
Dual Fractions Proteomic Analysis of Silica Nanoparticle Interactions with Protein Extracts

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Proteomics Analysis
2.3. Spectral Count Normalisation
2.4. Statistical Analysis
3. Results
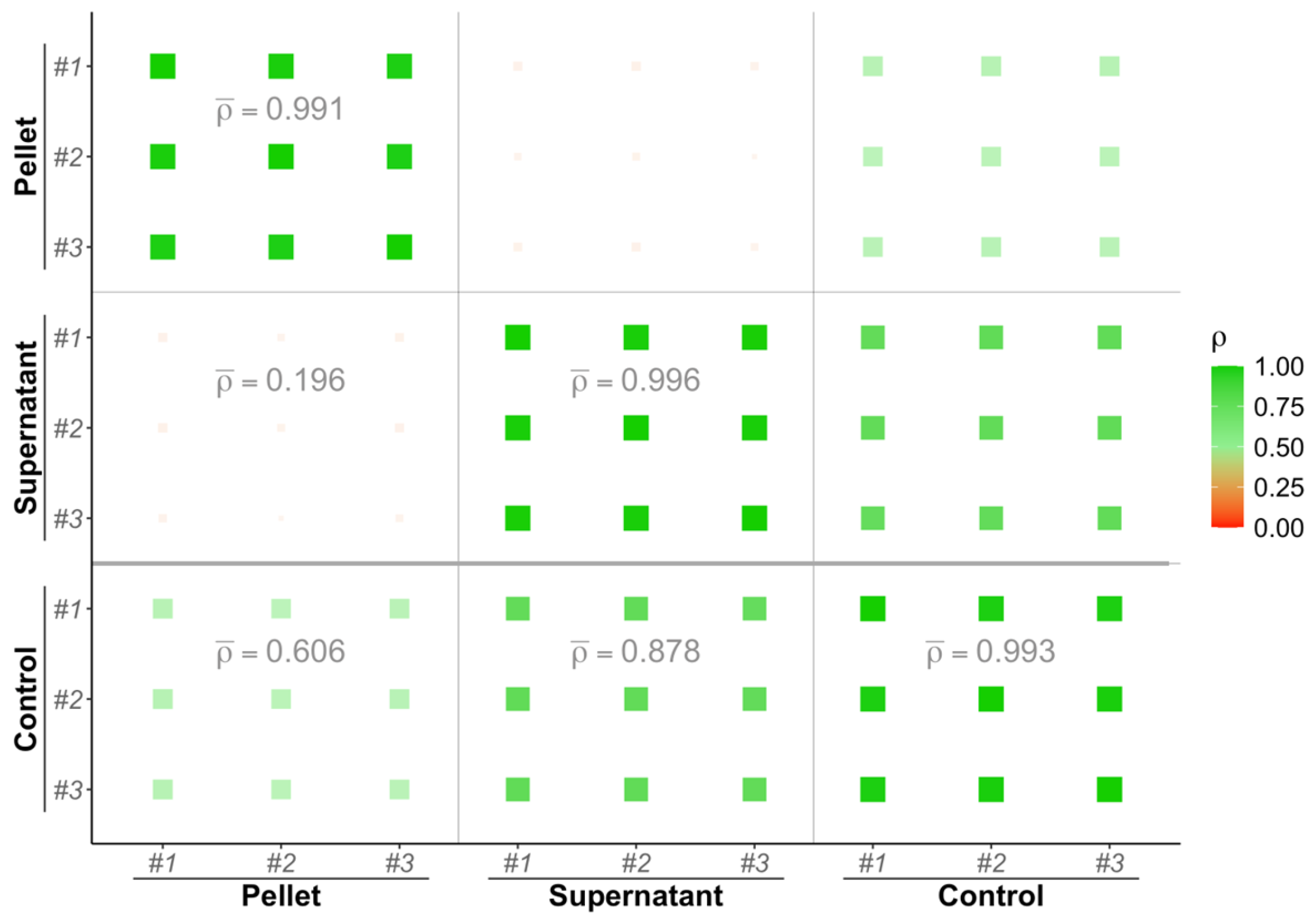
3.1. Biological Fractions Complementarity
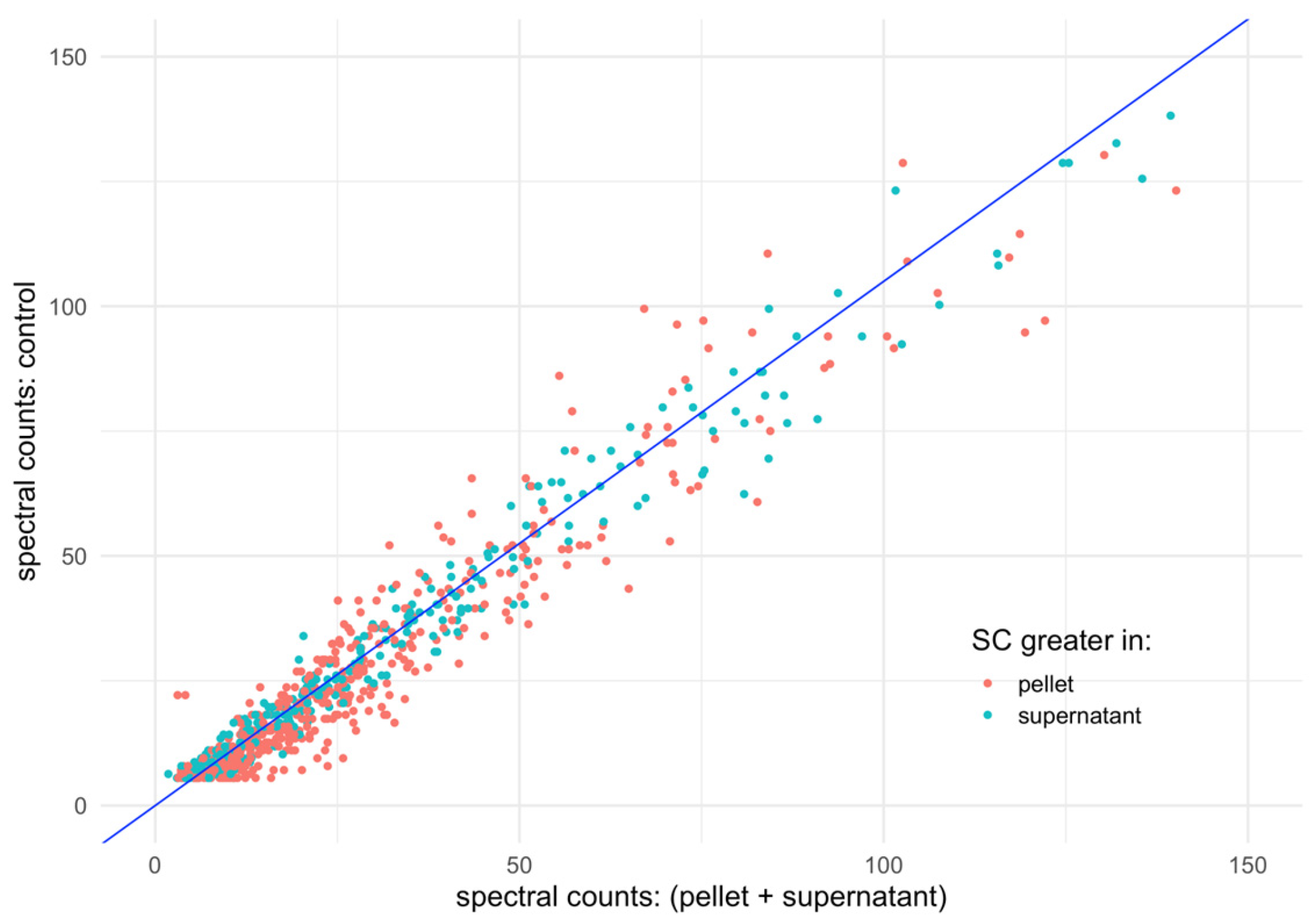
3.2. Spectral Counts Distribution Complementarity
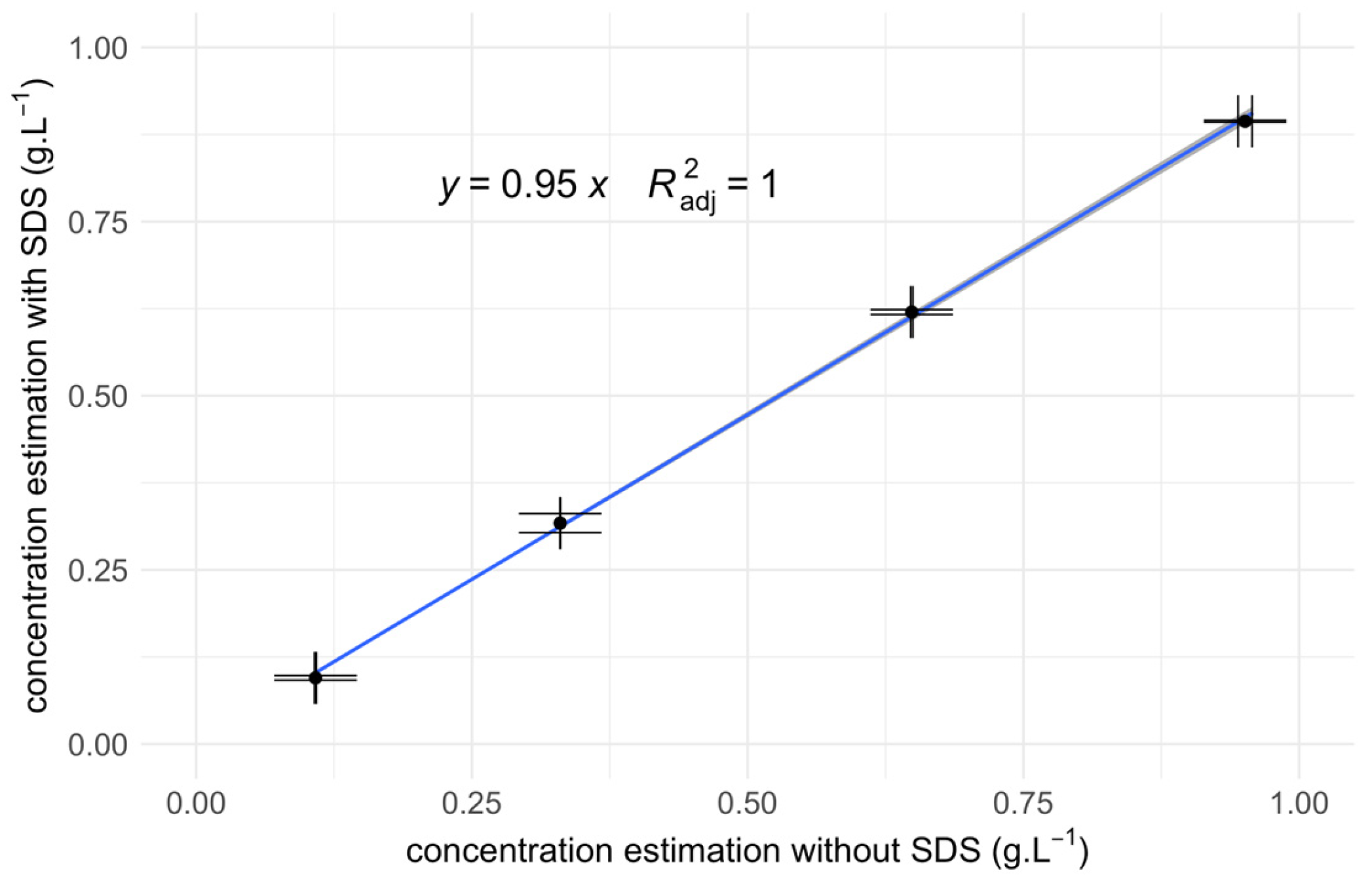
3.3. Regression Analysis
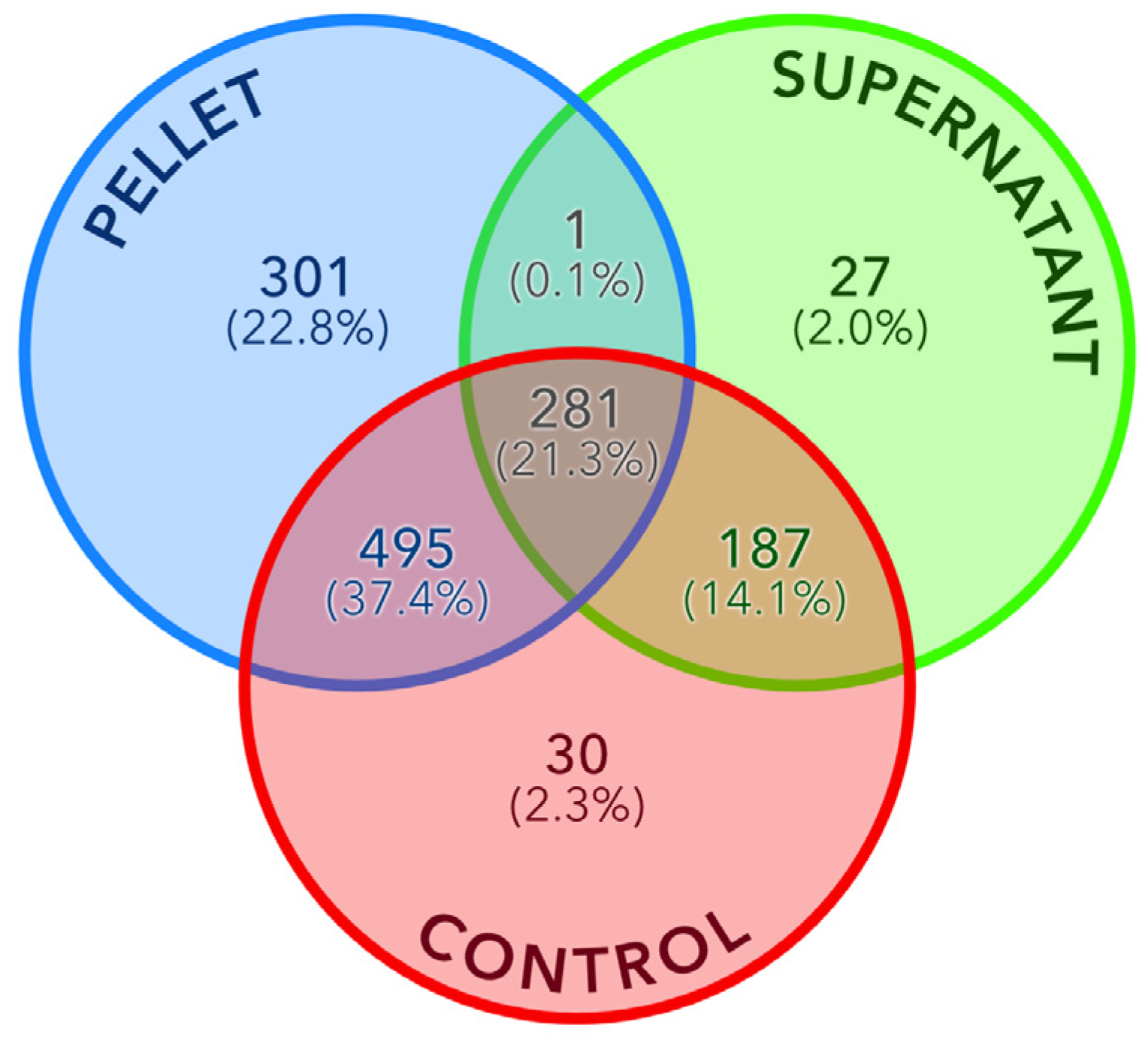
3.4. Differential Analysis between Fractions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nayl, A.A.; Abd-Elhamid, A.I.; Aly, A.A.; Bräse, S. Recent Progress in the Applications of Silica-Based Nanoparticles. RSC Adv. 2022, 12, 13706–13726. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Yu, D.; Feng, J.; You, H.; Bai, Y.; He, J.; Cao, H.; Che, Q.; Guo, J.; Su, Z. Toxicity Evaluation of Silica Nanoparticles for Delivery Applications. Drug Deliv. Transl. Res. 2023, 13, 2213–2238. [Google Scholar] [CrossRef]
- Huang, Y.; Li, P.; Zhao, R.; Zhao, L.; Liu, J.; Peng, S.; Fu, X.; Wang, X.; Luo, R.; Wang, R.; et al. Silica Nanoparticles: Biomedical Applications and Toxicity. Biomed. Pharmacother. Biomed. Pharmacother. 2022, 151, 113053. [Google Scholar] [CrossRef]
- Chen, L.; Liu, J.; Zhang, Y.; Zhang, G.; Kang, Y.; Chen, A.; Feng, X.; Shao, L. The Toxicity of Silica Nanoparticles to the Immune System. Nanomed. 2018, 13, 1939–1962. [Google Scholar] [CrossRef] [PubMed]
- Klein, G.; Mathé, C.; Biola-Clier, M.; Devineau, S.; Drouineau, E.; Hatem, E.; Marichal, L.; Alonso, B.; Gaillard, J.-C.; Lagniel, G.; et al. RNA Binding Proteins Are a Major Target of Silica Nanoparticles in Cell Extracts. Nanotoxicology 2016, 10, 1–30. [Google Scholar] [CrossRef]
- Marichal, L.; Klein, G.; Armengaud, J.; Boulard, Y.; Chédin, S.; Labarre, J.; Pin, S.; Renault, J.P.; Aude, J.C. Protein Corona Composition of Silica Nanoparticles in Complex Media: Nanoparticle Size Does Not Matter. Nanomaterials 2020, 10, 240. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J. Protein–Nanoparticle Interaction: Corona Formation and Conformational Changes in Proteins on Nanoparticles. Int. J. Nanomed. 2020, 15, 5783–5802. [Google Scholar] [CrossRef]
- Mathé, C.; Devineau, S.; Aude, J.-C.; Lagniel, G.; Chédin, S.; Legros, V.; Mathon, M.-H.; Renault, J.-P.; Pin, S.; Boulard, Y.; et al. Structural Determinants for Protein Adsorption/Non-Adsorption to Silica Surface. PLoS ONE 2013, 8, e81346. [Google Scholar] [CrossRef]
- Teeguarden, J.G.; Webb-Robertson, B.-J.; Waters, K.M.; Murray, A.R.; Kisin, E.R.; Varnum, S.M.; Jacobs, J.M.; Pounds, J.G.; Zanger, R.C.; Shvedova, A.A. Comparative Proteomics and Pulmonary Toxicity of Instilled Single-Walled Carbon Nanotubes, Crocidolite Asbestos, and Ultrafine Carbon Black in Mice. Toxicol. Sci. Off. J. Soc. Toxicol. 2011, 120, 123–135. [Google Scholar] [CrossRef]
- Schvartz, M.; Saudrais, F.; Devineau, S.; Aude, J.-C.; Chédin, S.; Henry, C.; Millán-Oropeza, A.; Perrault, T.; Pieri, L.; Pin, S.; et al. A Proteome Scale Study Reveals How Plastic Surfaces and Agitation Promote Protein Aggregation. Sci. Rep. 2023, 13, 1227. [Google Scholar] [CrossRef]
- Sanchez-Guzman, D.; Giraudon--Colas, G.; Marichal, L.; Boulard, Y.; Wien, F.; Degrouard, J.; Baeza-Squiban, A.; Pin, S.; Renault, J.P.; Devineau, S. In Situ Analysis of Weakly Bound Proteins Reveals Molecular Basis of Soft Corona Formation. ACS Nano 2020, 14, 9073–9088. [Google Scholar] [CrossRef] [PubMed]
- Giraudon--Colas, G.; Devineau, S.; Marichal, L.; Barruet, E.; Zitolo, A.; Renault, J.-P.; Pin, S. How Nanoparticles Modify Adsorbed Proteins: Impact of Silica Nanoparticles on the Hemoglobin Active Site. Int. J. Mol. Sci. 2023, 24, 3659. [Google Scholar] [CrossRef] [PubMed]
- Scopes, R.K. Measurement of Protein by Spectrophotometry at 205 Nm. Anal. Biochem. 1974, 59, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.; Haller, L.; Blein-Nicolas, M.; Zivy, M.; Canette, A.; Verbrugghe, M.; Mézange, C.; Boulay, M.; Gardan, R.; Samson, S.; et al. Identification of Hanks-Type Kinase PknB-Specific Targets in the Streptococcus Thermophilus Phosphoproteome. Front. Microbiol. 2019, 10, 1329. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using Lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Goodrich, B.; Gabry, J.; Ali, I.; Brilleman, S. Rstanarm: Bayesian Applied Regression Modeling via Stan; R Foundation for Statistical Computing: Vienna, Austria, 2023. [Google Scholar]
- Gronau, Q.F.; Singmann, H.; Wagenmakers, E.-J. Bridgesampling: An R Package for Estimating Normalizing Constants. J. Stat. Softw. 2020, 92, 1–29. [Google Scholar] [CrossRef]
- Morey, R.D.; Rouder, J.N. BayesFactor: Computation of Bayes Factors for Common Designs; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Blume, J.E.; Manning, W.C.; Troiano, G.; Hornburg, D.; Figa, M.; Hesterberg, L.; Platt, T.L.; Zhao, X.; Cuaresma, R.A.; Everley, P.A.; et al. Rapid, Deep and Precise Profiling of the Plasma Proteome with Multi-Nanoparticle Protein Corona. Nat. Commun. 2020, 11, 3662. [Google Scholar] [CrossRef]
- Partikel, K.; Korte, R.; Stein, N.C.; Mulac, D.; Herrmann, F.C.; Humpf, H.-U.; Langer, K. Effect of Nanoparticle Size and PEGylation on the Protein Corona of PLGA Nanoparticles. Eur. J. Pharm. Biopharm. 2019, 141, 70–80. [Google Scholar] [CrossRef]
- Biola-Clier, M.; Gaillard, J.-C.; Rabilloud, T.; Armengaud, J.; Carriere, M. Titanium Dioxide Nanoparticles Alter the Cellular Phosphoproteome in A549 Cells. Nanomaterials 2020, 10, 185. [Google Scholar] [CrossRef]
- Hirsh, S.L.; McKenzie, D.R.; Nosworthy, N.J.; Denman, J.A.; Sezerman, O.U.; Bilek, M.M.M. The Vroman Effect: Competitive Protein Exchange with Dynamic Multilayer Protein Aggregates. Colloids Surf. B Biointerfaces 2013, 103, 395–404. [Google Scholar] [CrossRef]
- Schvartz, M.; Saudrais, F.; Devineau, S.; Chédin, S.; Jamme, F.; Leroy, J.; Rakotozandriny, K.; Taché, O.; Brotons, G.; Pin, S.; et al. Role of the Protein Corona in the Colloidal Behavior of Microplastics. Langmuir 2023, 39, 4291–4303. [Google Scholar] [CrossRef] [PubMed]
- Millán-Oropeza, A.; Blein-Nicolas, M.; Monnet, V.; Zivy, M.; Henry, C. Comparison of Different Label-Free Techniques for the Semi-Absolute Quantification of Protein Abundance. Proteomes 2022, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Zubarev, R.A. The Challenge of the Proteome Dynamic Range and Its Implications for In-Depth Proteomics. Proteomics 2013, 13, 723–726. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fraction | Replica 1 | Replica 2 | Replica 3 | Average | % of Control |
|---|---|---|---|---|---|
| Control | 20,346 | 33,223 | 33,791 | 29,120 ± 7604 | 100.0% |
| Pellet | 15,629 | 17,503 | 16,068 | 16,400 ± 980 | 56.3% |
| Supernatant | 12,453 | 9768 | 15,939 | 12,720 ± 3094 | 43.7% |
| Fraction | Average Proteins Concentration |
|---|---|
| Pellet | 0.408 ± 0.005 g.L−1 |
| Supernatant | 0.318 ± 0.003 g.L−1 |
| Control | 0.629 ± 0.004 g.L−1 |
| Parameter | Coefficient | Confidence Interval | t-Student | p-Value |
|---|---|---|---|---|
| Intercept (α) | −0.09 ± 0.40 | [−0.88, 0.70] | 29,120 | 0.83 |
| SC Pellet (β1) | 1.07 ± 0.01 | [1.04, 1.09] | 16,400 | <0.001 |
| SC Supernatant (β2) | 1.05 ± 0.01 | [1.03, 1.06] | 12,720 | <0.001 |
| Bayes Factor (BF) Evidence |  Pellet Fraction Pellet Fraction | Supernatant Fraction  | ||
|---|---|---|---|---|
| Enriched | Depleted | Enriched | Depleted | |
| BF ≥ 3: substantial | 302 | 83 | 56 | 265 |
| BF ≥ 10: strong | 139 | 55 | 11 | 159 |
| BF ≥ 30: very strong | 54 | 27 | 2 | 88 |
| Depleted in the Supernatant |  Enriched in the Pellet & Detected in the Supernatant Enriched in the Pellet & Detected in the Supernatant
 | |||
|---|---|---|---|---|
| 302 | 179 | |||
| 265 | 124 | 41.1% | 124 | 69.3% |
| Depleted in the Pellet |  Enriched in the Supernatant & Detected in the Pellet
Enriched in the Supernatant & Detected in the Pellet
 | |||
|---|---|---|---|---|
| 56 | 36 | |||
| 83 | 32 | 57.1% | 32 | 88.9% |
| Unbound | Impacted | Bound | |||
|---|---|---|---|---|---|
| Very High | High | Indirect | Direct | High | Very High |
| 24 (4.3%) | 32 (5.8%) | 141 (25.5%) | 55 (9.9%) | 124 (22.4%) | 178 (32.1%) |
| 56 (10.1%) | 196 (35.4%) | 302 (54.5%) | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schvartz, M.; Saudrais, F.; Boulard, Y.; Renault, J.-P.; Henry, C.; Chédin, S.; Pin, S.; Aude, J.-C. Dual Fractions Proteomic Analysis of Silica Nanoparticle Interactions with Protein Extracts. Materials 2024, 17, 4909. https://doi.org/10.3390/ma17194909
Schvartz M, Saudrais F, Boulard Y, Renault J-P, Henry C, Chédin S, Pin S, Aude J-C. Dual Fractions Proteomic Analysis of Silica Nanoparticle Interactions with Protein Extracts. Materials. 2024; 17(19):4909. https://doi.org/10.3390/ma17194909
Chicago/Turabian StyleSchvartz, Marion, Florent Saudrais, Yves Boulard, Jean-Philippe Renault, Céline Henry, Stéphane Chédin, Serge Pin, and Jean-Christophe Aude. 2024. "Dual Fractions Proteomic Analysis of Silica Nanoparticle Interactions with Protein Extracts" Materials 17, no. 19: 4909. https://doi.org/10.3390/ma17194909
APA StyleSchvartz, M., Saudrais, F., Boulard, Y., Renault, J.-P., Henry, C., Chédin, S., Pin, S., & Aude, J.-C. (2024). Dual Fractions Proteomic Analysis of Silica Nanoparticle Interactions with Protein Extracts. Materials, 17(19), 4909. https://doi.org/10.3390/ma17194909