
Difference in Structure and Electronic Properties of Oxygen Vacancies in α-Quartz and α-Cristobalite Phases of SiO2

 , , and
, , and 
Abstract
1. Introduction
2. Background
2.1. O Vacancy Configurations in SiO
2.2. Properties of -Cristobalite
3. Materials and Methods
4. Results of Calculations
4.1. Pristine Crystals

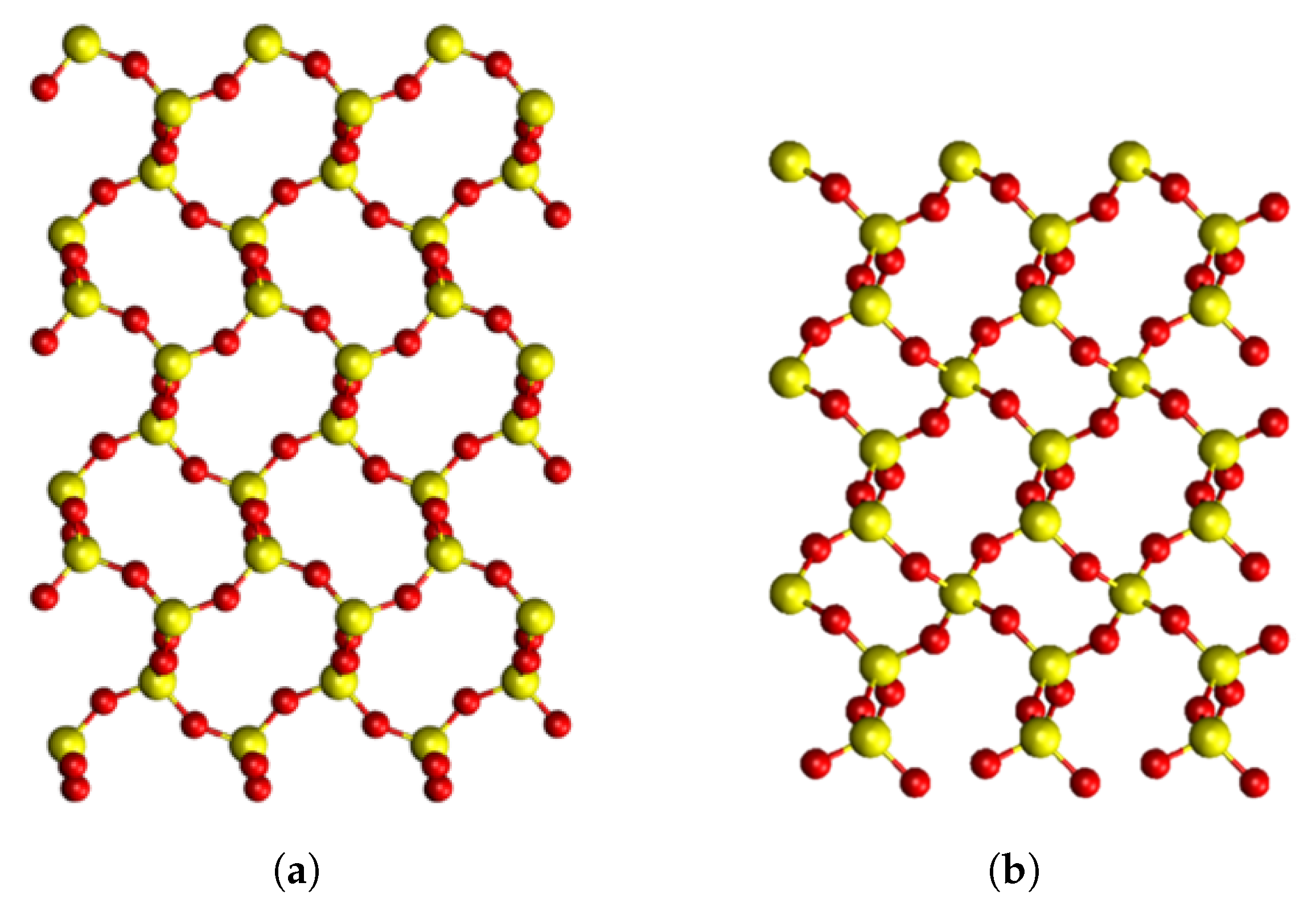{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | -Cristobalite | -Quartz | ||
|---|---|---|---|---|
| This Work | Literature Data | This Work | Literature Data | |
| Lattice Vectors | ||||
| a = b | 5.05 | 4.97 [87] | 4.93 | 4.91 [88] |
| c | 7.08 | 6.93 [87] | 5.43 | 5.40 [88] |
| Bond length | ||||
| Si-O | 1.604–1.609 | 1.600–1.607 [87] | 1.608 | 1.604 [88] |
| Si-O | 1.604–1.609 | 1.600–1.607 [87] | 1.612 | 1.613 [88] |
| Bond Angles | ||||
| O-Si-O | 108–111.8 | 108.1–111.3 [89] | 109.2–110.5 | 109.0–109.5 [88] |
| Si-O-Si | 150–153 | 146.6 [87] | 144.8–145.1 | 143.7 [88] |
| Density | 2.35 | 2.18–2.37 [44,45] | 2.41 | 2.47–2.70 [44,45,90] |
| Band gap | 8.57 | 8.54 [44] | 8.5 | 9.65 [75] |
4.2. Oxygen Vacancies
4.2.1. Geometric Structure and Stability
Neutral Vacancies
Positively Charged Vacancies
Negatively Charged Vacancies
4.2.2. Charge Transition Levels
4.2.3. Optical Absorption
4.2.4. EPR Parameters
5. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT | Density Functional Theory |
| -C | -cristobalite |
| -Q | -quartz |
| a-SiO | Amorphous silicon dioxide |
| CT | Charge transition |
| ODC | Oxygen deficient center |
| EPR | Electron paramagnetic resonance |
| Opal-CT | Opal cristobalite tridymite |
| LDA | Local density approximation |
| GTH | Goedecker-Teter-Hutter |
| BFGS | Broyden–Fletcher–Goldfarb–Shanno |
| XC | Exchange-correlation |
| PBE | Perdew-Burke-Ernzerhof |
| ADMM | Auxiliary density matrix method |
| TD-DFT | Time-dependent density functional theory |
| TC-LRC | Truncated coulomb long-range correction |
| NEB | Nudged elastic band |
| CI-NEB | Climbing image nudged elastic band |
| GGA | Generalized gradient approximation |
| HSE | Heyd-Scuseria-Ernzerhof |
| VB | Valence band |
| CB | Conduction band |
| CBM | Conduction band minimum |
| CTL | Charge transition level |
| TS | Transition state |
| OA | Optical absorption |
| AE | all-electron |
References
- Feigl, F.J.; Fowler, W.B.; Yip, K.L. Oxygen Vacancy Model for the E′-center in SiO2. Solid State Commun. 1974, 14, 225–229. [Google Scholar] [CrossRef]
- Pacchioni, G.; Ierano, G.; Márquez, A.M. Optical absorption and nonradiative decay mechanism of E′ center in silica. Phys. Rev. Lett. 1998, 81, 377–380. [Google Scholar] [CrossRef]
- Sushko, P.V.; Mukhopadhyay, S.; Mysovsky, A.S.; Sulimov, V.B.; Taga, A.; Shluger, A. Structure and properties of defects in amorphous silica: New insights from embedded cluster calculations. J. Phys. Condens. Matter 2005, 17, S2115. [Google Scholar] [CrossRef]
- Boero, M.; Pasquarello, A.; Sarnthein, J.; Car, R. Structure and Hyperfine Parameters of E1′ Centers in α-Quartz and in Vitreous SiO2. Phys. Rev. Lett. 1997, 78, 887–890. [Google Scholar] [CrossRef]
- Blöchl, P.E. First-principles calculations of defects in oxygen-deficient silica exposed to hydrogen. Phys. Rev. B 2000, 62, 6158–6179. [Google Scholar] [CrossRef]
- Lu, Z.Y.; Nicklaw, C.J.; Fleetwood, D.; Schrimpf, R.D.; Pantelides, S. Structure, Properties, and Dynamics of Oxygen Vacancies in Amorphous SiO2. Phys. Rev. Lett. 2002, 89, 285505. [Google Scholar] [CrossRef]
- Chadi, D.J. Negative-U property of the oxygen vacancy defect in SiO2 and its implication for the E′ center in α-quartz. Appl. Phys. Lett. 2003, 83, 437–439. [Google Scholar] [CrossRef]
- Giacomazzi, L.; Martin-Samos, L.; Boukenter, A.; Ouerdane, Y.; Girard, S.; Richard, N. EPR parameters of E′ centers in v-SiO2 from first-principles calculations. Phys. Rev. B 2014, 90, 014108. [Google Scholar] [CrossRef]
- Skuja, L. Optically active oxygen-deficiency-related centers in amorphous silicon dioxide. J.-Non-Cryst. Solid. 1998, 239, 16–48. [Google Scholar] [CrossRef]
- Rudra, J.K.; Fowler, W.B. Oxygen vacancy and the E′1 center in crystalline SiO2. Phys. Rev. B 1987, 35, 8223–8230. [Google Scholar] [CrossRef]
- Sushko, P.V.; Mukhopadhyay, S.; Stoneham, A.M.; Shluger, A.L. Oxygen vacancies in amorphous silica: Structure and distribution of properties. Microelectron. Eng. 2005, 80, 292–295. [Google Scholar] [CrossRef]
- Kimmel, A.; Sushko, P.; Shluger, A.; Bersuker, G. Positive and Negative Oxygen Vacancies in Amorphous Silica. ECS Trans. 2009, 19, 3–17. [Google Scholar] [CrossRef]
- Munde, M.S.; Gao, D.Z.; Shluger, A.L. Diffusion and aggregation of oxygen vacancies in amorphous silica. J. Phys. Condens. Matter 2017, 29, 245701. [Google Scholar] [CrossRef] [PubMed]
- Ganduglia-Pirovano, M.V.; Hofmann, A.; Sauer, J. Oxygen vacancies in transition metal and rare earth oxides: Current state of understanding and remaining challenges. Surf. Sci. Rep. 2007, 62, 219–270. [Google Scholar] [CrossRef]
- Jivanescu, M.; Stesmans, A.; Afanas’ev, V.V. Multifrequency ESR analysis of the Eδ′ defect in a-SiO2. Phys. Rev. B 2011, 83, 094118. [Google Scholar] [CrossRef]
- Arrigoni, M.; Madsen, G.K.H. Evolutionary computing and machine learning for discovering of low-energy defect configurations. Npj Comput. Mater. 2021, 7, 71. [Google Scholar] [CrossRef]
- Milardovich, D.; Jech, M.; Waldhoer, D.; El-Sayed, A.M.B.; Grasser, T. Machine Learning Prediction of Defect Structures in Amorphous Silicon Dioxide. In Proceedings of the IEEE 51st European Solid-State Device Research Conference (ESSDER2021), Grenoble, France, 13–22 September 2021; pp. 239–242. [Google Scholar] [CrossRef]
- Qiao, Z.; Liu, Q.; Zhang, S.; Wu, Y. The mineralogical characteristics between opaline silica in bentonite and α-cristobalite. Solid State Sci. 2019, 96, 105948. [Google Scholar] [CrossRef]
- Önal, M.; Kahraman, S.; Sarikaya, Y. Differentiation of α-cristobalite from opals in bentonites from Turkey. Appl. Clay Sci. 2007, 35, 25–30. [Google Scholar] [CrossRef]
- Griscom, D.L. Trapped-electron centers in pure and doped glassy silica: A review and synthesis. J.-Non-Cryst. Solid. 2011, 357, 1945–1962. [Google Scholar] [CrossRef]
- Imai, H.; Arai, K.; Hosono, H.; Abe, Y. Two types of oxygen-deficient centers in synthetic silica glass. Phys. Rev. B 1988, 38, 12773–12775. [Google Scholar] [CrossRef]
- Weeks, R.A. Paramagnetic resonance of lattice defects in irradiated quartz. J. Appl. Phys. 1956, 27, 1376–1381. [Google Scholar] [CrossRef]
- Weeks, R.A.; Magruder, R.H.; Stesmans, A. Review of some experiments in the 50 year saga of the E′ center and suggestions for future research. J.-Non-Cryst. Solid. 2008, 354, 208–216. [Google Scholar] [CrossRef]
- Mashkovtsev, R.I.; Pan, Y. Nature of paramagnetic defects in alpha-quartz: Progresses in the first decade of the 21st century. In New Developments in Quartz Research: Varieties, Crystal Chemistry and Uses in Technology; Nova Science Publishers: New York, NY, USA, 2013; pp. 66–104. [Google Scholar]
- Mashkovtsev, R.I.; Nepomnyashchikh, A.I.; Zhaboedov, A.P.; Paklin, A.S. EPR study of the E′ defects in optical glasses and cristobalite. Europhys. Lett. 2021, 133, 14003. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Sushko, P.V.; Stoneham, A.M.; Shluger, A.L. Modeling of the structure and properties of oxygen vacancies in amorphous silica. Phys. Rev. B 2004, 70, 195203. [Google Scholar] [CrossRef]
- Buscarino, G.; Agnello, S.; Gelardi, F.; Parlato, A. Electron paramagnetic resonance investigation on the hyperfine structure of the Eδ′ center in amorphous silicon dioxide. J.-Non-Cryst. Solid. 2007, 353, 518–521. [Google Scholar] [CrossRef]
- Fowler, W.B.; Edwards, A.H. Theory of defects and defect processes in silicon dioxide. J.-Non-Cryst. Solid. 1997, 222, 33–41. [Google Scholar] [CrossRef]
- Mysovsky, A.S.; Sushko, P.V.; Mukhopadhyay, S.; Edwards, A.H.; Shluger, A.L. Calibration of embedded-cluster method for defect studies in amorphous silica. Phys. Rev. B 2004, 69, 085202. [Google Scholar] [CrossRef]
- Kayama, M.; Nagaoka, H.; Niihara, T. Lunar and martian silica. Minerals 2018, 8, 8070267. [Google Scholar] [CrossRef]
- Brearley, A.J.; Jones, R.H. Chondritic meteorites. In Planetary Materials; Reviews in Mineralogy; De Gruyter: Berlin, Germany, 1998; Volume 36, pp. 3.1–3.398. [Google Scholar]
- Schipper, C.I.; Rickard, W.D.; Reddy, S.M.; Saxey, D.W.; Castro, J.M.; Fougerouse, D.; Quadir, Z.; Conway, C.; Prior, D.J.; Lilly, K. Volcanic SiO2-cristobalite: A natural product of chemical vapor deposition. Am. Mineral. 2020, 105, 510–524. [Google Scholar] [CrossRef]
- Smith, D.K. Opal, cristobalite, and tridymite: Noncrystallinity versus crystallinity, nomenclature of the silica minerals and bibliography. Power Diffr. 1998, 13, 2–19. [Google Scholar] [CrossRef]
- Correcher, V.; Garcia-Guinea, J.; Bustillo, M.A.; Garcia, R. Study of the thermoluminescence emission of a natural α-cristobalite. Radiat. Eff. Defects Solid. 2009, 164, 59–67. [Google Scholar] [CrossRef]
- Dağlar, S.; Kahya, N.D.; Ustunisik, G.; Onal, M. Thermal Crystallization Kinetics of an Opal-like Biogenic Silica. Silicon 2022, 14, 7211–7217. [Google Scholar] [CrossRef]
- Dera, P.; Lazarz, J.D.; Prakapenka, V.B.; Barkley, M.; Downs, R.T. New insights into the high-pressure polymorphism of SiO2 cristobalite. Phys. Chem. Miner. 2011, 38, 517–529. [Google Scholar] [CrossRef]
- Salvadó, M.A.; Pertierra, P.; Morales-García, A.; Menéndez, J.M.; Recio, J.M. Understanding chemical changes across the α-cristobalite to stishovite transition path in silica. J. Phys. Chem. C 2013, 117, 8950–8958. [Google Scholar] [CrossRef]
- Parise, J.B.; Yeganeh-Haeri, A.; Weidner, D.J.; Jorgensen, J.D.; Saltzberg, M.A. Pressure-induced phase transition and pressure dependence of crystal structure in low (α) and Ca/Al-doped cristobalite. J. Appl. Phys. 1994, 75, 1361–1367. [Google Scholar] [CrossRef]
- Hatch, D.; Ghose, S. The α-β phase transition in cristobalite, SiO2. Phys. Chem. Miner. 1991, 17, 554–562. [Google Scholar] [CrossRef]
- Garg, N.; Sharma, S.M. Classical molecular dynamical simulations of high pressure behavior of alpha cristobalite (SiO2). J. Phys. Condens. Matter 2007, 19, 456201. [Google Scholar] [CrossRef]
- Cernok, A.; Marquardt, K.; Caracas, R.; Bykova, E.; Habler, G.; Liermann, H.P.; Hanfland, M.; Mezouar, M.; Bobocioiu, E.; Dubrovinsky, L. Compressional pathways of α-cristobalite, structure of cristobalite X-I, and towards the understanding of seifertite formation. Nat. Commun. 2017, 8, 15647. [Google Scholar] [CrossRef]
- Shelton, H.; Bi, T.; Zurek, E.; Smith, J.; Dera, P. The Ideal Crystal Structure of Cristobalite X-I: A Bridge in SiO2 Densification. J. Phys. Chem. C 2018, 122, 17437–17446. [Google Scholar] [CrossRef]
- Güler, E.; Uğur, G.; Uğur, Ş.; Güler, M. A theoretical study for the band gap energies of the most common silica polymorphs. Chin. J. Phys. 2020, 65, 472–480. [Google Scholar] [CrossRef]
- Warmbier, R.; Mohammed, F.; Quandt, A. Optical and other material properties of SiO2 from ab initio studies. Opt. Eng. 2014, 53, 071808. [Google Scholar] [CrossRef]
- Sevik, C.; Bulutay, C. Theoretical study of the insulating oxides and nitrides: SiO2, GeO2, Al2O3, Si3N4, and Ge3N4. J. Mater. Sci. 2007, 42, 6555–6565. [Google Scholar] [CrossRef]
- Xu, Y.N.; Ching, W.Y. Electronic and optical properties of all polymorphic forms of silicon dioxide. Phys. Rev. B 1991, 44, 11048–11059. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.; Kawamoto, K.; Kageshima, H.; Uematsu, M.; Nakamura, K.; Ito, T. A first-principles study of O2 incorporation and its diffusion in compressively strained high-density silicon oxides. Thin Solid Films 2006, 508, 311–314. [Google Scholar] [CrossRef]
- Nagao, K.; Neaton, B.; Ashcroft, W. First-principles study of adhesion at Cu/SiO2 interfaces. Phys. Rev. B 2003, 68, 125403. [Google Scholar] [CrossRef]
- Rimola, A.; Costa, D.; Sodupe, M.; Lambert, J.F.; Ugliengo, P. Silica surface features and their role in the adsorption of biomolecules: Computational modeling and experiments. Chem. Rev. 2013, 113, 4216–4313. [Google Scholar] [CrossRef]
- Huang, L.; Kieffer, J. Amorphous-amorphous transitions in silica glass. I. Reversible transitions and thermomechanical anomalies. Phys. Rev. B 2004, 69, 224203. [Google Scholar] [CrossRef]
- Emami, F.S.; Puddu, V.; Berry, R.J.; Varshney, V.; Patwardhan, S.V.; Perry, C.C.; Heinz, H. Erratum: Force Field and a Surface Model Database for Silica to Simulate Interfacial Properties in Atomic Resolution. Chem. Mater. 2016, 26, 2647–2658. [Google Scholar] [CrossRef]
- Yang, J.; Meng, S.; Xu, L.; Wang, E.G. Water adsorption on hydroxylated silica surfaces studied using the density functional theory. Phys. Rev. B 2005, 71, 035413. [Google Scholar] [CrossRef]
- Valencia, E. Calculation of the adsorption potential energy of water vapour on α-cristobalite. J. Chem. Soc. Faraday Trans. 1994, 90, 2555–2559. [Google Scholar] [CrossRef]
- Shan, T.R.; Devine, B.D.; Phillpot, S.R.; Sinnott, S.B. Molecular dynamics study of the adhesion of Cu/SiO2 interfaces using a variable-charge interatomic potential. Phys. Rev. B 2011, 83, 115327. [Google Scholar] [CrossRef]
- Tang, C.; Cai, L. The effect of surface heterogeneity between α-quartz and α-cristobalite on adsorption behaviors toward Cu2+ solution. Colloids Surfaces A Physicochem. Eng. Asp. 2021, 609, 125651. [Google Scholar] [CrossRef]
- Vandevondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef]
- VandeVondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef] [PubMed]
- Goedecker, S.; Teter, M. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [Google Scholar] [CrossRef] [PubMed]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of crystals with the quasi-Newton method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Guidon, M.; Hutter, J.; VandeVondele, J. Robust periodic Hartree-Fock exchange for large-scale simulations using Gaussian basis sets. J. Chem. Theory Comput. 2009, 5, 3010–3021. [Google Scholar] [CrossRef]
- Guidon, M.; Hutter, J.; Vandevondele, J. Auxiliary density matrix methods for Hartree-Fock exchange calculations. J. Chem. Theory Comput. 2010, 6, 2348–2364. [Google Scholar] [CrossRef]
- Zhang, S.B.; Northrup, J.E. Chemical Potential Dependence of Defect Formation Energies in GaAs: Application to Ga Self-Diffusion. Phys. Rev. Lett. 1991, 67, 2339–2342. [Google Scholar] [CrossRef]
- Makov, G.; Payne, M.C. Periodic boundary conditions in ab initio calculations. Phys. Rev. B 1995, 51, 4014–4022. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.T.; Hine, N.D. Anisotropic charge screening and supercell size convergence of defect formation energies. Phys. Rev. B 2013, 87, 094111. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. Climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Iannuzzi, M.; Chassaing, T.; Wallman, T.; Hutter, J.; Hutter, J. Ground and Excited State Density Functional Calculations with the Gaussian and Augmented-Plane-Wave Method. Chimia 2005, 59, 499–503. [Google Scholar] [CrossRef]
- Strand, J.; Chulkov, S.K.; Watkins, M.B.; Shluger, A.L. First principles calculations of optical properties for oxygen vacancies in binary metal oxides. J. Chem. Phys. 2019, 150, 044702. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Lippert, G.; Hutter, J.; Parrinello, M. The Gaussian and augmented-plane-wave density functional method for ab initio molecular dynamics simulations. Theor. Chem. Acc. 1999, 103, 124–140. [Google Scholar] [CrossRef]
- Jensen, F. The optimum contraction of basis sets for calculating spin–spin coupling constants. Theor. Chem. Acc. 2010, 126, 371–382. [Google Scholar] [CrossRef]
- Weber, V.; Iannuzzi, M.; Giani, S.; Hutter, J.; Declerck, R.; Waroquier, M. Magnetic linear response properties calculations with the Gaussian and augmented-plane-wave method. J. Chem. Phys. 2009, 131, 014106. [Google Scholar] [CrossRef]
- Bart, F.; Gautier, J.; Henriot, M. (011¯0) α-quartz Surface: A LEED, XANES and ELS Study. Surf. Sci. 1992, 274, 317–328. [Google Scholar] [CrossRef]
- Bart, F.; Gautier, M.; Jollet, F.; Duraud, J. Electronic structure of the (0001) and (101¯0) quartz surfaces and of their defects as observed by reflection electron energy loss spectroscopy (REELS). Surf. Sci. 1994, 306, 342–358. [Google Scholar] [CrossRef]
- Garvie, L.A.J.; Rezb, P.; Alvarezb, J.R.; Buseck’, P.R. Interband Transitions of Crystalline and Amorphous SiO2: An Electron Energy-Loss Spectroscopy (EELS) Study of the Low-Loss Region. Solid State Commun. 1998, 106, 303–307. [Google Scholar] [CrossRef]
- Garvie, L.A.J.; Rez, P.; Alvarez, J.R.; Buseck, P.R.; Craven, A.J.; Brydson, R. Bonding in α-quartz (SiO2): A view of the unoccupied states. Am. Mineral. 2000, 85, 732–738. [Google Scholar] [CrossRef]
- Kresse, G.; Marsman, M.; Hintzsche, L.E.; Flage-Larsen, E. Optical and electronic properties of Si3N4 and α-SiO2. Phys. Rev. B 2012, 85, 045205. [Google Scholar] [CrossRef]
- El-Sayed, A.M.; Watkins, M.B.; Afanas’ev, V.V.; Shluger, A.L. Nature of intrinsic and extrinsic electron trapping in SiO2. Phys. Rev. B 2014, 89, 125201. [Google Scholar] [CrossRef]
- Maj, S. Energy gap and density in SiO2 polymorphs. Phys. Chem. Miner. 1988, 15, 271–273. [Google Scholar] [CrossRef]
- Li, Y.P.; Ching, W.Y. Band structures of all polycrystalline forms of silicon dioxide. Phys. Rev. B 1985, 31, 2172–2179. [Google Scholar] [CrossRef]
- Gnani, E.; Reggiani, S.; Colle, R.; Rudan, M. Band-Structure Calculations Of SiO2 by Means of Hartree-Fock and density-functional techniques. IEEE Trans. Electron Devices 2000, 47, 1795–1803. [Google Scholar] [CrossRef]
- Mozzi, R.L.; Warren, B.E. Structure of Vitreous Silica. J. Appl. Crystallogr. 1969, 2, 164–172. [Google Scholar] [CrossRef]
- El-Sayed, A.M.; Watkins, M.B.; Shluger, A.L.; Afanas’ev, V.V. Identification of intrinsic electron trapping sites in bulk amorphous silica from ab initio calculations. Microelectron. Eng. 2013, 109, 68–71. [Google Scholar] [CrossRef]
- DiStefano, T.; Eastman, D. The Band Edge of Amorphous SiO2 by Photoinjection and Photoconductivity Measurements. Solid State Commun. 1971, 9, 2259–2261. [Google Scholar] [CrossRef]
- Nithianandam, V.J.; Schnatterly, S.E. Soft-x-ray emission and inelastic electron-scattering study of the electronic excitations in amorphous and crystalline silicon dioxide. Phys. Rev. B 1998, 38, 5547–5553. [Google Scholar] [CrossRef]
- Weinberg, Z.A.; Rubloff, G.W.; Bassous, E. Transmission, photoconductivity, and the experimental band gap of thermally grown SiO2 films. Phys. Rev. B 1979, 19, 3107–3117. [Google Scholar] [CrossRef]
- Lee, S.; Xu, H. Using powder XRD and pair distribution function to determine anisotropic atomic displacement parameters of orthorhombic tridymite and tetragonal cristobalite. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2019, 75, 160–167. [Google Scholar] [CrossRef]
- Baur, W.H. In search of the crystal structure of low quartz. Z. Krist. 2009, 224, 580–592. [Google Scholar] [CrossRef]
- Pluth, J.J.; Smith, J.V.; Faber, J. Crystal structure of low cristobalite at 10, 293, and 473 K: Variation of framework geometry with temperature. J. Appl. Phys. 1985, 57, 1045–1049. [Google Scholar] [CrossRef]
- Anderson, O.L.; Schreiber, E. The Relation between Refractive Index and Density of Minerals Related to the Earth’s Mantle. J. Geophys. Res. 1965, 70, 1463–1471. [Google Scholar] [CrossRef]
- Capron, N.; Carniato, S.; Lagraa, A.; Boureau, G.; Pasturel, A. Local density approximation and generalized gradient approximation calculations for oxygen and silicon vacancies in silica. J. Chem. Phys. 2000, 112, 9543–9548. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Sushko, P.V.; Edwards, A.H.; Shluger, A.L. Calculation of relative concentrations of E′ centres in amorphous silica. J.-Non-Cryst. Solid. 2004, 345-346, 703–709. [Google Scholar] [CrossRef]
- Raghavachari, K.; Ricci, D.; Pacchioni, G. Optical properties of point defects in SiO2 from time-dependent density functional theory. J. Chem. Phys. 2002, 116, 825–831. [Google Scholar] [CrossRef]
- Guzzi, M.; Piot, F.; Spinolo, G.; Vedda, A.; Azzoni, C.B.; Paleari, A. Neutron irradiation effects in quartz: Optical absorption and electron paramagnetic resonance. J. Phys. Condens. Matter 1992, 4, 8635–8648. [Google Scholar] [CrossRef]
- Griscom, D.; Fowler, W.B. Electron-transfer model for E′-center optical absorption in SiO2. In The Physics of MOS Insulators, Proceedings of the International Topical Conference on the Physics of MOS Insulators; Pergamon Press: Oxford, UK, 1980; pp. 97–101. [Google Scholar]
- Trukhin, A.N. Radiation processes in oxygen-deficient silica glasses: Is ODC(I) a precursor of E′-center? J.-Non-Cryst. Solid. 2006, 352, 3002–3008. [Google Scholar] [CrossRef]
- Griscom, D.L. Optical properties and structure of defects in silica glass. J. Ceram. Soc. Jpn. 1991, 99, 923–942. [Google Scholar] [CrossRef]
- Hosono, H.; Abe, Y.; Imagawa, H.; Imai, H.; Arai, K. Experimental evidence for the Si-Si bond model of the 7.6-eV band in SiO2 glass. Phys. Rev. B 1991, 44, 12043–12045. [Google Scholar] [CrossRef] [PubMed]
- Henderson, B.; Chen, Y.; Sibley, W.A. Temperature Dependence of Luminescence of F’ and F Centers in CaO. Phys. Rev. B 1972, 6, 4060–4068. [Google Scholar] [CrossRef]
- Perlson, B.D.; Weil, J.A. Electron paramagnetic resonance studies of the E′ centers in alpha-quartz. Can. J. Phys. 2008, 86, 871–881. [Google Scholar] [CrossRef]
- Zhang, L.; Leisure, R.G. The Eδ′ and triplet-state centers in x-irradiated high-purity amorphous SiO2. J. Appl. Phys. 1996, 80, 3744–3749. [Google Scholar] [CrossRef]
- Afanas’ev, V.; Stesmans, A. Charge state of paramagnetic E′ centre in thermal SiO2 layers on silicon. J. Phys. Condens. Matter 2000, 12, 2285–2290. [Google Scholar] [CrossRef]
- Ling, S.; El-Sayed, A.M.; Lopez-Gejo, F.; Watkins, M.B.; Afanas’ev, V.V.; Shluger, A.L. A computational study of Si–H bonds as precursors for neutral E0 centres in amorphous silica and at the Si/SiO2 interface. Microelectron. Eng. 2013, 109, 310–313. [Google Scholar] [CrossRef]






| Bond | -Cristobalite | -Quartz | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pristine | Pristine | ||||||||||
| Si-Si | 3.11 | 2.38 | 2.88 | 4.59 | 2.39 | 3.09 | 2.41 | 3.00 | 4.45 | 5.38 | 2.47 |
| Si-O | 5.31 | 5.54 | 5.22 | 4.83 | 5.63 | 3.66 | 4.10 | 3.71 | 1.83 | 1.84 | 4.07 |
| Si-O | 1.61 | 1.61–1.63 | 1.57–1.58 | 1.62–1.63 | 1.67–1.70 | 1.61 | 1.63 | 1.58 | 1.62 | 1.61–1.64 | 1.69–1.73 |
| Si-O | 1.61 | 1.62–1.63 | 1.57–1.58 | 1.54–1.55 | 1.67–1.71 | 1.61 | 1.62 | 1.57 | 1.58 | 1.57–1.59 | 1.66–1.68 |
| Cutoff Radius (Å) | Phase | Defect | Peak Energy (eV) | Oscillator Strength | Transition Type |
|---|---|---|---|---|---|
| 7.5 | -C | 7.62 | 0.22 | ||
| 7.72 | 0.20 | ||||
| 8.13 | 0.16 | ||||
| 8.4 | 0.13 | ||||
| 7.56 | 0.12 | ||||
| 7.72 | 0.11 | * | |||
| 6.4 | -Q | 6.18 | 0.12 | Si. → deloc. ring | |
| 2 | -C | 7.86 | 0.27 | ||
| 7.94 | 0.19 | ||||
| 8.20 | 0.13 | ||||
| 8.48 | 0.12 | ||||
| 6.27 | 0.15 | * | |||
| 7.86 | 0.12 | ||||
| -Q | 6.38 | 0.14 | Si → Si | ||
| 6.43 | 0.12 | Si. → deloc. ring |
| AE Basis | -Quartz (mT) | -Cristobalite (mT) | |||
|---|---|---|---|---|---|
| Silica | Si | Si | Si | Si | Si |
| pcJ-0 | 12.29 | 10.11 | 42.25 | 11.16 | 10.78 |
| pcJ-1 | 11.14 | 8.70 | 38.96 | 10.44 | 10.01 |
| pcJ-2 | 10.26 | 8.06 | 37.46 | 9.55 | 9.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milton, K.L.; Durrant, T.R.; Cobos Freire, T.; Shluger, A.L. Difference in Structure and Electronic Properties of Oxygen Vacancies in α-Quartz and α-Cristobalite Phases of SiO2. Materials 2023, 16, 1382. https://doi.org/10.3390/ma16041382
Milton KL, Durrant TR, Cobos Freire T, Shluger AL. Difference in Structure and Electronic Properties of Oxygen Vacancies in α-Quartz and α-Cristobalite Phases of SiO2. Materials. 2023; 16(4):1382. https://doi.org/10.3390/ma16041382
Chicago/Turabian StyleMilton, Katherine L., Thomas R. Durrant, Teofilo Cobos Freire, and Alexander L. Shluger. 2023. "Difference in Structure and Electronic Properties of Oxygen Vacancies in α-Quartz and α-Cristobalite Phases of SiO2" Materials 16, no. 4: 1382. https://doi.org/10.3390/ma16041382
APA StyleMilton, K. L., Durrant, T. R., Cobos Freire, T., & Shluger, A. L. (2023). Difference in Structure and Electronic Properties of Oxygen Vacancies in α-Quartz and α-Cristobalite Phases of SiO2. Materials, 16(4), 1382. https://doi.org/10.3390/ma16041382