [CrossRef]
- Kai, W.H.; Hua, L.; Zhao, L.; Inoue, Y. Synthesis of novel star shaped poly (epsilon-caprolactone) utilizing fullerene as the molecular core. Macromol. Rapid Commun. 2006, 27, 1702–1706. [Google Scholar] [CrossRef]
- Liu, M.X.; Guo, B.C.; Du, M.L.; Chen, F.; Jia, D.M. Halloysite nanotubes as a novel beta-nucleating agent for isotactic polypropylene. Polymer 2009, 50, 3022–3030. [Google Scholar] [CrossRef]
- Lu, K.B.; Grossiord, N.; Koning, C.E.; Miltner, H.E.; van Mele, B.; Loos, J. Carbon nanotube/isotactic polypropylene composites prepared by LaTex technology: Morphology analysis of CNT-induced nucleation. Macromolecules 2008, 41, 8081–8085. [Google Scholar] [CrossRef]
- Hou, Z.C.; Wang, K.; Zhao, P.; Zhang, Q.; Yang, C.Y.; Chen, D.Q.; Du, R.N.; Fu, Q. Structural orientation and tensile behavior in the extrusion-stretched sheets of polyp ropylene/multi-walled carbon nanotubes' composite. Polymer 2008, 49, 3582–3589. [Google Scholar] [CrossRef]
- Pujari, S.; Ramanathan, T.; Kasimatis, K.; Masuda, J.; Andrews, R.; Torkelson, J.M.; Brinson, L.C.; Burghardt, W.R. Preparation and characterization of multiwalled carbon nanotube dispersions in polypropylene: melt mixing versus solid-state shear pulverization. J. Polym. Sci. Pol. Phys. 2009, 47, 1426–1436. [Google Scholar] [CrossRef]
- Wu, D.F.; Sun, Y.R.; Zhang, M. Kinetics study on melt compounding of carbon nanotube/polypropylene nanocomposites. J. Polym. Sci. Pol. Phys. 2009, 47, 608–618. [Google Scholar] [CrossRef]
- Xu, D.H.; Wang, Z.G.; Douglas, J.F. Influence of carbon nanotube aspect ratio on normat stress differences in isotactic polypropylene nanocomposite melts. Macromolecules 2008, 41, 815–825. [Google Scholar] [CrossRef]
- Haggenmueller, R.; Du, F.M.; Fischer, J.E.; Winey, K.I. Interfacial in situ polymerization of single wall carbon nanotube/nylon-6,6 nanocomposites. Polymer 2006, 47, 2381–2388. [Google Scholar] [CrossRef]
- Li, L.Y.; Li, C.Y.; Ni, C.Y.; Rong, L.X.; Hsiao, B. Structure and crystallization behavior of Nylon 66/multi-walled carbon nanotube nanocomposites at low carbon nanotube contents. Polymer 2007, 48, 3452–3460. [Google Scholar] [CrossRef]
- Kang, M.; Myung, S.J.; Jin, H.J. Nylon 610 and carbon nanotube composite by in situ interfacial polymerization. Polymer 2006, 47, 3961–3966. [Google Scholar] [CrossRef]
- Zeng, H.L.; Gao, C.; Wang, Y.P.; Watts, P.C.P.; Kong, H.; Cui, X.W.; Yan, D.Y. In situ polymerization approach to multiwalled carbon nanotubes-reinforced nylon 1010 composites: Mechanical properties and crystallization behavior. Polymer 2006, 47, 113–122. [Google Scholar] [CrossRef]
- Sandler, J.K.W.; Pegel, S.; Cadek, M.; Gojny, F.; van Es, M.; Lohmar, J.; Blau, W.J.; Schulte, K.; Windle, A.H.; Shaffer, M.S.P. A comparative study of melt spun polyamide-12 fibres reinforced with carbon nanotubes and nanofibres. Polymer 2004, 45, 2001–2015. [Google Scholar] [CrossRef]
- Antoniadis, G.; Paraskevopoulos, K.M.; Bikiaris, D.; Chrissafis, K. Melt-crystallization mechanism of poly(ethylene terephthalate)/multi-walled carbon nanotubes prepared by in situ polymerization. J. Polym. Sci. Pol. Phys. 2009, 47, 1452–1466. [Google Scholar] [CrossRef]
- Mun, S.J.; Jung, Y.M.; Kim, J.C.; Chang, J.H. Poly(ethylene terephthalate) nanocomposite fibers with functionalized multiwalled carbon nanotubes via in-situ polymerization. J. Appl. Polym. Sci. 2008, 109, 638–646. [Google Scholar] [CrossRef]
- Liu, F.J.; Huang, L.M.; Wen, T.C.; Gopalan, A. Large-area network of polyaniline nanowires supported platinum nanocatalysts for methanol oxidation. Synth. Met. 2007, 157, 651–658. [Google Scholar] [CrossRef]
- Palmero, S.; Colina, A.; Muñoz, E.; Heras, A.; Ruiz, V.; López-Palacios, J. Layer-by-layer electrosynthesis of Pt–Polyaniline nanocomposites for the catalytic oxidation of methanol. Electrochem. Commun. 2009, 11, 122–125. [Google Scholar] [CrossRef]
- Tristany, M.; Moreno-Mañas, M.; Pleixats, R.; Chaudret, B.; Philippot, K.; Dieudonné, P.; Lecante, P. Formation of nanocomposites of platinum nanoparticles embedded into heavily fluorinated aniline and displaying long range organization. J. Mater. Chem. 2008, 18, 660–666. [Google Scholar] [CrossRef]
- Okamoto, M.; Fujigaya, T.; Nakashima, N. Design of an assembly of poly (benzimidazole), carbon nanotubes, and pt nanoparticles for a fuel-cell electrocatalyst with an ideal interfacial nanostructure. Small 2009, 5, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Sapurina, I.Y.; Kompan, M.E.; Zabrodskii, A.G.; Stejskal, J.; Trchova, M. Nanocomposites with mixed electronic and protonic conduction for electrocatalysis. Russ. J. Electrochem. 2007, 43, 528–536. [Google Scholar] [CrossRef]
- Compagnini, G. Noble metal particles for polymer-based nanostructured thin films. Appl. Surf. Sci. 2004, 226, 216–225. [Google Scholar] [CrossRef]
- Zhu, Y.; Kockrick, E.; Kaskel, S.; Ikoma, T.; Hanagata, N. Nanocasting route to ordered mesoporous carbon with FePt nanoparticles and its phenol adsorption property. J. Phys. Chem. C 2009, 113, 5998–6002. [Google Scholar] [CrossRef]
- Fuhrer, R.; Athanassiou, E.K.; Luechinger, N.A.; Stark, W.J. Crosslinking metal nanoparticles into the polymer backbone of hydrogels enables preparation of soft, magnetic field-driven actuators with muscle-like flexibility. Small 2009, 5, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Brayner, R.; Vaulay, M.J.; Fievet, F.; Coradin, T. Alginate-Mediated growth of Co, Ni, and CoNi nanoparticles: Influence of the biopolymer structure. Chem. Mater. 2007, 19, 1190–1198. [Google Scholar] [CrossRef]
- Burke, N.A.D.; Stover, H.D.H.; Dawson, F.P. Magnetic nanocomposites: Preparation and characterization of polymer-coated iron nanoparticles. Chem. Mater. 2002, 14, 4752–4761. [Google Scholar] [CrossRef]
- Forster, S.; Plantenberg, T. From self-organizing polymers to nanohybrid and biomaterials. Angew. Chem. Int. Edit. 2002, 41, 688–714. [Google Scholar] [CrossRef]
- Das, D.; Basu, S.; Paul, A. PVC-Cu composites with chemically deposited ultrafine copper particles. J. Mater. Sci. 1980, 15, 1719–1723. [Google Scholar] [CrossRef]
- Fekete, E.; Molnar, S.; Kim, G.M.; Michler, G.H.; Pukanszky, B. Aggregation, fracture initiation, and strength of PP/CaCO 3 composites. J. Macromol. Sci. B 1999, 38, 885–899. [Google Scholar] [CrossRef]
- King, S.; Hyunh, K.; Tannenbaum, R. Kinetics of nucleation, growth, and stabilization of cobalt oxide nanoclusters. J. Phys. Chem. B 2003, 107, 12097–12104. [Google Scholar] [CrossRef]
- Kiss, A.; Fekete, E.; Pukanszky, B. Aggregation of CaCO3 particles in PP composites: Effect of surface coating. Compos. Sci. Technol. 2007, 67, 1574–1583. [Google Scholar] [CrossRef]
- Osman, M.A.; Suter, U.W. Surface treatment of calcite with fatty acids: Structure and properties of the organic monolayer. Chem. Mater. 2002, 14, 4408–4415. [Google Scholar] [CrossRef]
- Raj, R.G.; Kokta, B.V.; Dembele, F.; Sanschagrain, B. Compounding of cellulose fibers with polypropylene: Effect of fiber treatment on dispersion in the polymer matirx. J. Appl. Polym. Sci. 1989, 38, 1987–1996. [Google Scholar] [CrossRef]
- Luechinger, N.A.; Booth, N.; Heness, G.; Bandyopadhyay, S.; Grass, R.N.; Stark, W.J. Surfactant-Free, Melt-Processable metal-polymer hybrid materials: Use of graphene as a dispersing agent. Adv. Mater. 2008, 20, 3044–3049. [Google Scholar] [CrossRef]
- Ghose, S.; Watson, K.A.; Working, D.C.; Connell, J.W.; Smith, J.G.; Sun, Y.P. Thermal conductivity of ethylene vinyl acetate copolymer/nanofiller blends. Compos. Sci. Technol. 2008, 68, 1843–1853. [Google Scholar] [CrossRef]
- Sahiner, N. In situ metal particle preparation in cross-linked poly (2-acrylamido-2-methyl-1-propansulfonic acid) hydrogel networks. Colloid Polym. Sci. 2006, 285, 283–292. [Google Scholar] [CrossRef]
- Chelebaeva, E.; Guari, Y.; Larionova, J.; Trifonov, A.; Guérin, C. Soluble ligand-stabilized cyano-bridged coordination polymer nanoparticles. Chem. Mater. 2008, 20, 1367–1375. [Google Scholar] [CrossRef]
- Zhao, W.; Zhang, Q.; Chen, T.; Lu, T. Preparation and thermal decomposition of PS/Ni microspheres. Mater. Chem. Phys. 2009, 113, 428–434. [Google Scholar] [CrossRef]
- Compton, J.; Thompson, D.; Kranbuehl, D.; Ohl, S.; Gain, O.; David, L.; Espuche, E. Hybrid films of polyimide containing in situ generated silver or palladium nanoparticles: Effect of the particle precursor and of the processing conditions on the morphology and the gas permeability. Polymer 2006, 47, 5303–5313. [Google Scholar] [CrossRef]
- Wada, Y.; Kobayashi, T.; Yamasaki, H.; Sakata, T.; Hasegawa, N.; Mori, H.; Tsukahara, Y. Nanohybrid polymer prepared by successive polymerization of methacrylate monomer containing silver nanoparticles in situ prepared under microwave irradiation. Polymer 2007, 48, 1441–1444. [Google Scholar] [CrossRef]
- Gao, C.; Vo, C.D.; Jin, Y.Z.; Li, W.; Armes, S.P. Multihydroxy polymer-functionalized carbon nanotubes: Synthesis, derivatization, and metal loading. Macromolecules 2005, 38, 8634–8648. [Google Scholar] [CrossRef]
- Deshmukh, R.D.; Composto, R.J. Surface segregation and formation of silver nanoparticles created in situ in poly (methyl methacrylate) films. Chem. Mater. 2007, 19, 745–754. [Google Scholar] [CrossRef]
- Mbhele, Z.H.; Salemane, M.G.; Van Sittert, C.; Nedeljkovic, J.M.; Djokovic, V.; Luyt, A.S. Fabrication and characterization of silver polyvinyl alcohol nanocomposites. Chem. Mater. 2003, 15, 5019–5024. [Google Scholar] [CrossRef]
- Mukherjee, P.; Nandi, A.K. Electronic properties of poly (o-methoxy aniline)-silver nanocomposite thin films: Influence of nanoparticle size and density. J. Mater. Chem. 2009, 19, 781–786. [Google Scholar]
- Lee, J.Y.; Liao, Y.; Nagahata, R.; Horiuchi, S. Effect of metal nanoparticles on thermal stabilization of polymer/metal nanocomposites prepared by a one-step dry process. Polymer 2006, 47, 7970–7979. [Google Scholar] [CrossRef]
- Wang, H.L.; Gao, J.; Sansinena, J.M.; McCarthy, P. Fabrication and characterization of polyaniline monolithic actuators based on a novel configuration: integrally skinned asymmetric membrane. Chem. Mater. 2002, 14, 2546–2552. [Google Scholar] [CrossRef]
- Mei, Y.; Lu, Y.; Polzer, F.; Ballauff, M.; Drechsler, M. Catalytic activity of palladium nanoparticles encapsulated in spherical polyelectrolyte brushes and core shell microgels. Chem. Mater. 2007, 19, 1062–1069. [Google Scholar] [CrossRef]
- French, B.L.; Davis, L.M.; Munzinger, E.S.; Slavin, J.W.J.; Christy, P.C.; Thompson, D.W.; Southward, R.E. Palladium polyimide nanocomposite membranes: Synthesis and characterization of reflective and electrically conductive surface-metallized films. Chem. Mater. 2005, 17, 2091–2100. [Google Scholar] [CrossRef]
- Krebs, F.C.; Nyberg, R.B.; Jorgensen, M. Influence of residual catalyst on the properties of conjugated polyphenylenevinylene materials: Palladium nanoparticles and poor electrical performance. Chem. Mater. 2004, 16, 1313–1318. [Google Scholar] [CrossRef]
- Goldenberg, L.M.; Sakhno, O.V.; Smirnova, T.N.; Helliwell, P.; Chechik, V.; Stumpe, J. Holographic composites with gold nanoparticles: Nanoparticles promote polymer segregation. Chem. Mater. 2008, 20, 4619–4627. [Google Scholar] [CrossRef]
- Hata, H.; Kobayashi, Y.; Salama, M.; Malek, R.; Mallouk, T.E. pH-dependent intercalation of gold nanoparticles into a synthetic fluoromica modified with poly (allylamine). Chem. Mater. 2007, 19, 6588–6596. [Google Scholar] [CrossRef]
- Li, D.; He, Q.; Cui, Y.; Li, J. Fabrication of pH-responsive nanocomposites of gold nanoparticles/poly (4-vinylpyridine). Chem. Mater. 2007, 19, 412–417. [Google Scholar] [CrossRef]
- Uhlenhaut, D.I.; Smith, P.; Caseri, W. Color Switching in Gold-Polysiloxane Elastomeric Nanocomposites. Adv. Mater. 2006, 18, 1653–1656. [Google Scholar] [CrossRef]
- Marinakos, S.M.; Shultz, D.A.; Feldheim, D.L. Gold nanoparticles as templates for the synthesis of hollow nanometer-sized conductive polymer capsules. Adv. Mater. 1999, 11, 34–37. [Google Scholar] [CrossRef]
- Park, J.H.; Lim, Y.T.; Park, O.O.; Kim, J.K.; Yu, J.W.; Kim, Y.C. Polymer/Gold nanoparticle nanocomposite light-emitting diodes: Enhancement of electroluminescence stability and quantum efficiency of blue-light-emitting polymers. Chem. Mater. 2004, 16, 688–692. [Google Scholar] [CrossRef]
- Tanaka, M.; Fujita, R.; Nishide, H. Alternate network film of thiol group-terminated polythiophene and gold nanoparticle. Polymer 2007, 48, 5884–5888. [Google Scholar] [CrossRef]
- Cioffi, N.; Losito, I.; Torsi, L.; Farella, I.; Valentini, A.; Sabbatini, L.; Zambonin, P.G.; Bleve-Zacheos, T. Analysis of the Surface Chemical Composition and Morphological Structure of Vapor-Sensing Gold Fluoropolymer Nanocomposites. Chem. Mater. 2002, 14, 804–811. [Google Scholar] [CrossRef]
- Laicer, C.S.T.; Mrozek, R.A.; Taton, T.A. Domain nucleation dictates overall nanostructure in composites of block copolymers and model nanorods. Polymer 2007, 48, 1316–1328. [Google Scholar] [CrossRef]
- Guo, Z.; Lin, H.; Karki, A.B.; Wei, S.; Young, D.P.; Park, S.; Willis, J.; Hahn, T.H. Facile monomer stabilization approach to fabricate iron/vinyl ester resin nanocomposites. Compos. Sci. Technol. 2008, 68, 2551–2556. [Google Scholar] [CrossRef]
- Baker, C.; Ismat Shah, S.; Hasanain, S.K. Magnetic behavior of iron and iron-oxide nanoparticle/polymer composites. J. Magn. Magn. Mater. 2004, 280, 412–418. [Google Scholar] [CrossRef]
- Ushakov, N.M.; Kochubei, V.I.; Zapsis, K.V.; Kosobudskii, I.D. Optical properties of metal-polymer nanocomposites based on iron and high-pressure polyethylene. Opt. Spectrosc. 2004, 96, 798–803. [Google Scholar] [CrossRef]
- Huang, X.; Jiang, P.; Kim, C.; Ke, Q.; Wang, G. Preparation, microstructure and properties of polyethylene aluminum nanocomposite dielectrics. Compos. Sci. Technol. 2008, 68, 2134–2140. [Google Scholar] [CrossRef]
- Huang, X.; Kim, C.; Jiang, P.; Yin, Y.; Li, Z. Influence of aluminum nanoparticle surface treatment on the electrical properties of polyethylene composites. J. Appl. Phys. 2009, 105, 014105. [Google Scholar] [CrossRef]
- Huang, X.; Kim, C.; Ma, Z.; Jiang, P.; Yin, Y.; Li, Z. Correlation between rheological, electrical, and microstructure characteristics in polyethylene/aluminum nanocomposites. J. Polym. Sci. Pol. Phys. 2008, 46, 2143–2154. [Google Scholar] [CrossRef]
- Mallick, K.; Witcomb, M.J.; Scurrell, M.S. In situ synthesis of copper nanoparticles and poly (o-toluidine): A metal–polymer composite material. Eur. Polym. J. 2006, 42, 670–675. [Google Scholar] [CrossRef]
- Untereker, D.; Lyu, S.; Schley, J.; Martinez, G.; Lohstreter, L. Maximum conductivity of packed nanoparticles and their polymer composites. ACS Appl. Mater. Interfaces 2009, 1, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, N.; Torsi, L.; Ditaranto, N.; Tantillo, G.; Ghibelli, L.; Sabbatini, L.; Bleve-Zacheo, T.; D'Alessio, M.; Zambonin, P.G.; Traversa, E. Copper nanoparticle/polymer composites with antifungal and bacteriostatic properties. Chem. Mater. 2005, 17, 5255–5262. [Google Scholar] [CrossRef]
- Sormana, J.-L.; Chattopadhyay, S.; Meredith, J.C. Mechanical and thermal properties of poly(urethane urea) nanocomposites prepared with diamine-modified Laponite®. J. Nanomater. 2008, 2008, 1–9. [Google Scholar] [CrossRef]
- Mishra, A.K.; Chattopadhyay, S.; Nando, G.B.; Devadoss, E. Synthesis and characterization of elastomeric polyurethane-Laponite® nanocomposite. Des. Monomers Polym. 2008, 11, 394–407. [Google Scholar] [CrossRef]
- Mishra, A.K.; Nando, G.B.; Chattopadhyay, S. Exploring preferential association of Laponite® and cloisite with soft and hard segments in TPU-Clay nanocomposite prepared by solution mixing technique. J. Polym. Sci. Pol. Phys. 2008, 46, 2341–2354. [Google Scholar] [CrossRef]
- Korley, L.T.J.; Liff, S.M.; Kumar, N.; McKinley, G.H.; Hammond, P.T. Preferential Association of Segment Blocks in Polyurethane Nanocomposites. Macromolecules 2006, 39, 7030–7036. [Google Scholar] [CrossRef]
- Fang, F.; Kim, J.; Choi, H.; Kim, C. Synthesis and electrorheological response of nano-sized Laponite® stabilized poly(methyl methacrylate) spheres. Colloid Polym. Sci. 2009, 287, 745–749. [Google Scholar] [CrossRef]
- Chang, C.-C.; Hou, S.-S. Intercalation of poly(methyl methacrylate) into tyramine-modified layered silicates through hydrogen-bonding interaction. Eur. Polym. J. 2008, 44, 1337–1345. [Google Scholar] [CrossRef]
- Wang, J.; Wheeler, P.; Jarrett, W.; Mathias, L. Synthesis and characterization of dual-functionalized Laponite® clay for acrylic nanocomposites. J. Appl. Polym. Sci. 2007, 106, 1497–1506. [Google Scholar]
- Wheeler, P.A.; Wang, J.; Mathias, L.J. Poly(methyl methacrylate)/Laponite® Nanocomposites: Exploring Covalent and Ionic Clay Modifications. Chem. Mater. 2006, 18, 3937–3945. [Google Scholar] [CrossRef]
- Konn, C.; Morel, F.; Beyou, E.; Chaumont, P.; Bourgeat-Lami, E. Nitroxide-Mediated Polymerization of Styrene Initiated from the Surface of Laponite® Clay Platelets. Macromolecules 2007, 40, 7464–7472. [Google Scholar] [CrossRef]
- Negrete-Herrera, N.; Putaux, J.-L.; David, L.; Bourgeat-Lami, E. Polymer/Laponite® composite colloids through emulsion polymerization: Influence of the clay modification level on particle morphology. Macromolecules 2006, 39, 9177–9184. [Google Scholar] [CrossRef]
- Negrete-Herrera, N.; Putaux, J.-L.; David, L.; De Haas, F.; Bourgeat-Lami, E. Polymer/Laponite® composite latexes: Particle morphology, film microstructure, and properties. Macromol. Rapid Commun. 2007, 28, 1567–1573. [Google Scholar] [CrossRef]
- Ruggerone, R.; Plummer, C.J.G.; Herrera, N.N.; Bourgeat-Lami, E.; Månson, J.-A.E. Highly filled polystyrene-Laponite® nanocomposites prepared by emulsion polymerization. Eur. Polym. J. 2009, 45, 621–629. [Google Scholar] [CrossRef]
- Caruso, R.A.; Susha, A.; Caruso, F. Multilayered titania, silica, and Laponite® nanoparticle coatings on polystyrene colloidal templates and resulting inorganic hollow spheres. Chem. Mater. 2001, 13, 400–409. [Google Scholar] [CrossRef]
- Lorthioir, C.; Lauprêtre, F.; Soulestin, J.; Lefebvre, J.-M. Segmental dynamics of poly(ethylene oxide) chains in a model polymer/clay intercalated phase: Solid-State NMR investigation. Macromolecules 2009, 42, 218–230. [Google Scholar] [CrossRef]
- Loyens, W.; Jannasch, P.; Maurer, F.H.J. Poly(ethylene oxide)/Laponite® nanocomposites via melt-compounding: Effect of clay modification and matrix molar mass. Polymer 2005, 46, 915–928. [Google Scholar] [CrossRef]
- Gournis, D.; Floudas, G. Hairy Plates: Poly(ethylene oxide)-b-polyisoprene Copolymers in the Presence of Laponite® Clay. Chem. Mater. 2004, 16, 1686–1692. [Google Scholar] [CrossRef]
- Mitchell, C.A.; Krishnamoorti, R. Rheological properties of diblock copolymer/layered-silicate nanocomposites. J. Polym. Sci. Pol. Phys. 2002, 40, 1434–1443. [Google Scholar] [CrossRef]
- Agrawal, S.K.; Sanabria-DeLong, N.; Tew, G.N.; Bhatia, S.R. Nanoparticle-reinforced associative network hydrogels. Langmuir 2008, 24, 13148–13154. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Tirelli, N.; Cellesi, F.; Saunders, B.R. Temperature-triggered gelation of aqueous Laponite® dispersions containing a cationic poly(n-isopropyl acrylamide) graft copolymer. Langmuir 2009, 25, 490–496. [Google Scholar] [CrossRef] [PubMed]
- De Lisi, R.; Lazzara, G.; Lombardo, R.; Milioto, S.; Muratore, N.; Turco Liveri, M.L. Adsorption of triblock copolymers and their homopolymers at Laponite® clay/solution interface. Role played by the copolymer nature. Phys. Chem. Chem. Phys. 2005, 7, 3994–4001. [Google Scholar] [CrossRef] [PubMed]
- De Lisi, R.; Gradzielski, M.; Lazzara, G.; Milioto, S.; Muratore, N.; Prevost, S. Aqueous Laponite® clay dispersions in the presence of poly(ethylene oxide) or poly(propylene oxide) oligomers and their triblock copolymers. J.Phys.Chem. B 2008, 112, 9328–9336. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.; Cosgrove, T. Small-angle neutron scattering study of adsorbed pluronic tri-block copolymers on Laponite®. Langmuir 2005, 21, 9176–9182. [Google Scholar] [CrossRef] [PubMed]
- De Lisi, R.; Lazzara, G.; Milioto, S.; Muratore, N. Laponite® clay in homopolymer and tri-block copolymer matrices. J. Therm. Anal. Calorim. 2007, 87, 61–67. [Google Scholar] [CrossRef]
- De Lisi, R.; Lazzara, G.; Milioto, S.; Muratore, N. Aqueous nonionic copolymer-functionalized Laponite® clay. a thermodynamic and spectrophotometric study to characterize its behavior toward an organic material. Langmuir 2006, 22, 8056–8062. [Google Scholar] [CrossRef] [PubMed]
- Dan, C.H.; Lee, M.H.; Kim, Y.D.; Min, B.H.; Kim, J.H. Effect of clay modifiers on the morphology and physical properties of thermoplastic polyurethane/clay nanocomposites. Polymer 2006, 47, 6718–6730. [Google Scholar] [CrossRef]
- Lee, H.-T.; Lin, L.-H. Waterborne polyurethane/clay nanocomposites: Novel effects of the clay and its interlayer ions on the morphology and physical and electrical properties. Macromolecules 2006, 39, 6133–6141. [Google Scholar] [CrossRef]
- Osman, M.A.; Mittal, V.; Morbidelli, M.; Suter, U.W. Polyurethane adhesive nanocomposites as gas permeation barrier. Macromolecules 2003, 36, 9851–9858. [Google Scholar] [CrossRef]
- Plummer, C.J.G.; Rodlert, M.; Bucaille, J.-L.; Grunbauer, H.J.M.; Månson, J.-A.E. Correlating the rheological and mechanical response of polyurethane nanocomposites containing hyperbranched polymers. Polymer 2005, 46, 6543–6553. [Google Scholar] [CrossRef]
- Chen-Yang, Y.W.; Lee, Y.K.; Chen, Y.T.; Wu, J.C. High improvement in the properties of exfoliated PU/clay nanocomposites by the alternative swelling process. Polymer 2007, 48, 2969–2979. [Google Scholar] [CrossRef]
- Tien, Y.I.; Wei, K.H. High-tensile-property layered silicates/polyurethane nanocomposites by using reactive silicates as pseudo chain extenders. Macromolecules 2001, 34, 9045–9052. [Google Scholar] [CrossRef]
- Tarkin-Tas, E.; Goswami, S.K.; Nayak, B.R.; Mathias, L.J. Highly exfoliated poly(ε-caprolactone)/organomontmorillonite nanocomposites prepared in situ polymerization. J. Appl. Polym. Sci. 2008, 107, 976–984. [Google Scholar] [CrossRef]
- Liao, L.; Zhang, C.; Gong, S. Preparation of poly(-caprolactone)/clay nanocomposites by microwave-assisted in situ ring-opening polymerization. Macromol. Rapid Comm. 2007, 28, 1148–1154. [Google Scholar] [CrossRef]
- Kiersnowski, A.; Gutmann, J.S.; Piglowski, J. Influence of organic modifiers on morphology and crystallization of poly(ε-caprolactone)/synthetic clay intercalated nanocomposites. J. Polym. Sci. Pol. Phys. 2007, 45, 2350–2367. [Google Scholar] [CrossRef]
- Yu, Z.; Yin, J.; Yan, S.; Xie, Y.; Ma, J.; Chen, X. Biodegradable poly(l-lactide)/poly([var epsilon]-caprolactone)-modified montmorillonite nanocomposites: Preparation and characterization. Polymer 2007, 48, 6439–6447. [Google Scholar] [CrossRef]
- Wu, T.; Xie, T.; Yang, G. Preparation and characterization of poly([epsilon]-caprolactone)/Na+-MMT nanocomposites. Appl. Clay Sci. 2009, 45, 105–110. [Google Scholar] [CrossRef]
- Feng, S.-S.; Mei, L.; Anitha, P.; Gan, C.W.; Zhou, W. Poly(lactide)-vitamin E derivative/montmorillonite nanoparticle formulations for the oral delivery of Docetaxel. Biomaterials 2009, 30, 3297–3306. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.-Y.; Ren, J.; Dong, B. Melt rheology of polylactide/montmorillonite nanocomposites. J. Polym. Sci. Pol. Phys. 2007, 45, 3189–3196. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, J.; Wolcott, M.P. Comparison of polylactide/nano-sized calcium carbonate and polylactide/montmorillonite composites: Reinforcing effects and toughening mechanisms. Polymer 2007, 48, 7632–7644. [Google Scholar] [CrossRef]
- Lee, Y.H.; Lee, J.H.; An, I.-G.; Kim, C.; Lee, D.S.; Lee, Y.K.; Nam, J.-D. Electrospun dual-porosity structure and biodegradation morphology of Montmorillonite reinforced PLLA nanocomposite scaffolds. Biomaterials 2005, 26, 3165–3172. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Ranganathan, B.; Feng, S.-S. Multifunctional poly(d,l-lactide-co-glycolide)/montmorillonite (PLGA/MMT) nanoparticles decorated by Trastuzumab for targeted chemotherapy of breast cancer. Biomaterials 2008, 29, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Urbanczyk, L.; Ngoundjo, F.; Alexandre, M.; Jérôme, C.; Detrembleur, C.; Calberg, C. Synthesis of polylactide/clay nanocomposites by in situ intercalative polymerization in supercritical carbon dioxide. Eur. Polym. J. 2009, 45, 643–648. [Google Scholar] [CrossRef]
- Dong, Y.; Feng, S.-S. Poly(d,l-lactide-co-glycolide)/montmorillonite nanoparticles for oral delivery of anticancer drugs. Biomaterials 2005, 26, 6068–6076. [Google Scholar] [CrossRef] [PubMed]
- Loyens, W.; Maurer, H.J.F.; Jannasch, P. Melt-compounded salt-containing poly(ethylene oxide)/clay nanocomposites for polymer electrolyte membranes. Polymer 2005, 46, 7334–7345. [Google Scholar] [CrossRef]
- Malwitz, M.M.; Dundigalla, A.; Ferreiro, V.; Butler, P.D.; Henk, M.C.; Schmidt, G. Layered structures of shear-oriented and multilayered PEO/silicate nanocomposite films. Phys. Chem. Chem. Phys. 2004, 6, 2977–2982. [Google Scholar] [CrossRef]
- Stefanescu, E.A.; Stefanescu, C.; Negulescu, I.I.; Daly, W.H. Effect of ionic species on the structure and properties of salt-containing PEO-montmorillonite nanocomposites. Macromol. Chem. Phys. 2008, 209, 2320–2330. [Google Scholar] [CrossRef]
- Stefanescu, E.A.; Petrovan, S.; Daly, W.H.; Negulescu, I.I. Elongational rheology of polymer/clay dispersions: determination of orientational extent in elongational flow processes. Macromol. Mater. Eng. 2008, 293, 303–309. [Google Scholar] [CrossRef]
- Sun, Y.-h.; Luo, Y.-F.; Jia, D.-M. Preparation and characterization of polypropylene/solid-state organomodified montmorillonite nanocomposites. Polym. Compos. 2008, 29, 357–363. [Google Scholar] [CrossRef]
- Rohlmann, C.O.; Failla, M.D.; Quinzani, L.M. Linear viscoelasticity and structure of polypropylene-montmorillonite nanocomposites. Polymer 2006, 47, 7795–7804. [Google Scholar] [CrossRef]
- Qin, H.; Zhang, S.; Liu, H.; Xie, S.; Yang, M.; Shen, D. Photo-oxidative degradation of polypropylene/montmorillonite nanocomposites. Polymer 2005, 46, 3149–3156. [Google Scholar] [CrossRef]
- Nowacki, R.; Monasse, B.; Piorkowska, E.; Galeski, A.; Haudin, J.M. Spherulite nucleation in isotactic polypropylene based nanocomposites with montmorillonite under shear. Polymer 2004, 45, 4877–4892. [Google Scholar] [CrossRef]
- Chow, W.S.; Tham, W.L. Effects of antistatic agent on the mechanical, morphological and antistatic properties of polypropylene/organo-montmorillonite nanocomposites. Express Polym. Lett. 2009, 3, 116–125. [Google Scholar] [CrossRef]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stefanescu, E.A.; Daranga, C.; Stefanescu, C. Insight into the Broad Field of Polymer Nanocomposites: From Carbon Nanotubes to Clay Nanoplatelets, via Metal Nanoparticles. Materials 2009, 2, 2095-2153. https://doi.org/10.3390/ma2042095
Stefanescu EA, Daranga C, Stefanescu C. Insight into the Broad Field of Polymer Nanocomposites: From Carbon Nanotubes to Clay Nanoplatelets, via Metal Nanoparticles. Materials. 2009; 2(4):2095-2153. https://doi.org/10.3390/ma2042095
Chicago/Turabian StyleStefanescu, Eduard A., Codrin Daranga, and Cristina Stefanescu. 2009. "Insight into the Broad Field of Polymer Nanocomposites: From Carbon Nanotubes to Clay Nanoplatelets, via Metal Nanoparticles" Materials 2, no. 4: 2095-2153. https://doi.org/10.3390/ma2042095
APA StyleStefanescu, E. A., Daranga, C., & Stefanescu, C. (2009). Insight into the Broad Field of Polymer Nanocomposites: From Carbon Nanotubes to Clay Nanoplatelets, via Metal Nanoparticles. Materials, 2(4), 2095-2153. https://doi.org/10.3390/ma2042095