

Nano-Silicon@Exfoliated Graphite/Pyrolytic Polyaniline Composite of a High-Performance Cathode for Lithium Storage

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
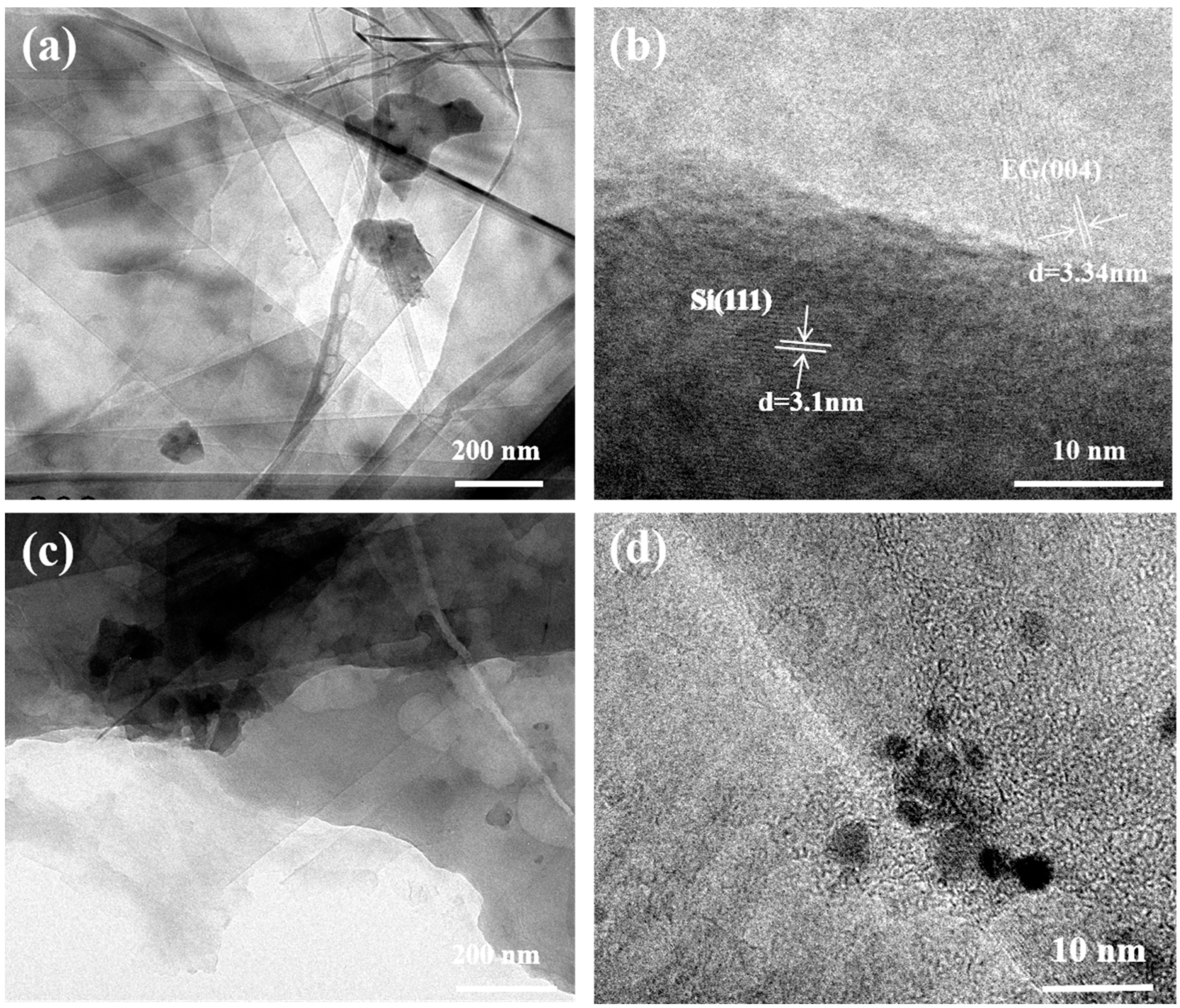
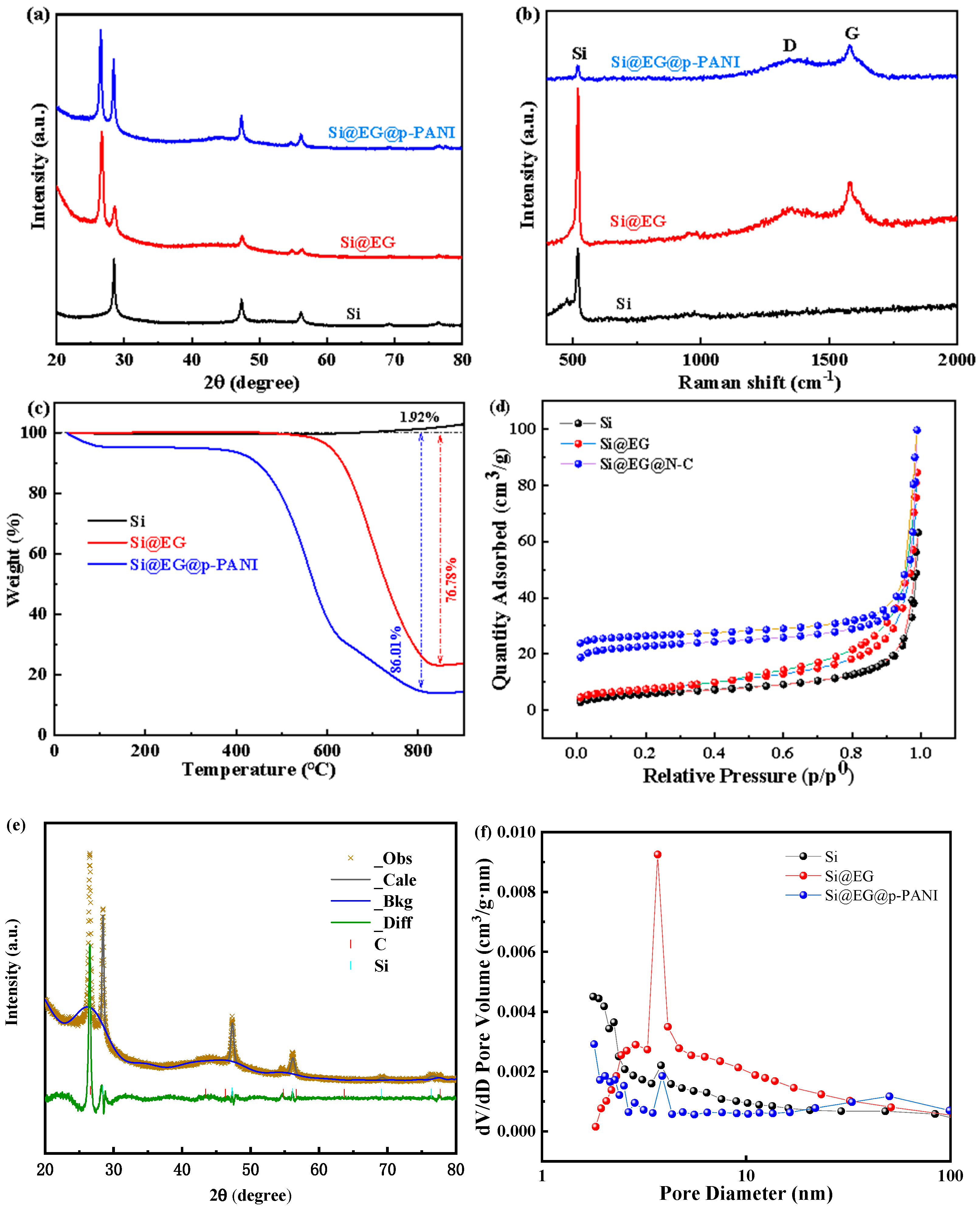
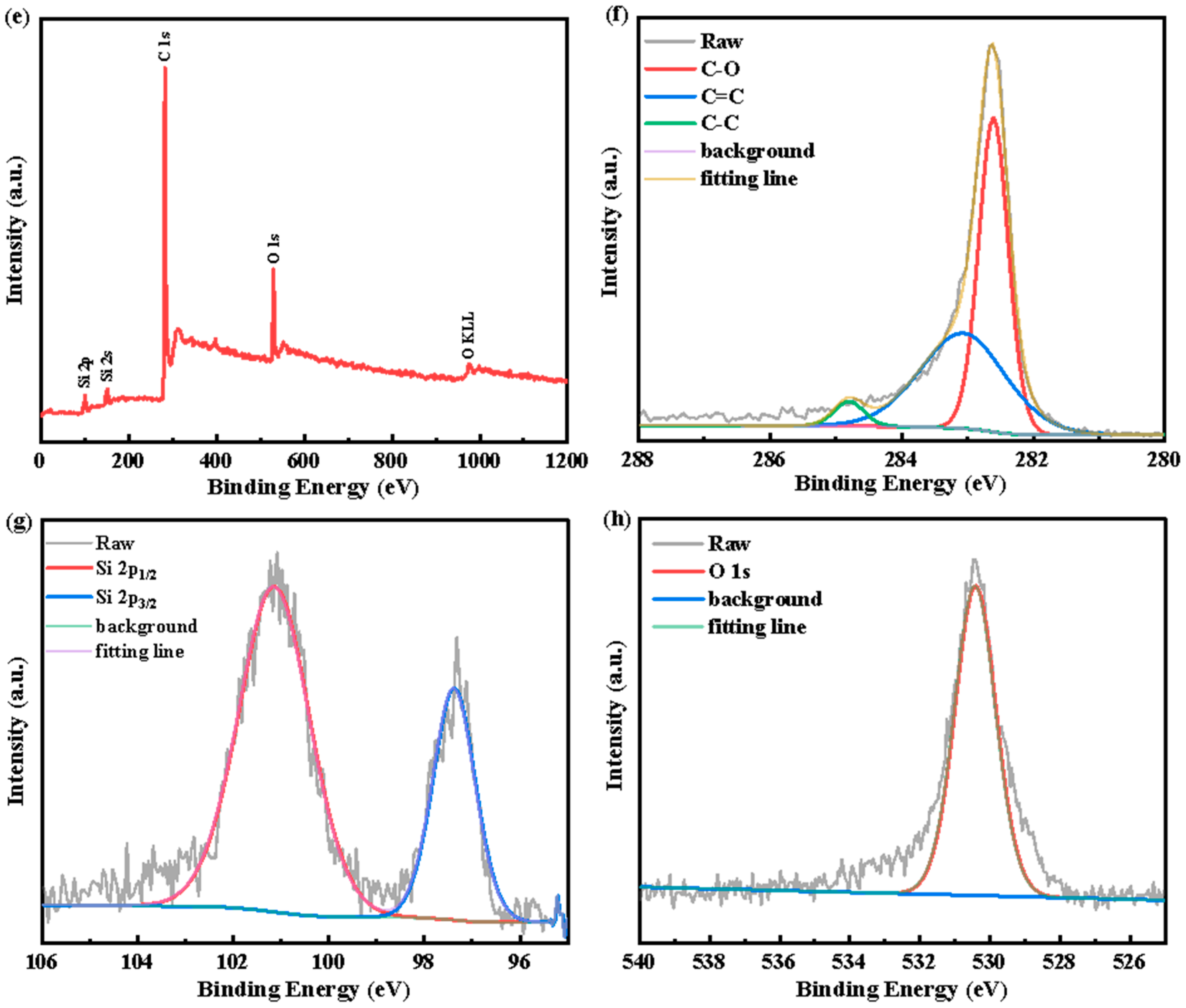
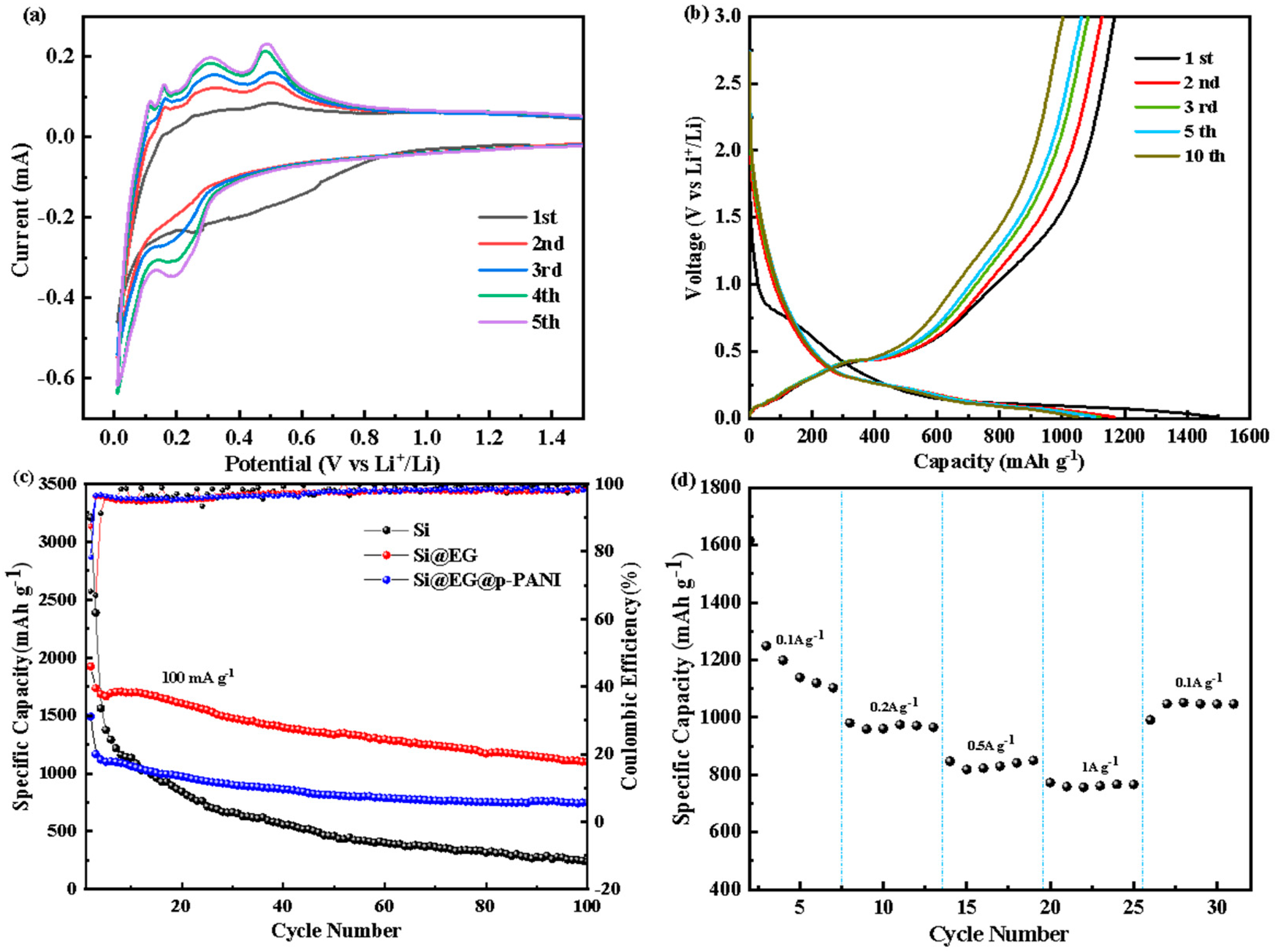
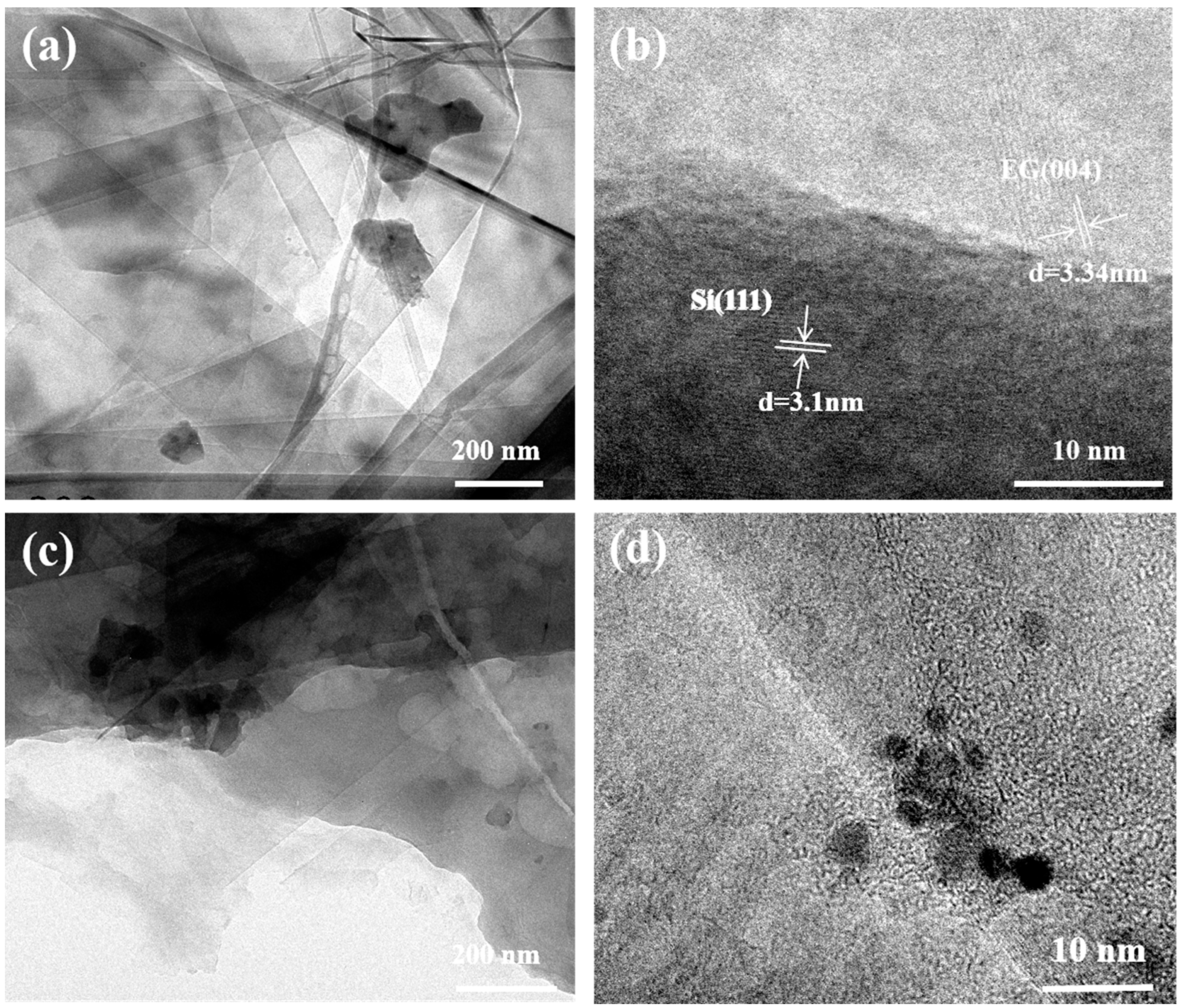
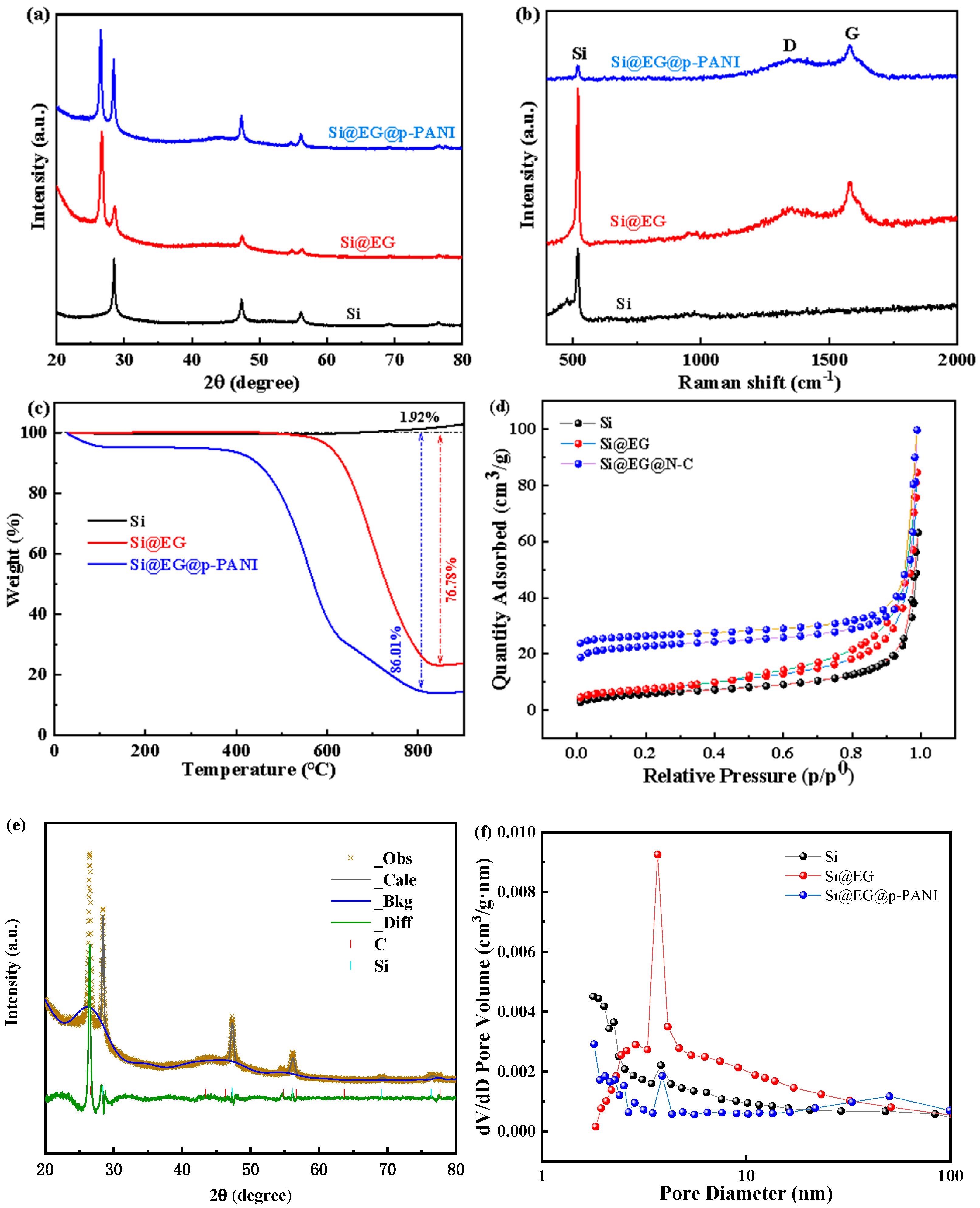
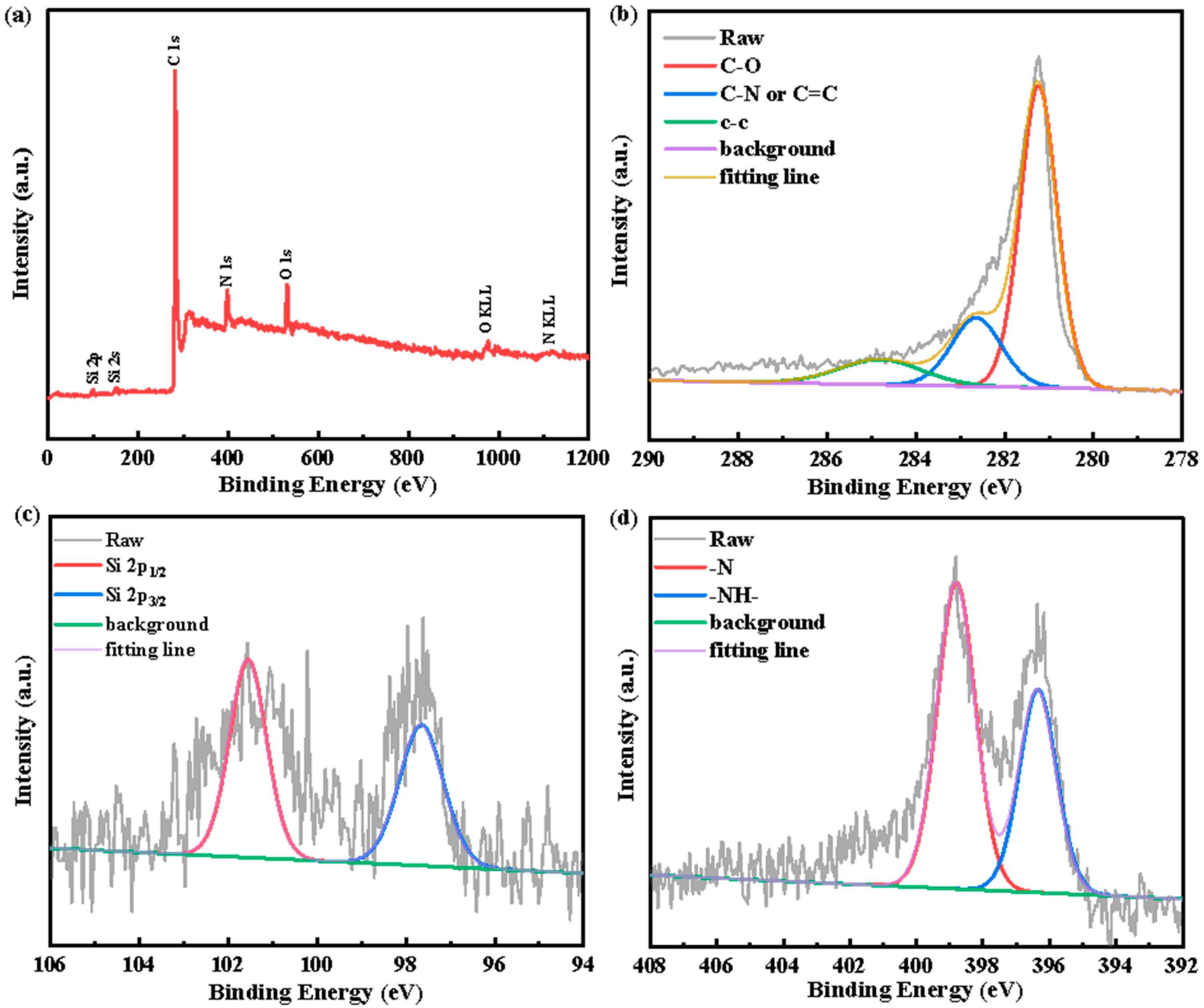
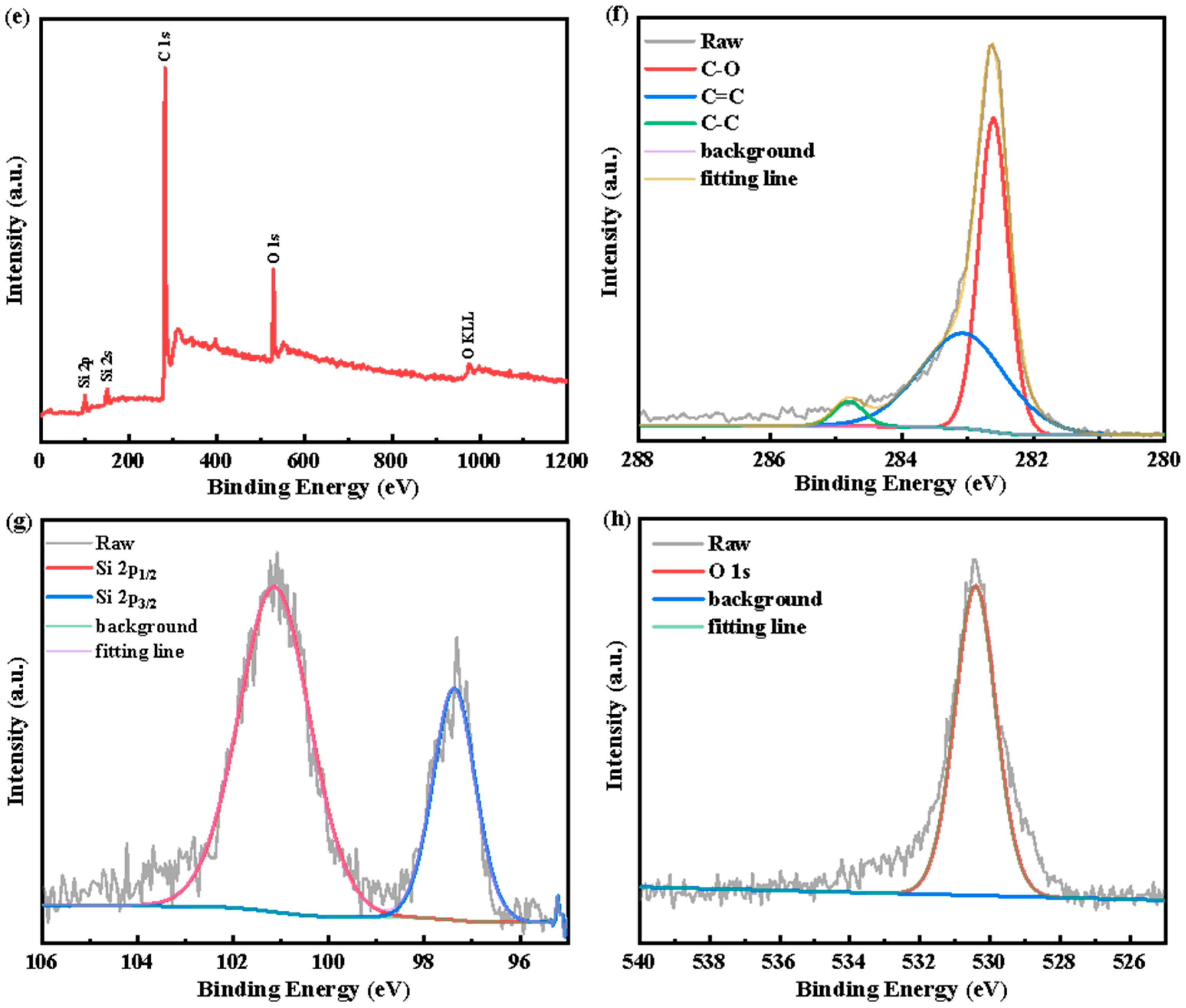
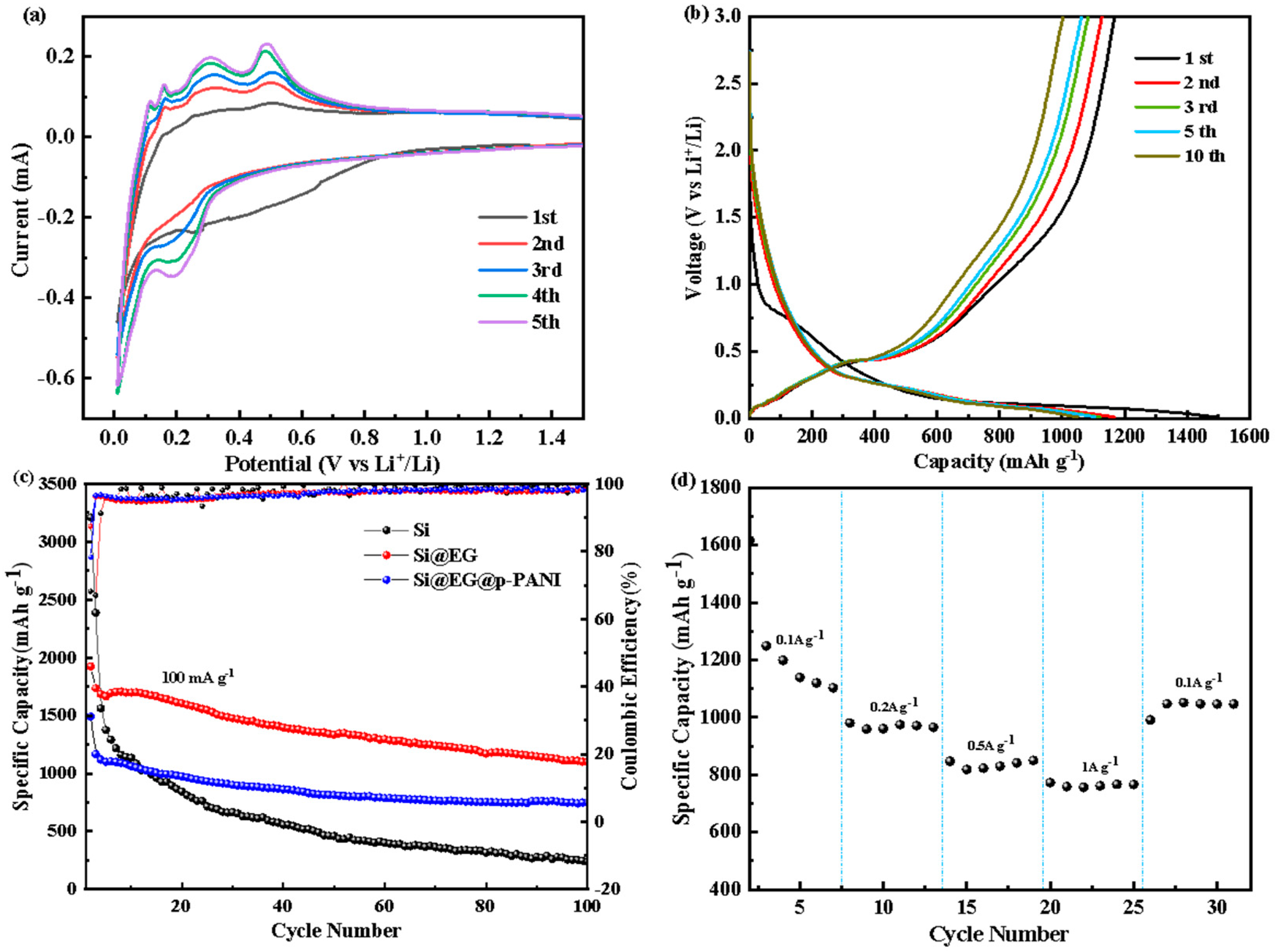
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kinoshita, K.; Zaghib, K. Negative electrodes for Li-ion batteries. J. Power Sources 2002, 110, 416–423. [Google Scholar] [CrossRef]
- Armand, M.; Tarascon, J.M. Building better batteries. Nature 2008, 451, 652–657. [Google Scholar] [CrossRef]
- Zhu, Z.Q.; Chen, X.D. Artificial interphase engineering of electrode materials to improve the overall performance of lithium-ion batteries. Nano Res. 2017, 10, 4115–4138. [Google Scholar] [CrossRef]
- Obrovac, M.N. Si-alloy negative electrodes for Li-ion batteries. Curr. Opin. Electrochem. 2018, 9, 8–17. [Google Scholar] [CrossRef]
- Wu, H.; Cui, Y. Designing nanostructured Si anodes for high energy lithium ion batteries. Nano Today 2012, 7, 414–429. [Google Scholar] [CrossRef]
- Obrovac, M.N.; Chevrier, V.L. Alloy Negative Electrodes for Li-Ion Batteries. Chem. Rev. 2014, 114, 11444–11502. [Google Scholar] [CrossRef]
- Li, J.; Dahn, J.R. An in-situ X-ray diffraction study of the reaction of Li with crystalline Si. J. Electrochem. Soc. 2007, 154, A156–A161. [Google Scholar] [CrossRef]
- Liu, X.H.; Huang, J.Y. In situ TEM electrochemistry of anode materials in lithium ion batteries. Energy Environ. Sci. 2011, 4, 3844–3860. [Google Scholar] [CrossRef]
- Kim, H.; Han, B.; Choo, J.; Cho, J. Three-Dimensional Porous Silicon Particles for Use in High-Performance Lithium Secondary Batteries. Angew. Chem. Int. Ed. 2008, 47, 10151–10154. [Google Scholar] [CrossRef] [PubMed]
- Magasinski, A.; Dixon, P.; Hertzberg, B.; Kvit, A.; Ayala, J.; Yushin, G. High-performance lithium-ion anodes using a hierarchical bottom-up approach. Nat. Mater. 2010, 9, 461. [Google Scholar] [CrossRef]
- Liu, J.; Kopold, P.; van Aken, P.A.; Maier, J.; Yu, Y. Energy Storage Materials from Nature through Nanotechnology: A Sustainable Route from Reed Plants to a Silicon Anode for Lithium-Ion Batteries. Angew. Chem. Int. Ed. 2015, 54, 9632–9636. [Google Scholar] [CrossRef] [PubMed]
- Kasavajjula, U.; Wang, C.S.; Appleby, A.J. Nano- and bulk-silicon-based insertion anodes for lithium-ion secondary cells. J. Power Sources 2007, 163, 1003–1039. [Google Scholar] [CrossRef]
- Jeong, M.G.; Islam, M.; Du, H.L.; Lee, Y.S.; Sun, H.H.; Choi, W.; Lee, J.K.; Chung, K.Y.; Jung, H.G. Nitrogen-doped Carbon Coated Porous Silicon as High Performance Anode Material for Lithium-Ion Batterie. Electrochim. Acta 2016, 209, 299–307. [Google Scholar] [CrossRef]
- Zhou, Z.W.; Liu, Y.T.; Xie, X.M.; Ye, X.Y. Constructing Novel Si@SnO2 Core-Shell Heterostructures by Facile Self-Assembly of SnO2 Nanowires on Silicon Hollow Nanospheres for Large, Reversible Lithium Storage. ACS Appl. Mater. Interfaces 2016, 8, 7092–7100. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, L.Y.; Eberman, K.W.; Turner, R.L.; Krause, L.J.; Dahn, J.R. Colossal reversible volume changes in lithium alloys. Electrochem. Solid-State Lett. 2001, 4, A137–A140. [Google Scholar] [CrossRef]
- Yang, D.D.; Shi, J.; Shi, J.H.; Yang, H.B. Simple synthesis of Si/Sn@C-G anodes with enhanced electrochemical properties for Li-ion batteries. Electrochim. Acta 2018, 259, 1081–1088. [Google Scholar] [CrossRef]
- Wang, F.; Song, C.; Zhao, B.; Sun, L.; Du, H. One-pot solution synthesis of carbon coated silicon nanoparticles as an anode material for lithium-ion batteries. Chem. Commun. 2020, 56, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Wu, Q.; Li, J.; Xiao, X.; Lott, A.; Lu, W.; Sheldon, B.W.; Wu, J. Silicon-Based Nanomaterials for Lithium-Ion Batteries: A Review. Adv. Energy Mater. 2014, 4, 1300882. [Google Scholar] [CrossRef]
- He, Y.; Jiang, L.; Chen, T.; Xu, Y.; Jia, H.; Yi, R.; Xue, D.; Song, M.; Genc, A.; Bouchet-Marquis, C.; et al. Progressive growth of the solid–electrolyte interphase towards the Si anode interior causes capacity fading. Nat. Nanotechnol. 2021, 16, 1113–1120. [Google Scholar] [CrossRef]
- Yu, Y.; Gu, L.; Zhu, C.; Tsukimoto, S.; van Aken, P.A.; Maier, J. Reversible storage of lithium in silver-coated three-dimensional macroporous silicon. Adv. Mater. 2010, 22, 2247–2250. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Hu, X.; Hou, Y.; Wen, Z. Tunable Synthesis of Yolk-Shell Porous Silicon@Carbon for Optimizing Si/C-Based Anode of Lithium-Ion Batteries. ACS Appl. Mater. Interfaces 2017, 9, 42084–42092. [Google Scholar] [CrossRef]
- Huang, R.; Xie, Y.; Chang, Q.; Xiong, J.; Guan, S.; Yuan, S.; Jiang, G. PANI-Encapsulated Si Nanocomposites with a Chemical Bond Linkage in the Interface Exhibiting Higher Electrochemical Stability as Anode Materials for Lithium-Ion Batteries. Nano 2019, 14, 1950078. [Google Scholar] [CrossRef]
- Kong, J.; Yee, W.A.; Wei, Y.; Yang, L.; Ang, J.M.; Phua, S.L.; Wong, S.Y.; Zhou, R.; Dong, Y.; Li, X.; et al. Silicon nanoparticles encapsulated in hollow graphitized carbon nanofibers for lithium-ion battery anodes. Nanoscale 2013, 5, 2967–2973. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, D.; Qiu, X.; Ma, Y.; Kong, D.; Mullen, K.; Li, X.; Zhi, L. Stable high-capacity and high-rate silicon-based lithium battery anodes upon two-dimensional covalent encapsulation. Nat. Commun. 2020, 11, 3826. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, Y.; Du, C.; Ren, Y.; Mu, T.; Zuo, P.; Yin, G.; Ma, Y.; Cheng, X.; Gao, Y. Polyaniline-encapsulated silicon on three-dimensional carbon nanotubes foam with enhanced electrochemical performance for lithium-ion batteries. J. Power Sources 2018, 381, 156–163. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, S.; Ruan, K.; Zhang, H.; Gu, J. Highly thermally conductive carbon nanotubes pillared exfoliated graphite/polyimide composites. NPJ Flex. Electron. 2021, 5, 16. [Google Scholar] [CrossRef]
- Gao, X.; Li, J.; Xie, Y.; Guan, D.; Yuan, C. A multilayered silicon-reduced graphite oxide electrode for high performance lithium-ion batteries. ACS Appl. Mater. Interfaces 2015, 7, 7855–7862. [Google Scholar] [CrossRef] [PubMed]
- Watthage, S.C.; Song, Z.; Shrestha, N.; Phillips, A.B.; Liyanage, G.K.; Roland, P.J.; Ellingson, R.J.; Heben, M.J. Enhanced Grain Size, Photoluminescence, and Photoconversion Efficiency with Cadmium Addition during the Two-Step Growth of CH3NH3PbI3. ACS Appl. Mater. Interfaces 2017, 9, 2334–2341. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Wu, M.; Feng, T.; Chen, C.; Tu, C.; Li, X.; Duan, C.; Xia, D.; Wang, D. High rate capability and long cycling life of graphite-coated silicon composite anodes for lithium ion batteries. Electrochim. Acta 2017, 256, 259–266. [Google Scholar] [CrossRef]
- Cong, R.; Park, H.H.; Jo, M.; Lee, H.; Lee, C.S. Synthesis and Electrochemical Performance of Electrostatic Self-Assembled Nano-Silicon@N-Doped Reduced Graphite Oxide/Carbon Nanofibers Composite as Anode Material for Lithium-Ion Batteries. Molecules 2021, 26, 4831. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Bai, Y.; Wang, M.; Wang, G.; Ma, Y.; Li, L.; Xiao, B.; Zheng, J. Self-assembly encapsulation of Si in N-doped reduced graphite oxide as lithium-ion battery anode with significantly enhanced electrochemical performance. Sustain. Energy Fuels 2019, 3, 1427–1438. [Google Scholar] [CrossRef]
- Men, Y.; Ambrogi, M.; Han, B.; Yuan, J. Fast Conversion of Ionic Liquids and Poly(Ionic Liquid)s into Porous Nitrogen-Doped Carbons in Air. Int. J. Mol. Sci. 2016, 17, 532. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Z.; Jia, R.; Yang, Y.; Zhu, F.; Yang, Z.; Huang, Y.; Li, X.; Xu, W. Facile electrostatic self-assembly of silicon/reduced graphite oxide porous composite by silica assist as high performance anode for Li-ion battery. Appl. Surf. Sci. 2018, 456, 379–389. [Google Scholar] [CrossRef]
- Goswami, S.; Nandy, S.; Fortunato, E.; Martins, R. Polyaniline and its composites engineering: A class of multifunctional smart energy materials. J. Solid State Chem. 2023, 317, 123679. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Method | Capacity [mAh g−1] | Reference |
|---|---|---|---|
| 3D porous bulk Si particles | reduction of SiCl4 with sodium naphthalide | 2800 (2 A g−1) | [9] |
| CN@P-Si | nitrogen-doped carbon coating | 2000 (0.8 A g−1) | [13] |
| Si@C | in situ synthesized viaa facile one-pot solution | 1120 (2 A g−1) | [17] |
| p-SiNPs@HC | hydrothermal | 1400 (0.2 A g−1) | [21] |
| Si/Sn@C-G | ball milling and annealing process | 612 (0.1 A g−1) | [16] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Q.; Zhu, Y.; Duan, H.; Zhu, L.; Zhang, Y.; Xu, H.; Egun, I.L.; He, H. Nano-Silicon@Exfoliated Graphite/Pyrolytic Polyaniline Composite of a High-Performance Cathode for Lithium Storage. Materials 2023, 16, 1584. https://doi.org/10.3390/ma16041584
Wu Q, Zhu Y, Duan H, Zhu L, Zhang Y, Xu H, Egun IL, He H. Nano-Silicon@Exfoliated Graphite/Pyrolytic Polyaniline Composite of a High-Performance Cathode for Lithium Storage. Materials. 2023; 16(4):1584. https://doi.org/10.3390/ma16041584
Chicago/Turabian StyleWu, Qian, Yinghong Zhu, Haojie Duan, Lin Zhu, Yuting Zhang, Hongqiang Xu, Ishioma Laurene Egun, and Haiyong He. 2023. "Nano-Silicon@Exfoliated Graphite/Pyrolytic Polyaniline Composite of a High-Performance Cathode for Lithium Storage" Materials 16, no. 4: 1584. https://doi.org/10.3390/ma16041584
APA StyleWu, Q., Zhu, Y., Duan, H., Zhu, L., Zhang, Y., Xu, H., Egun, I. L., & He, H. (2023). Nano-Silicon@Exfoliated Graphite/Pyrolytic Polyaniline Composite of a High-Performance Cathode for Lithium Storage. Materials, 16(4), 1584. https://doi.org/10.3390/ma16041584