
A Micromechanical Study of Interactions of Cyanate Ester Monomer with Graphene or Boron Nitride Monolayer




Abstract
:1. Introduction
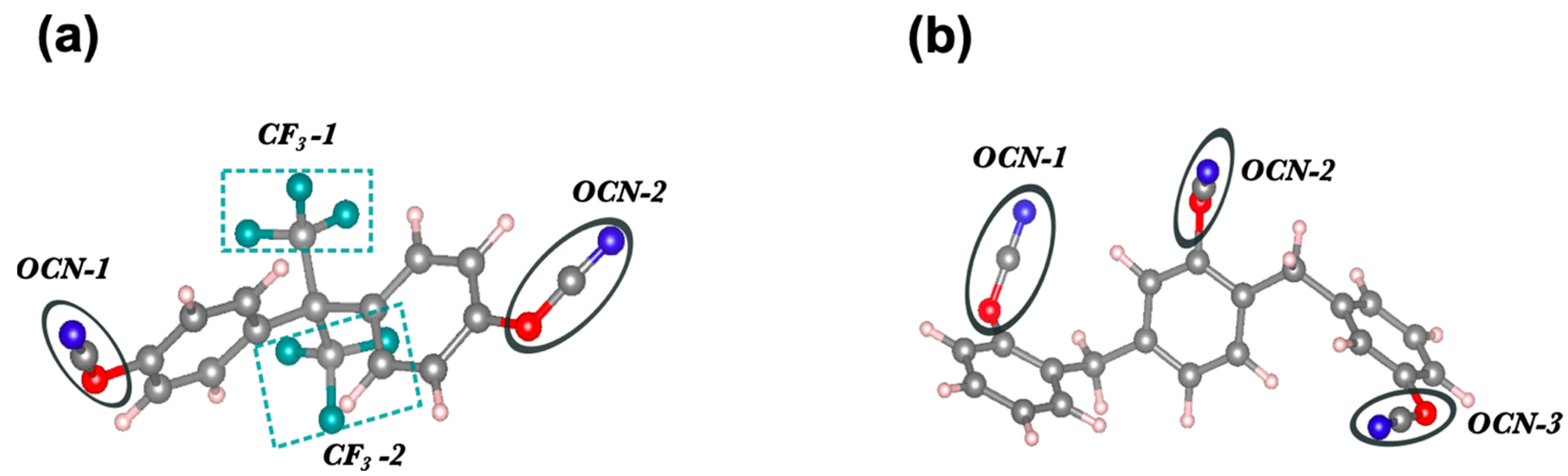
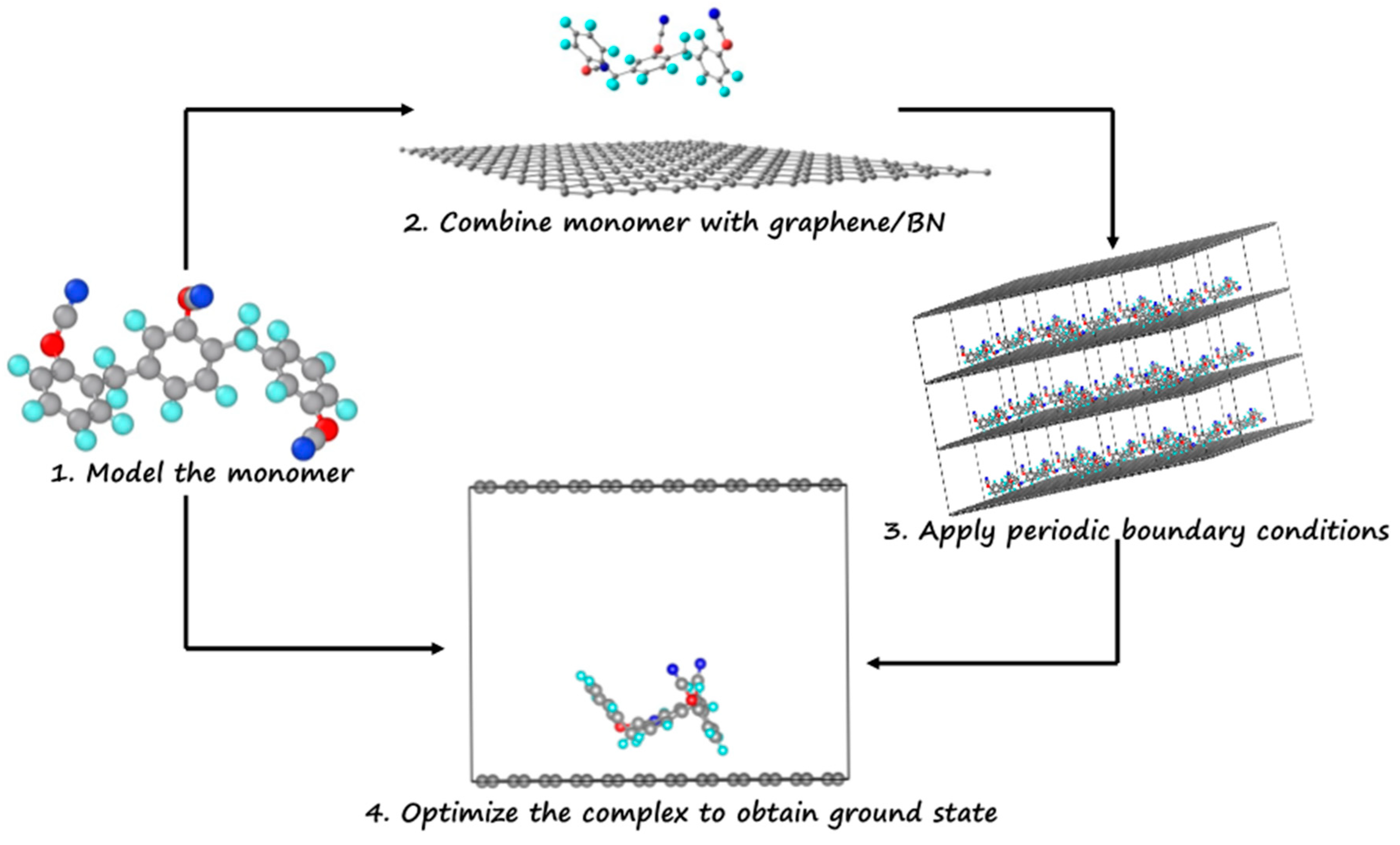
2. Computational Details
3. Results and Discussion
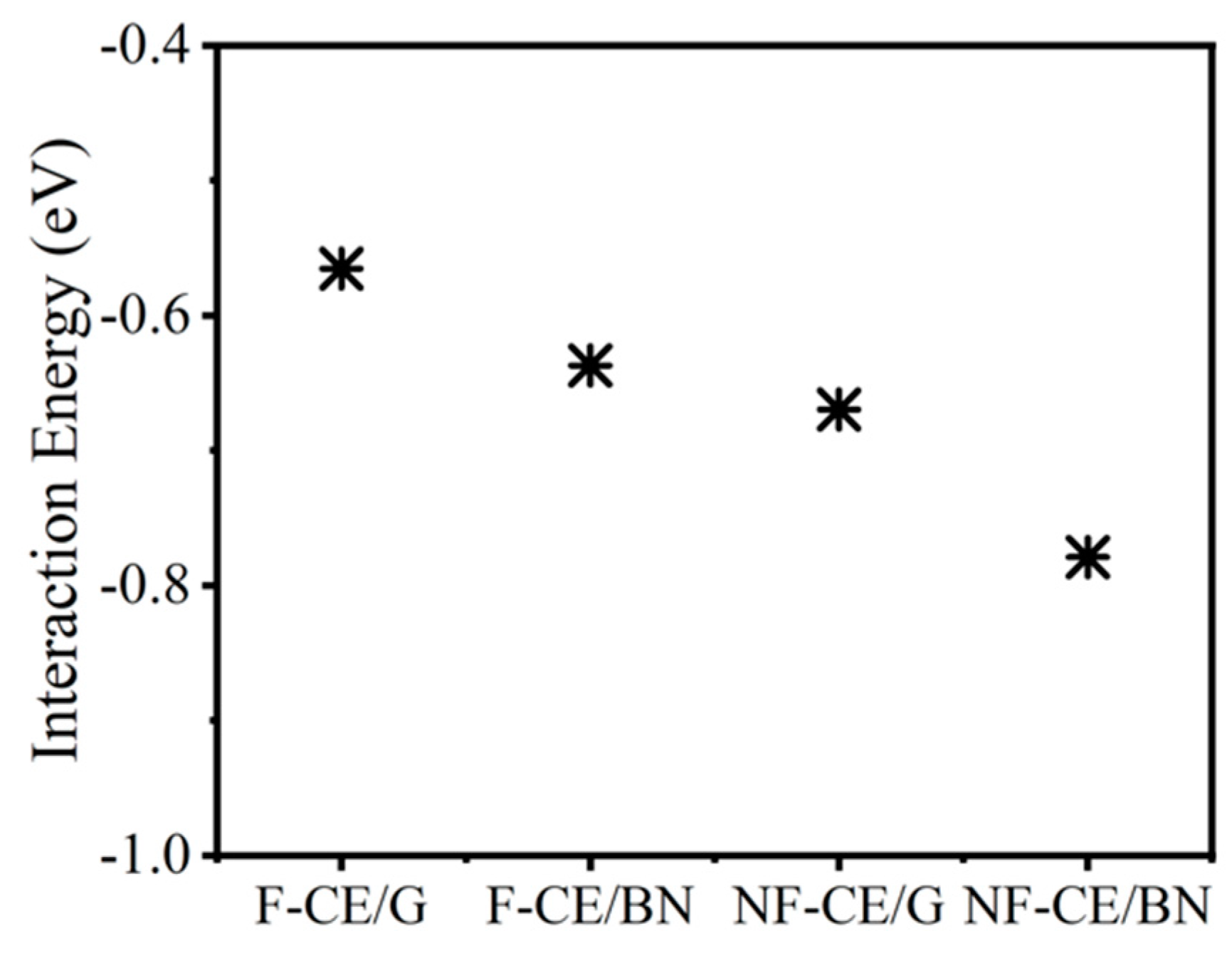
3.1. Interaction Energy
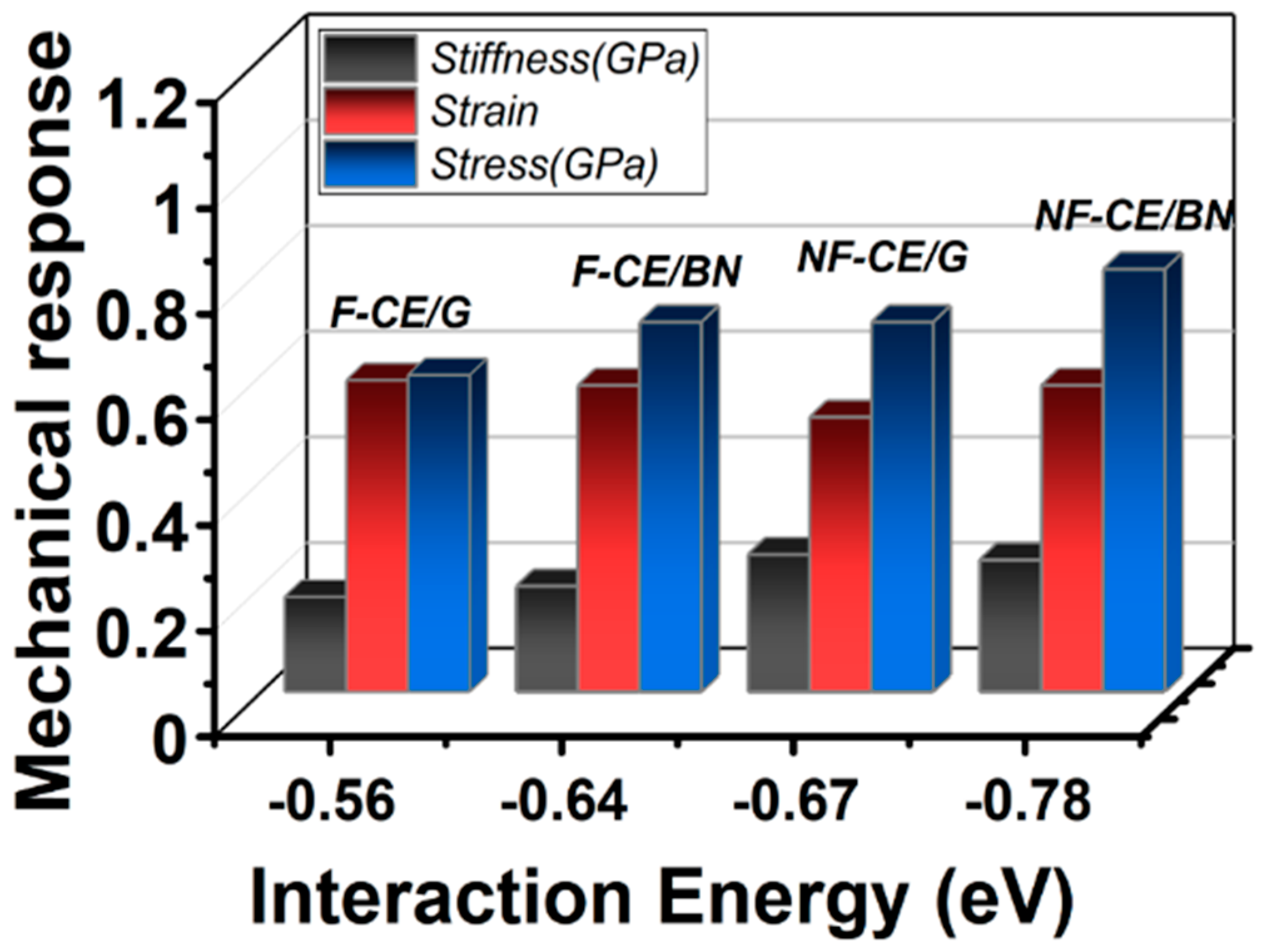
3.2. Mechanical Response
4. Summary
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nayak, N.V. Composite materials in aerospace applications. Int. J. Sci. Res. Publ. 2014, 4, 1–10. [Google Scholar]
- Kesarwani, S. Polymer composites in the aviation sector. J. Int. J. Eng. Res. Technol. 2017, 6, 518–525. [Google Scholar] [CrossRef]
- Rana, S.; Fangueiro, R. Advanced composites in aerospace engineering. In Advanced Composite Materials for Aerospace Engineering; Elsevier: Amsterdam, The Netherlands, 2016; pp. 1–15. [Google Scholar]
- Available online: https://www.nasa.gov/offices/oct/home/roadmaps/index.html (accessed on 20 August 2021).
- Kinloch, I.A.; Suhr, J.; Lou, J.; Young, R.J.; Ajayan, P.M. Composites with carbon nanotubes and graphene: An outlook. Science 2018, 362, 547–553. [Google Scholar] [CrossRef]
- Downes, R.D.; Hao, A.; Park, J.G.; Su, Y.F.; Liang, R.; Jensen, B.D.; Siochi, E.J.; Wise, K.E. Geometrically constrained self-assembly and crystal packing of flattened and aligned carbon nanotubes. Carbon 2015, 93, 953–966. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Q.; Holesinger, T.G.; Arendt, P.N.; Huang, J.; Kirven, P.D.; Clapp, T.G.; DePaula, R.F.; Liao, X.; Zhao, Y.; et al. Ultrastrong, stiff, and lightweight carbon-nanotube fibers. Adv. Mater. 2007, 19, 4198–4201. [Google Scholar] [CrossRef]
- Papageorgiou, D.G.; Kinloch, I.A.; Young, R.J. Mechanical properties of graphene and graphene-based nanocomposites. Prog. Mater. Sci. 2017, 90, 75–127. [Google Scholar] [CrossRef]
- Choi, D.; Wang, Q.; Azuma, Y.; Majima, Y.; Warner, J.H.; Miyata, Y.; Shinohara, H.; Kitaura, R. Fabrication and characterization of fully flattened carbon nanotubes: A new graphene nanoribbon analogue. Sci. Rep. 2013, 3, 1617. [Google Scholar] [CrossRef]
- Sachdeva, G.; Lobato, A.; Pandey, R.; Odegard, G.M. Mechanical Response of Polymer Epoxy/BMI Composites with Graphene and a Boron Nitride Monolayer from First Principles. ACS Appl. Polym. Mater. 2021, 3, 1052–1059. [Google Scholar] [CrossRef]
- Ottiger, P.; Pfaffen, C.; Leist, R.; Leutwyler, S.; Bachorz, R.A.; Klopper, W. Strong N−H··· π hydrogen bonding in amide− benzene interactions. J. Phys. Chem. B 2009, 113, 2937–2943. [Google Scholar] [CrossRef]
- Kandelbauer, A. Cyanate ester resins. In Handbook of Thermoset Plastics; Elsevier: Amsterdam, The Netherlands, 2022; pp. 587–617. [Google Scholar]
- Wooster, T.J.; Abrol, S.; Hey, J.M.; MacFarlane, D.R. Thermal, mechanical, and conductivity properties of cyanate ester composites. Compos. Part A Appl. Sci. Manuf. 2004, 35, 75–82. [Google Scholar] [CrossRef]
- Nair, C.R.; Mathew, D.; Ninan, K. Cyanate ester resins, recent developments. In New Polymerization Techniques and Synthetic Methodologies; Springer: Berlin/Heidelberg, Germany, 2001; pp. 1–99. [Google Scholar]
- Hamerton, I.; Hay, J.N. Recent technological developments in cyanate ester resins. High-Perform. Polym. 1998, 10, 163–174. [Google Scholar] [CrossRef]
- Brand, R.; Harrison, E. Development of Tough, Moisture Resistant Laminating Resins; General Dynamics Convair Div: San Diego, CA, USA, 1982. [Google Scholar]
- Fang, T.; Shimp, D.A. Polycyanate esters: Science and applications. Prog. Polym. Sci. 1995, 20, 61–118. [Google Scholar] [CrossRef]
- Hamerton, I. Chemistry and Technology of Cyanate Ester Resins; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Zhong, X.; Yap, Y.K.; Pandey, R.; Karna, S.P. First-principles study of strain-induced modulation of energy gaps of graphene/BN and BN bilayers. Phys. Rev. B 2011, 83, 193403. [Google Scholar] [CrossRef]
- Hod, O. Graphite and hexagonal boron-nitride have the same interlayer distance. Why? J. Chem. Theory Comput. 2012, 8, 1360–1369. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Sokalski, W.A.; Dyguda, E.; Leszczyński, J. Quantitative classification of covalent and noncovalent H-bonds. J. Phys. Chem. B 2006, 110, 6444–6446. [Google Scholar] [CrossRef]
- Chandra, Y.; Scarpa, F.; Adhikari, S.; Zhang, J.; Flores, E.S.; Peng, H.X. Pullout strength of graphene and carbon nanotube/epoxy composites. Compos. Part B Eng. 2016, 102, 1–8. [Google Scholar] [CrossRef]
- Barber, A.H.; Cohen, S.R.; Wagner, H.D. Measurement of carbon nanotube-polymer interfacial strength. Appl. Phys. Lett. 2003, 82, 4140–4142. [Google Scholar] [CrossRef]
- Cooper, C.A.; Cohen, S.R.; Barber, A.H.; Wagner, H.D. Detachment of nanotubes from a polymer matrix. Appl. Phys. Lett. 2002, 81, 3873–3875. [Google Scholar] [CrossRef]
- DiFrancia, C.; Ward, T.C.; Claus, R.O. The single-fiber pull-out test. 1: Review and interpretation. Compos. Part A Appl. Sci. Manuf. 1996, 27, 597–612. [Google Scholar] [CrossRef]
- Chorfi, H.; Lobato, Á.; Boudjada, F.; Salvadó, M.A.; Franco, R.; Baonza, V.G.; Recio, J.M. Computational modeling of tensile stress effects on the structure and stability of prototypical covalent and layered materials. Nanomaterials 2019, 9, 1483. [Google Scholar] [CrossRef]
- Tang, W.; Sanville, E.; Henkelman, G. A Grid-Based Bader Anal. Algorithm Without Lattice Bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef]
- Nasrabadi, A.T.; Foroutan, M. Interactions between polymers and single-walled boron nitride nanotubes: A molecular dynamics simulation approach. J. Phys. Chem. B 2010, 114, 15429–15436. [Google Scholar] [CrossRef]
- Gou, J.; Fan, B.; Song, G.; Khan, A. Study of affinities between the single-walled nanotube and epoxy resin using molecular dynamics simulation. Int. J. Nanosci. 2006, 5, 131–144. [Google Scholar] [CrossRef]
- Cozzi, F.; Ponzini, F.; Annunziata, R.; Cinquini, M.; Siegel, J.S. Polar Interactions between Stacked π Systems in Fluorinated 1, 8-Diarylnaphthalenes: Importance of Quadrupole Moments in Molecular Recognition. Angew. Chem. Int. Ed. Engl. 1995, 34, 1019–1020. [Google Scholar] [CrossRef]
- Hunter, C.A.; Sanders, J.K. The nature of. pi.-. pi. Interactions. J. Am. Chem. Soc. 1990, 112, 5525–5534. [Google Scholar] [CrossRef]
- Anslyn, E.V.; Dougherty, D.A. Modern Physical Organic Chemistry; University Science Books: Melville, NY, USA, 2006. [Google Scholar]
- Sinnokrot, M.O.; Valeev, E.F.; Sherrill, C.D. Estimates of the ab initio limit for π−π interactions: The benzene dimer. J. Am. Chem. Soc. 2002, 124, 10887–10893. [Google Scholar] [CrossRef]
- Lv, C.; Xue, Q.; Xia, D.; Ma, M.; Xie, J.; Chen, H. Effect of chemisorption on the interfacial bonding characteristics of graphene-polymer composites. J. Phys. Chem. C 2010, 114, 6588–6594. [Google Scholar] [CrossRef]
- Liang, Y.-Y.; Xu, J.Z.; Liu, X.Y.; Zhong, G.J.; Li, Z.M. Role of surface chemical groups on carbon nanotubes in nucleation for polymer crystallization: Interfacial interaction and steric effect. Polymer 2013, 54, 6479–6488. [Google Scholar] [CrossRef]
- Castner, D.G.; Grainger, D.W. Fluorinated Surfaces, Coatings, and Films; ACS Publications: Washington, DC, USA, 2001. [Google Scholar]
- Brosh, E.; Makov, G.; Shneck, R.Z. The spinodal constraint on the equation of state of expanded fluids. J. Phys. Condens. Matter 2003, 15, 2991. [Google Scholar] [CrossRef]
- Pisani, W.A.; Radue, M.S.; Patil, S.U.; Odegard, G.M. Interfacial modeling of flattened CNT composites with cyanate ester and PEEK polymers. Compos. Part B Eng. 2021, 211, 108672. [Google Scholar] [CrossRef]
- Kresse, G.; Gil, A.; Sautet, P. Significance of single-electron energies for the description of CO on Pt (111). Phys. Rev. B 2003, 68, 073401. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | ∆E (eV) | Population (%) | Q (e) | |
|---|---|---|---|---|
| Graphene | AroCy-F10 (F-CE) | −0.56 | 11.4 | 0.03 |
| Graphene | Primaset PT-30 (NF-CE) | −0.67 | 13.6 | 0.04 |
| BN monolayer | AroCy-F10 (F-CE) | −0.64 | 11.4 | 0.04 |
| BN monolayer | Primaset PT-30 (NF-CE) | −0.78 | 13.6 | 0.04 |
| Complex | (Out-of-Plane) Separation Point εc (%) | Transverse Strength fc (nN) | |
|---|---|---|---|
| Graphene | AroCy-F10 (fluorinated) | 5.9 ± 0.3 | 0.57 ± 0.01 |
| Graphene | Primaset PT-30 (non-fluorinated) | 5.2 ± 0.3 | 0.71 ± 0.03 |
| BN monolayer | AroCy-F10 (fluorinated) | 6.4 ± 0.2 | 0.63 ± 0.01 |
| BN monolayer | Primaset PT-30 (non-fluorinated) | 5.8 ± 0.3 | 0.80 ± 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sachdeva, G.; Lobato, Á.; Pandey, R.; Odegard, G.M. A Micromechanical Study of Interactions of Cyanate Ester Monomer with Graphene or Boron Nitride Monolayer. Materials 2024, 17, 108. https://doi.org/10.3390/ma17010108
Sachdeva G, Lobato Á, Pandey R, Odegard GM. A Micromechanical Study of Interactions of Cyanate Ester Monomer with Graphene or Boron Nitride Monolayer. Materials. 2024; 17(1):108. https://doi.org/10.3390/ma17010108
Chicago/Turabian StyleSachdeva, Geeta, Álvaro Lobato, Ravindra Pandey, and Gregory M. Odegard. 2024. "A Micromechanical Study of Interactions of Cyanate Ester Monomer with Graphene or Boron Nitride Monolayer" Materials 17, no. 1: 108. https://doi.org/10.3390/ma17010108
APA StyleSachdeva, G., Lobato, Á., Pandey, R., & Odegard, G. M. (2024). A Micromechanical Study of Interactions of Cyanate Ester Monomer with Graphene or Boron Nitride Monolayer. Materials, 17(1), 108. https://doi.org/10.3390/ma17010108