
Mechanism and Kinetics of the Phase Formation and Dissolution of NaxWO3 on a Pt Electrode in a Na2WO4–WO3 Melt

 ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
3. Experimental Results
3.1. Cyclic Voltammetry
3.2. Chronoamperometry
4. Models
4.1. Deposit Consisting of OTB and Tungsten
4.2. Deposit Containing Two OTBs
4.3. Inhomogeneity of Sodium Distribution in an OTB Crystal
4.4. Parameters and Simulation Procedure
5. Simulation Results and Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Labbe, P. Tungsten oxides, tungsten bronzes and tungsten bronze-type structures. Key Eng. Mater. 1992, 68, 293–339. [Google Scholar] [CrossRef]
- Bartha, L.; Kiss, A.B.; Szalay, T. Chemistry of Tungsten Oxide Bronzes. Int. J. Refract. Met. Hard Mater. 1995, 13, 77–91. [Google Scholar] [CrossRef]
- Lawrence, S.; Stevenson, S.; Mavadia, K.; Sermon, P. Solid-State Properties of Some Polycrystalline Alkali-Metal Tungsten Bronzes. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1987, 411, 95–121. [Google Scholar]
- Shanks, H.R.; Sidles, P.H.; Danielson, G.C. Electrical Properties of the Tungsten Bronzes. In Nonstoichiometric Compounds; Advances in Chemistry Series; Ward, R., Ed.; ACS: Washington, DC, USA, 1963; Volume 39, pp. 237–245. [Google Scholar]
- Ellerbeck, L.D.; Shanks, H.R.; Sidles, P.H.; Danielson, G.C. Electrical Resistivity of Cubic Sodium Tungsten Bronze. J. Chem. Phys. 1961, 35, 298–303. [Google Scholar] [CrossRef]
- Lekshmi, I.C.; Hegde, M.S. Synthesis and electrical properties of cubic NaxWO3 thin films across the metal–insulator transition. Mater. Res. Bull. 2005, 40, 1443–1450. [Google Scholar] [CrossRef][Green Version]
- Raj, S.; Matsui, H.; Souma, S.; Sato, T.; Takahashi, T.; Chakraborty, A.; Sarma, D.D.; Mahadevan, P.; Oishi, S.; McCarroll, W.H.; et al. Electronic structure of sodium tungsten bronzes NaxWO3 by high-resolution angle-resolved photoemission spectroscopy. Phys. Rev. B 2007, 75, 155116. [Google Scholar] [CrossRef]
- Sano, K.; Nitta, Y.; Ōno, Y. Transition Temperature of Superconductivity in Sodium Tungsten Bronze—Theoretical Study Based on First-principles Calculations. J. Phys. Soc. Jpn. 2020, 89, 113704. [Google Scholar] [CrossRef]
- Cogan, S.F.; Plante, T.D.; Parker, M.A.; Rauh, R.D. Free-electron electrochromic modulation in crystalline LixWO3. J. Appl. Phys. 1986, 60, 2735–2739. [Google Scholar] [CrossRef]
- Tegg, L.; Haberfehlner, G.; Kothleitner, G.; Kisi, E.; Keast, V.J. Crystal structures, electrical properties, and electron energy-loss spectroscopy of the sodium and potassium tetragonal tungsten bronzes. J. Alloys Compd. 2021, 868, 159200. [Google Scholar] [CrossRef]
- El-Sayed, A.M.; Mousa, S.M.A. Some properties of sodium tungsten bronzes as a function of sodium concentration. Ind. J. Chem. Technol. 2005, 12, 304–308. [Google Scholar]
- Wechter, M.A.; Shanks, H.R.; Carter, G.; Ebert, G.M.; Guglielmino, R.; Voigt, A.F. Use of metal tungsten bronze electrodes in chemical analysis. Anal. Chem. 1972, 44, 850–853. [Google Scholar] [CrossRef] [PubMed]
- Hahn, P.B.; Wechter, M.A.; Johnson, D.C.; Voigt, A.F. Sodium tungsten bronze as a potentiometric indicating electrode for dissolved oxygen in aqueous solution. Anal. Chem. 1973, 45, 1016–1021. [Google Scholar] [CrossRef]
- Griffith, C.S.; Luca, V.; Hanna, J.V.; Pike, K.J.; Smith, M.E.; Thorogood, G.S. Microcrystalline Hexagonal Tungsten Bronze. 1. Basis of Ion Exchange Selectivity for Cesium and Strontium. Inorg. Chem. 2009, 48, 5648–5662. [Google Scholar] [CrossRef] [PubMed]
- Egorin, A.M.; Dran’kov, A.N.; Didenko, N.V.; Tokar’, E.A.; Sokol’nitskaya, T.A.; Papynov, E.K.; Tananaev, I.G. Synthesis and sorption characteristics of tungsten oxides-based materials for Sr-90 removal from water media. J. Mater. Sci. 2020, 55, 9374–9384. [Google Scholar] [CrossRef]
- Zimmer, A.; Gilliot, M.; Tresse, M.; Broch, L.; Tillous, K.E.; Boulanger, C.; Stein, N.; Horwat, D. Coloration mechanism of electrochromic NaxWO3 thin films. Opt. Lett. 2019, 44, 1104–1107. [Google Scholar] [CrossRef] [PubMed]
- Tegg, L.; Cuskelly, D.; Keast, V.J. The sodium tungsten bronzes as plasmonic materials: Fabrication, calculation and characterization. Mater. Res. Express 2017, 4, 065703. [Google Scholar] [CrossRef]
- Tegg, L.; Cuskelly, D.; Keast, V.J. Plasmon Responses in the Sodium Tungsten Bronzes. Plasmonics 2018, 13, 437–444. [Google Scholar] [CrossRef]
- Azimirad, R.; Khademi, A.; Akhavan, J.; Moshfegh, A.Z. Growth of Na0.3WO3 nanorods for the field emission application. J. Phys. D Appl. Phys. 2009, 42, 205405. [Google Scholar] [CrossRef]
- Guo, C.; Yin, S.; Sato, T. Effects of Crystallization Atmospheres on the Near-Infrared Absorbtion and Electroconductive Properties of Tungsten Bronze Type MxWO3 (M = Na, K). J. Am. Ceram. Soc. 2012, 95, 1634–1639. [Google Scholar] [CrossRef]
- Kotvanova, M.K.; Pavlova, S.S.; Efremova, N.N. Nanosized crystals of oxide bronzes of titanium, molybdenum, tungsten as components of anticorrosive coatings. Izvestiya Vuzov. Khimiya Khimicheskaya Tekhnologiya 2013, 56, 88–91. (In Russian) [Google Scholar]
- Jie, S.; Guo, X.; Ouyang, Z. Tumor ablation using novel photothermal NaxWO3 nanoparticles against breast cancer osteolytic bone metastasis. Int. J. Nanomed. 2019, 14, 7353–7362. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhan, J.; Fan, W.; Cui, G.; Sun, H.; Zhuo, L.; Zhao, X.; Tang, B. Microcrystalline sodium tungsten bronze nanowire bundles as efficient visible light-responsive photocatalysts. Chem. Commun. 2010, 46, 8833–8835. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-M.; Naseem, S.; Chou, M.-H.; Wang, J.-H.; Jian, Y.-Q. Recent Advances in Tungsten-Oxide-Based Materials and Their Applications. Front. Mater. 2019, 6, 49. [Google Scholar] [CrossRef]
- Yi, L.; Zhao, W.; Huang, Y.; Wu, X.; Wang, J.; Zhang, G. Tungsten bronze Cs0.33WO3 nanorods modified by molybdenum for improved photocatalytic CO2 reduction directly from air. Sci. China Mater. 2020, 63, 2206–2214. [Google Scholar] [CrossRef]
- Vakarin, S.V.; Melyaeva, A.A.; Semerikova, O.L.; Kondratuk, V.S.; Pankratov, A.A.; Plaksin, S.V.; Porotnikova, N.M.; Zaykov, Y.P.; Petrov, L.A.; Mikushina, Y.V.; et al. Catalase activity of coarse grained and nanosized oxide tungsten bronzes obtained by electrolysis of molten salts. Russ. Chem. Bull. 2011, 60, 1985–1988. [Google Scholar] [CrossRef]
- Semerikova, O.L.; Vakarin, S.V.; Kosov, A.V.; Plaksin, S.V.; Pankratov, A.A.; Grishenkova, O.V.; Zaykov, Y.P.; Shishmakov, A.B.; Mikushina, Y.V.; Petrov, L.A. Electrochemical Synthesis of Nanohybrid Systems Based on Copper and the Oxide Tungsten Bronzes. J. Electrochem. Soc. 2019, 166, D792–D797. [Google Scholar] [CrossRef]
- Drobasheva, T.I.; Spitsyn, V.I. Tungsten and molybdenum bronzes with two alkaline elements. In Oxide Bronzes; Spitsyn, V.I., Ed.; Nauka: Moscow, Russia, 1982; pp. 40–75. (In Russian) [Google Scholar]
- Kaliev, K.A.; Baraboshkin, A.N. Electrocrystallization of transition metal oxide bronzes from molten salts. In Oxide Bronzes; Spitsyn, V.I., Ed.; Nauka: Moscow, Russia, 1982; pp. 137–175. (In Russian) [Google Scholar]
- Banks, E.; Fleischmann, C.W.; Meites, L. On the nature of the species reduced during the electrochemical synthesis of tungsten bronzes. J. Solid State Chem. 1970, 1, 372–375. [Google Scholar] [CrossRef]
- Meites, L.; Banks, E.; Fleischmann, C.W. Voltammetric behaviors of platinum electrodes and decomposition potentials of alkali tungstate and polytungstate melts. Anal. Chem. 1972, 44, 1133–1139. [Google Scholar] [CrossRef]
- Fredlein, R.A.; Damjanovic, A. Electrochemical deposition and dissolution of tungsten oxide bronzes. J. Solid State Chem. 1972, 4, 94–102. [Google Scholar] [CrossRef]
- Randin, J.-P. Electrochemical deposition of sodium tungsten bronzes. J. Electrochem. Soc. 1973, 120, 1325–1330. [Google Scholar] [CrossRef]
- Randin, J.-P. Kinetics of the electrochemical deposition and dissolution of sodium tungsten bronzes. Electrochim. Acta 1974, 19, 745–751. [Google Scholar] [CrossRef]
- Elwell, D.; DeMattei, R.C.; Zubeck, I.V.; Feigelson, R.S.; Huggins, R.A. DC resistances in electrolytic crystallization from molten salts. J. Cryst. Growth 1976, 33, 232–238. [Google Scholar] [CrossRef]
- Okada, K.; Miyake, M.; Iwai, S.; Ohno, H.; Furukawa, K. Structural Analysis of Molten Na2WO4. J. Chem. Soc. Faraday Trans. 2 1978, 74, 1141–1148. [Google Scholar] [CrossRef]
- Miyake, M.; Okada, K.; Iwai, S.; Ohno, H.; Furukawa, K. Structural analysis of molten Na2W2O7. J. Chem. Soc. Faraday Trans. 2 1978, 74, 1880–1884. [Google Scholar] [CrossRef]
- Baraboshkin, A.N.; Kaliev, K.A.; Aksentiev, A.G. Formation of metastable phases on the cathode in the initial period of electrolysis. Elektrokhimiya 1978, 14, 1836–1839. (In Russian) [Google Scholar]
- Afonichkin, V.K.; Leontiev, V.N.; Komarov, V.E. Equilibrium electrode potentials of tungsten in melts of the Na2WO4–WO3 system. Elektrokhimiya 1993, 29, 341–347. (In Russian) [Google Scholar]
- Vorozhbit, V.U.; Shurdumov, G.K.; Kaliev, K.A. Obtaining highly dispersed powders of oxide lithium-tungsten bronzes. Tsvetnye Met. 1995, 39–41. (In Russian) [Google Scholar]
- Voron’ko, Y.K.; Sobol’, A.A. Influence of Cations on the Vibrational Spectra and Structure of [WO4] Complexes in Molten Tungstates. Inorg. Mater. 2005, 41, 420–428. [Google Scholar] [CrossRef]
- Voronko, Y.K.; Sobol, A.A.; Shukshin, V.E. Raman scattering study of molten alkali-metal molybdates and tungstates rich in basic oxides. Inorg. Mater. 2014, 50, 837–843. [Google Scholar] [CrossRef]
- Wang, J.; You, J.L.; Wang, Y.Y.; Wang, M.; Jun, W.U. In-situ Raman Spectroscopic Study of the Molten Tungstates in Li2O-WO3 Binary System. Chin. J. Light Scatt. 2016, 28, 149–152. [Google Scholar]
- Wang, J.; You, J.; Wang, M.; Lu, L.; Wan, S.; Sobol, A.A. In-situ studies on the micro-structure evolution of A2W2O7 (A = Li, Na, K) during melting by high temperature Raman spectroscopy and density functional theory. Spectrochim. Acta A 2017, 185, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; You, J.L.; Sobol, A.A.; Lu, L.M.; Wang, M.; Wu, J.; Lv, X.M.; Wan, S.M. In-situ high temperature Raman spectroscopic study on the structural evolution of Na2W2O7 from the crystalline to molten states. J. Raman Spectrosc. 2017, 48, 298–304. [Google Scholar] [CrossRef]
- Kosov, A.V.; Semerikova, O.L.; Vakarin, S.V.; Grishenkova, O.V.; Vorob’ev, A.S.; Khudorozhkova, A.O.; Zaikov, Y.P. Ionic Equilibria in Polytungstate Melts. Processes 2022, 10, 2658. [Google Scholar] [CrossRef]
- Baraboshkin, A.N.; Tarasova, K.P.; Nazarov, B.A.; Martemyanova, Z.S. Study of the composition and structure of cathode deposits during the electrolysis of molten Na2WO4-WO3 mixtures. Electrochem. Molten Salt Solid Electrolytes Proc. Inst. Electrochem. AS USSR 1973, 19, 44–48. (In Russian) [Google Scholar]
- Fletcher, S.; Halliday, C.S.; Gates, D.; Westcott, M.; Lwin, T.; Nelson, G. The response of some nucleation/growth processes to triangular scans of potential. J. Electroanal. Chem. 1983, 159, 267–285. [Google Scholar] [CrossRef]
- Velmurugan, J.; Noël, J.-M.; Nogala, W.; Mirkin, M.V. Nucleation and growth of metal on nanoelectrodes. Chem. Sci. 2012, 3, 3307–3314. [Google Scholar] [CrossRef]
- Isaev, V.A.; Grishenkova, O.V.; Zaykov, Y.P. Theory of cyclic voltammetry for electrochemical nucleation and growth. J. Solid State Electrochem. 2018, 22, 2775–2778. [Google Scholar] [CrossRef]
- Kosov, A.V.; Grishenkova, O.V.; Semerikova, O.L.; Isaev, V.A.; Zaykov, Y.P. On the theory of cyclic voltammetry for multiple nucleation and growth: Scan rate influence. J. Electroanal. Chem. 2021, 883, 115056. [Google Scholar] [CrossRef]
- Grishenkova, O.V.; Kosov, A.V.; Semerikova, O.L.; Isaev, V.A.; Zaikov, Y.P. Theoretical and experimental cyclic voltammetry studies of the initial stages of electrocrystallization. Russ. Metall. (Met.) 2021, 2021, 1016–1022. [Google Scholar] [CrossRef]
- Scharifker, B.R.; Hills, G.J. Theoretical and experimental studies of multiple nucleation. Electrochim. Acta 1983, 28, 879–889. [Google Scholar] [CrossRef]
- Isaev, V.A.; Grishenkova, O.V.; Zaykov, Y.P. Analysis of the geometrical–probabilistic models of electrocrystallization. Russ. Metall. (Met.) 2016, 2016, 776–784. [Google Scholar] [CrossRef]
- Isaev, V.A.; Zaykov, Y.P.; Grishenkova, O.V.; Kosov, A.V.; Semerikova, O.L. Analysis of Potentiostatic Current Transients for Multiple Nucleation with Diffusion and Kinetic Controlled Growth. J. Electrochem. Soc. 2019, 166, D851–D856. [Google Scholar] [CrossRef]
- Scharifker, B.R.; Mostany, J. Three-dimensional nucleation with diffusion controlled growth. Part I. Number density of active sites and nucleation rates per site. J. Electroanal. Chem. 1984, 177, 13–23. [Google Scholar] [CrossRef]
- Abyaneh, M.Y. Formulation of a general model for nucleation and growth of electrodeposits. Electrochim. Acta 1991, 36, 727–732. [Google Scholar] [CrossRef]
- Altimari, P.; Pagnanelli, F. Electrochemical nucleation and three-dimensional growth under mixed kinetic-diffusion control: Analytical approximation of the current transient. Electrochim. Acta 2016, 205, 113–117. [Google Scholar] [CrossRef]
- Milchev, A.; Zapryanova, T. Nucleation and growth of copper under combined charge transfer and diffusion limitations: Part I. Electrochim. Acta 2006, 51, 2926–2933. [Google Scholar] [CrossRef]
- Milchev, A.; Zapryanova, T. Nucleation and growth of copper under combined charge transfer and diffusion limitations: Part II. Electrochim. Acta 2006, 51, 4916–4921. [Google Scholar] [CrossRef]
- Kosov, A.V.; Grishenkova, O.V.; Isaev, V.A.; Zaikov, Y. Simulation of Diffusion-Controlled Growth of Interdependent Nuclei under Potentiostatic Conditions. Materials 2022, 15, 3603. [Google Scholar] [CrossRef] [PubMed]
- Isaev, V.A.; Grishenkova, O.V.; Kosov, A.V.; Semerikova, O.L.; Zaikov, Y. Simulation of 3D electrochemical phase formation: Mixed growth control. Materials 2021, 14, 6330. [Google Scholar] [CrossRef]
- Kosov, A.V.; Grishenkova, O.V. Modeling and simulation of 3D electrochemical phase formation under mixed kinetic-diffusion growth control. J. Solid State Electrochem. 2023. [Google Scholar] [CrossRef]
- D’Ajello, P.C.T.; Munford, M.L.; Pasa, A.A. Transient equations for multiple nucleation on solid electrodes: A stochastic description. J. Chem. Phys. 1999, 111, 4267–4273. [Google Scholar] [CrossRef]
- D’Ajello, P.C.T.; Fiori, M.A.; Pasa, A.A.; Kipervaser, Z.G. Reaction-Diffusion Interplay in Electrochemical Deposition Processes. A Theoretical Approach. J. Electrochem. Soc. 2000, 147, 4562–4566. [Google Scholar] [CrossRef]
- Luo, G.; Li, D.; Yuan, G.; Li, N. Potentiostatic Current Transient for Multiple Nucleation: A Limited-Diffusion Process Description. J. Electrochem. Soc. 2018, 165, D147–D151. [Google Scholar] [CrossRef]
- Volgin, V.M.; Volgina, O.V.; Davydov, A.D. Finite difference method of simulation of non-steady-state ion transfer in electrochemical systems with allowance for migration. Comput. Biol. Chem. 2003, 27, 185–196. [Google Scholar] [CrossRef]
- Myland, J.C.; Oldham, K.B. Convolutive modelling in the absence of supporting electrolyte: Coping with migration and changing resistance in predicting voltammetry. J. Electroanal. Chem. 2002, 529, 66–74. [Google Scholar] [CrossRef]
- Chadwick, A.F.; Vardar, G.; DeWitt, S.; Sleightholme, A.E.S.; Monroe, C.W.; Siegel, D.J.; Thornton, K. Computational Model of Magnesium Deposition and Dissolution for Property Determination via Cyclic Voltammetry. J. Electrochem. Soc. 2016, 163, A1813–A1821. [Google Scholar] [CrossRef]
- Isaev, V.A. Electrochemical Phase Formation; UrO RAN: Ekaterinburg, Russia, 2007. (In Russian) [Google Scholar]
- Galus, Z. Fundamentals of Electrochemical Analysis, 2nd ed.; Ellis Horwood: New York, NY, USA, 1994. [Google Scholar]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; Wiley: New York, NY, USA, 2001. [Google Scholar]
- Brown, B.W.; Banks, E. The Sodium Tungsten Bronzes. J. Am. Chem. Soc. 1954, 76, 963–966. [Google Scholar] [CrossRef]
- Walkenbach, J. Excel 2013 Power Programming with VBA; Wiley: New York, NY, USA, 2013. [Google Scholar]
- Metso. HSC Chemistry. Available online: www.outotec.com/HSC (accessed on 15 July 2023).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
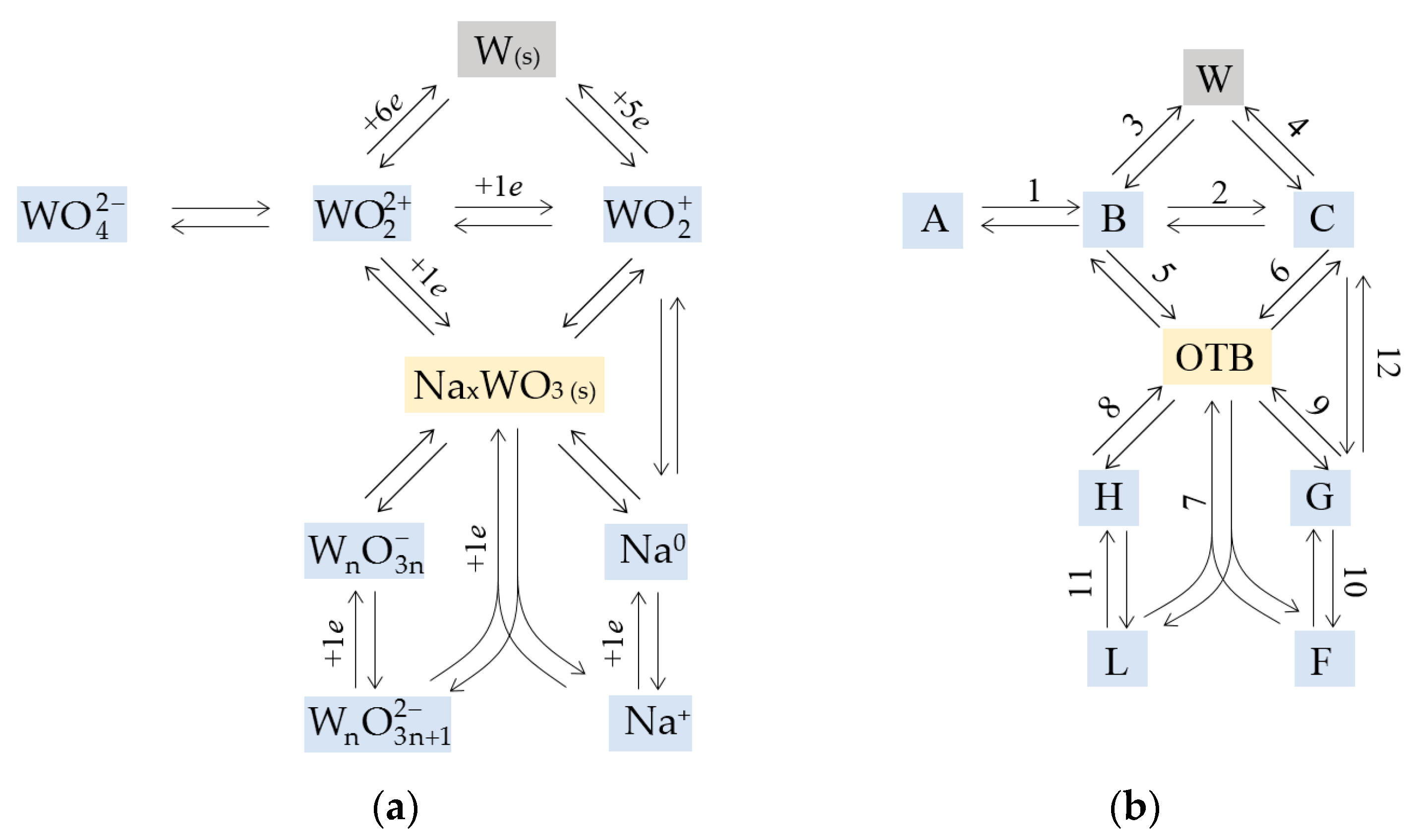{kind=link}
{kind=link}
{kind=link}
{kind=link}
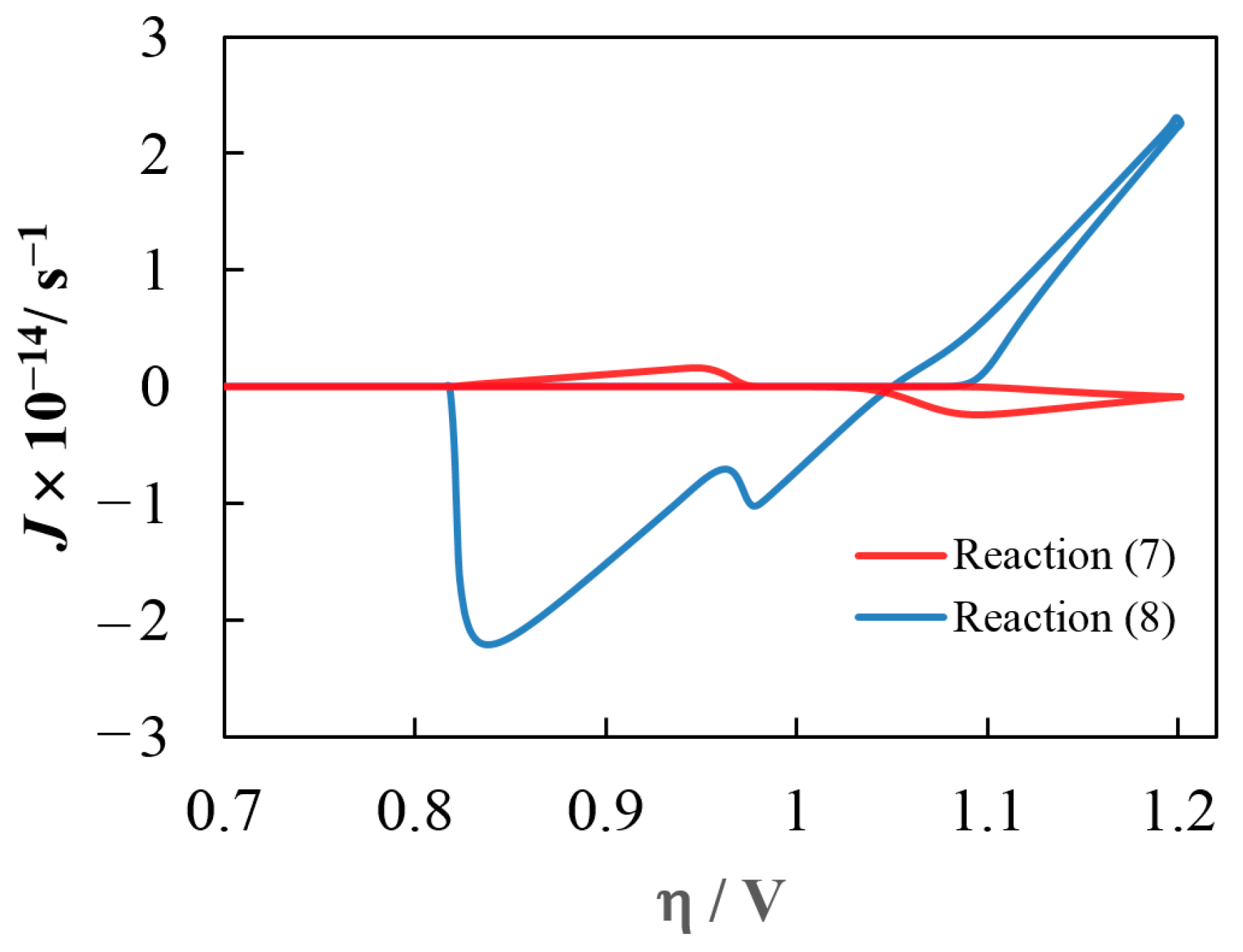{kind=link}
{kind=link}
| Spectrum | Concentration, at.% | |||
|---|---|---|---|---|
| O | Na | W | Pt | |
| 1 | 62.02 | 17.89 | 20.09 | - |
| 2 | 48.71 | 16.32 | 34.97 | - |
| 3 | 7.50 | - | - | 92.50 |
| 4 | 12.98 | - | 2.35 | 84.67 |
| 5 | 59.34 | 17.61 | 21.69 | 1.36 |
| 6 | 8.76 | - | 0.92 | 90.32 |
| 7 | 12.14 | - | 1.30 | 86.56 |
| 8 | 13.84 | - | 1.96 | 84.20 |
| 9 | 51.97 | 20.25 | 27.79 | - |
| 10 | 9.51 | - | 90.49 | - |
| 11 | 42.51 | - | 57.49 | - |
| 12 | 36.24 | - | 63.76 | - |
| Parameters | ν = 0.1 V·s–1 | ηλ = 1.2 V | Average Value | Standard Deviation | ||||
|---|---|---|---|---|---|---|---|---|
| ηλ, V | ν, V·s–1 | |||||||
| 1.13 | 1.15 | 1.20 | 0.2 | 0.3 | 0.4 | |||
| , cm2s−1 | 3.99 × 10−5 | 4.59 × 10−5 | 4.59 × 10−5 | 4.50 × 10−5 | 4.51 × 10−5 | 4.39 × 10−5 | 4.43 × 10−5 | 2.28 × 10−6 |
| , cm·s–1 | 4.59 × 1015 | 5.16 × 1015 | 5.31 × 1015 | 5.14 × 1015 | 5.17 × 1015 | 5.44 × 1015 | 5.13 × 1015 | 2.90 × 1014 |
| , cm·s –1 | 1.55 × 1020 | 4.79 × 1020 | 4.28 × 1020 | 4.71 × 1020 | 5.19 × 1020 | 5.77 × 1020 | 3.88 × 1020 | 1.42 × 1020 |
| , cm·s –1 | 1.05 × 10−11 | 1.18 × 10−11 | 1.21 × 10−11 | 1.17 × 10−11 | 1.18 × 10−11 | 1.24 × 10−11 | 1.17 × 10−11 | 6.57 × 10−13 |
| , cm·s –1 | 5.56 × 10−7 | 3.29 × 10−7 | 4.40 × 10−7 | 3.58 × 10−7 | 3.29 × 10−7 | 3.44 × 10−7 | 3.93 × 10−7 | 9.01 × 10−8 |
| , cm·s –1 | 3.35 × 1015 | 3.76 × 1015 | 3.87 × 1015 | 3.75 × 1015 | 3.77 × 1015 | 3.96 × 1015 | 3.74 × 1015 | 2.11 × 1014 |
| , cm·s –1 | 1.11 × 1020 | 8.94 × 1019 | 8.30 × 1019 | 9.28 × 1019 | 1.05 × 1020 | 1.23 × 1020 | 1.01 × 1020 | 1.50 × 1019 |
| , cm·s –1 | 1.52 × 1020 | 1.15 × 1020 | 1.00 × 1020 | 1.09 × 1020 | 1.26 × 1020 | 1.58 × 1020 | 1.27 × 1020 | 2.34 × 1019 |
| , cm·s –1 | 28.03 | 23.98 | 22.19 | 22.90 | 25.75 | 31.21 | 25.68 | 3.43 |
| , cm·s –1 | 60.35 | 83.63 | 82.19 | 102.62 | 93.33 | 96.97 | 86.52 | 15.01 |
| , cm·s –1 | 1.22 × 1020 | 1.74 × 1020 | 2.03 × 1020 | 2.51 × 1020 | 2.12 × 1020 | 1.99 × 1020 | 1.94 × 1020 | 4.32 × 1019 |
| , cm−2s−1 | 2.34 × 1015 | 2.36 × 1015 | 2.18 × 1015 | 2.35 × 1015 | 2.45 × 1015 | 2.47 × 1015 | 2.36 × 1015 | 1.04 × 1014 |
| ζ0/ζmax | 0.684 | 0.821 | 0.932 | 0.862 | 0.744 | 0.660 | – | – |
| Δζ/ζmax | 0.214 | 0.090 | 0.044 | 0.099 | 0.137 | 0.178 | – | – |
| NOTB, cm−2 | 5.04 × 103 | 3.66 × 103 | 3.47 × 103 | 6.13 × 103 | 8.41 × 103 | 1.58 × 104 | – | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kosov, A.V.; Grishenkova, O.V.; Semerikova, O.L.; Vakarin, S.V.; Zaikov, Y.P. Mechanism and Kinetics of the Phase Formation and Dissolution of NaxWO3 on a Pt Electrode in a Na2WO4–WO3 Melt. Materials 2023, 16, 7207. https://doi.org/10.3390/ma16227207
Kosov AV, Grishenkova OV, Semerikova OL, Vakarin SV, Zaikov YP. Mechanism and Kinetics of the Phase Formation and Dissolution of NaxWO3 on a Pt Electrode in a Na2WO4–WO3 Melt. Materials. 2023; 16(22):7207. https://doi.org/10.3390/ma16227207
Chicago/Turabian StyleKosov, Alexander V., Olga V. Grishenkova, Olga L. Semerikova, Sergey V. Vakarin, and Yuriy P. Zaikov. 2023. "Mechanism and Kinetics of the Phase Formation and Dissolution of NaxWO3 on a Pt Electrode in a Na2WO4–WO3 Melt" Materials 16, no. 22: 7207. https://doi.org/10.3390/ma16227207
APA StyleKosov, A. V., Grishenkova, O. V., Semerikova, O. L., Vakarin, S. V., & Zaikov, Y. P. (2023). Mechanism and Kinetics of the Phase Formation and Dissolution of NaxWO3 on a Pt Electrode in a Na2WO4–WO3 Melt. Materials, 16(22), 7207. https://doi.org/10.3390/ma16227207