

Impact of Mn Alloying on Phase Stabilities, Magnetic Properties and Electronic Structures in Fe

 ,
,  , and
, and
Abstract
:1. Introduction
2. Calculational Methods
3. Results and Discussion
3.1. Bcc-Structured
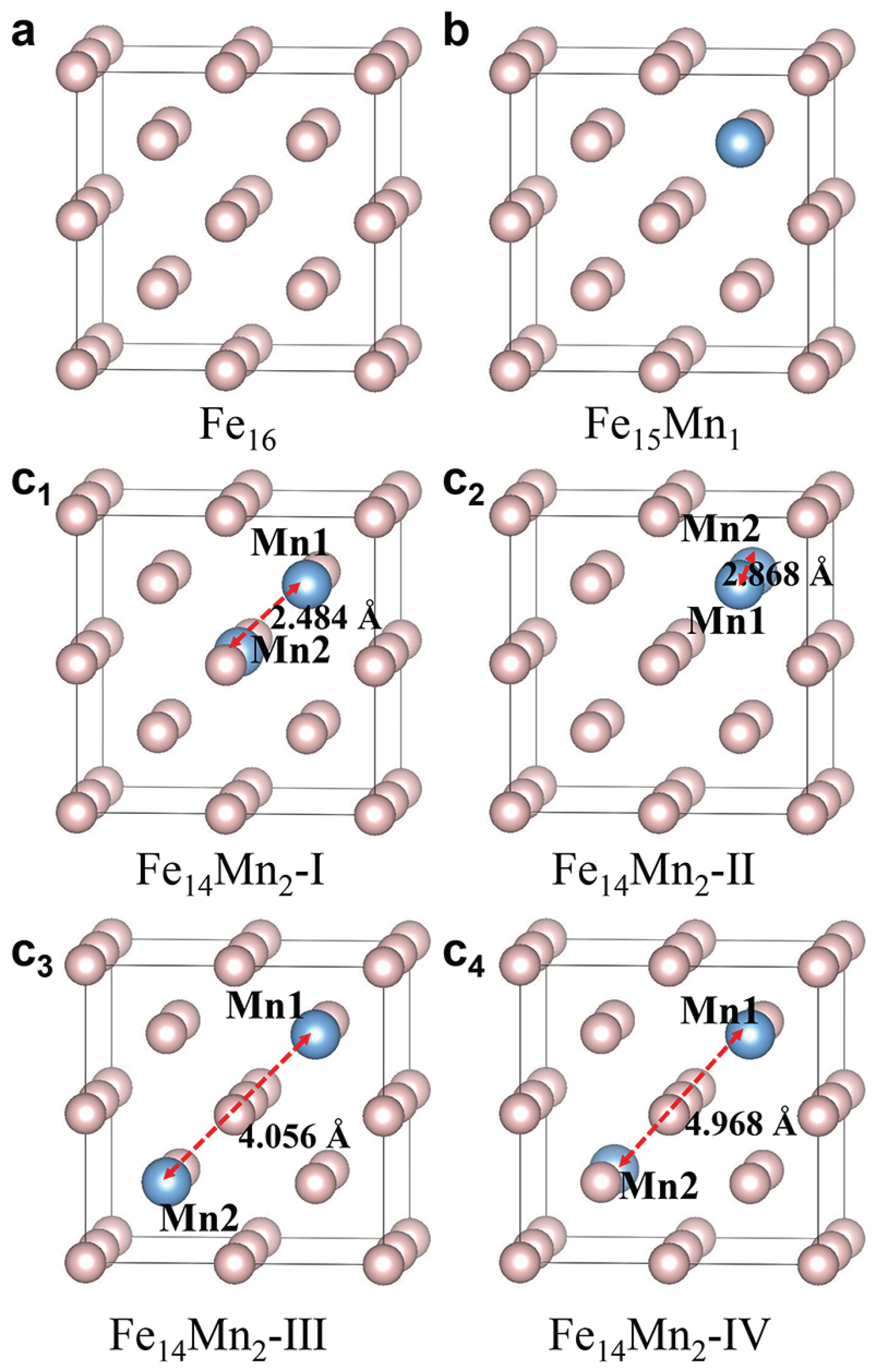
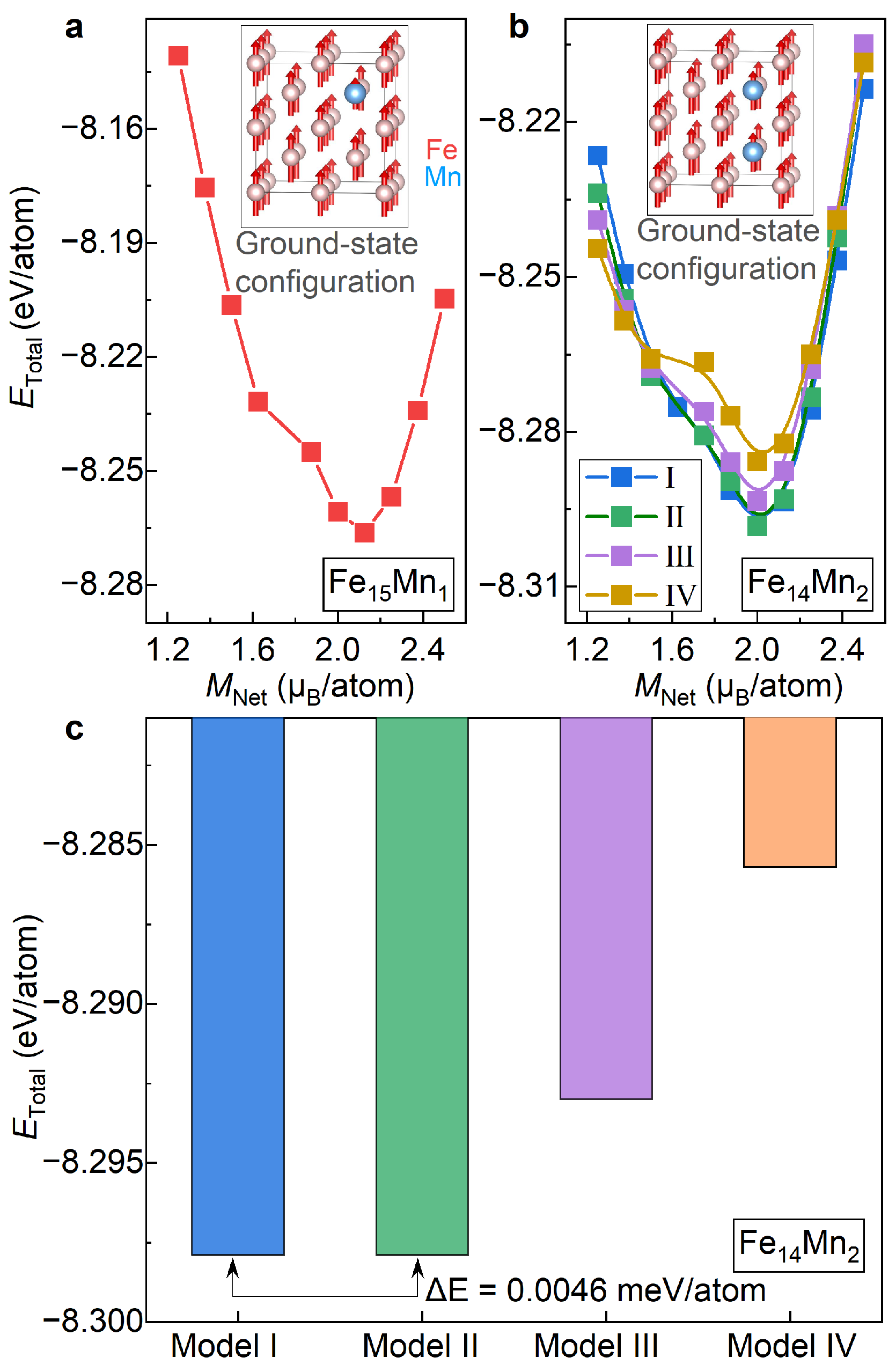
3.1.1. Preferred Structural Model
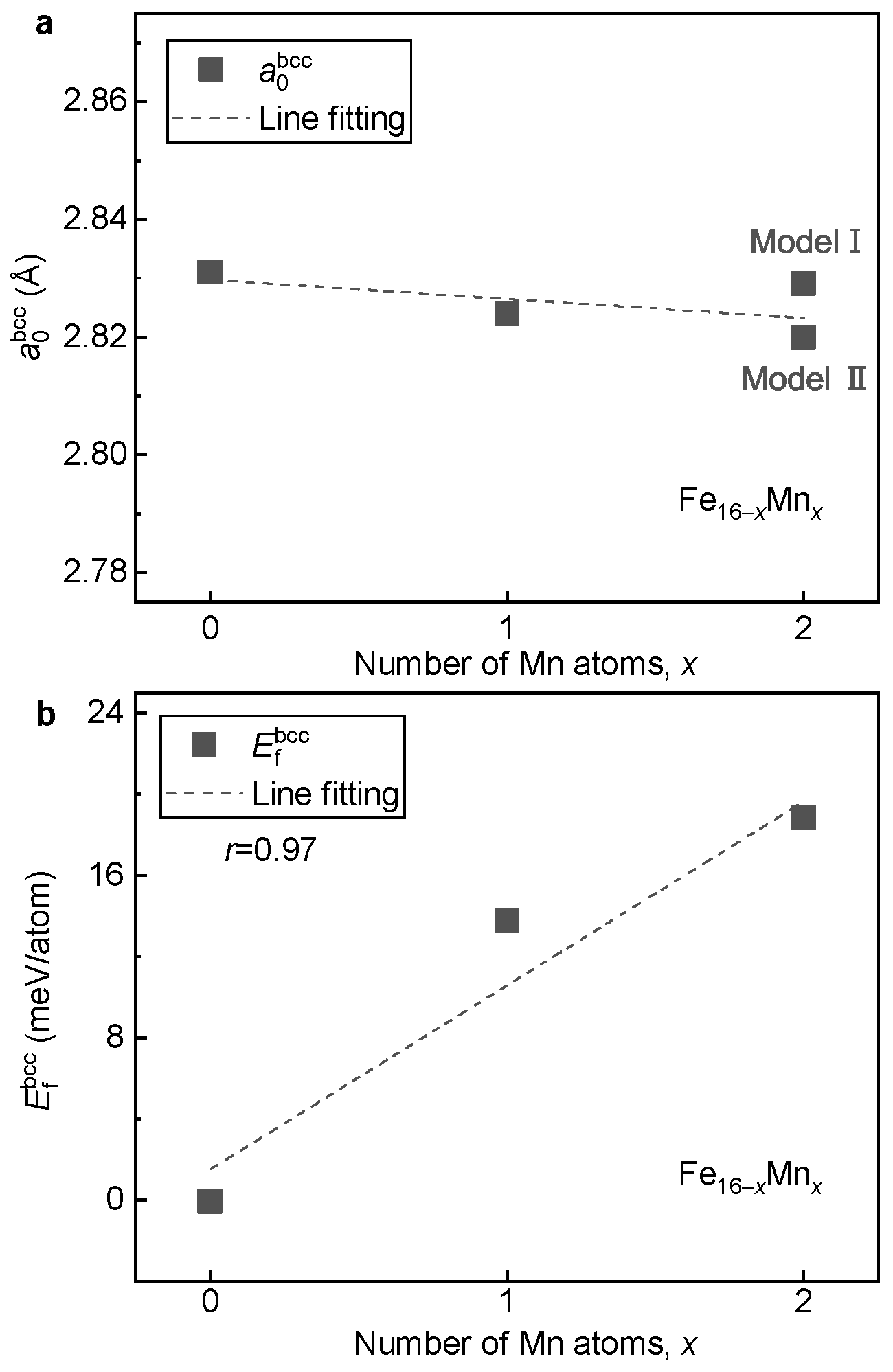
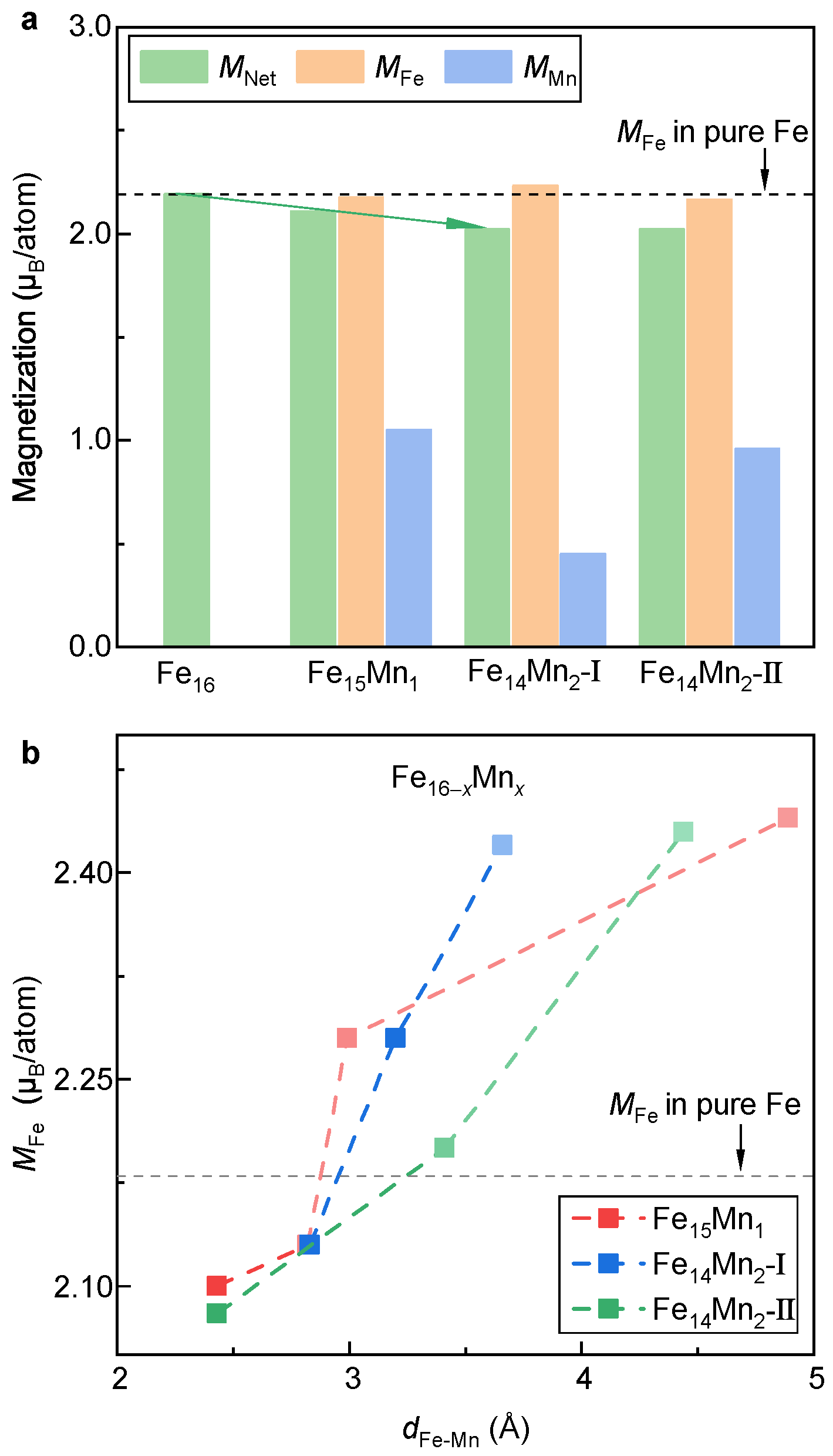
3.1.2. Lattice Stabilities and Magnetic Properties
3.1.3. Electronic Structures
3.2. Fcc-Structured
3.2.1. Preferred Structural Models
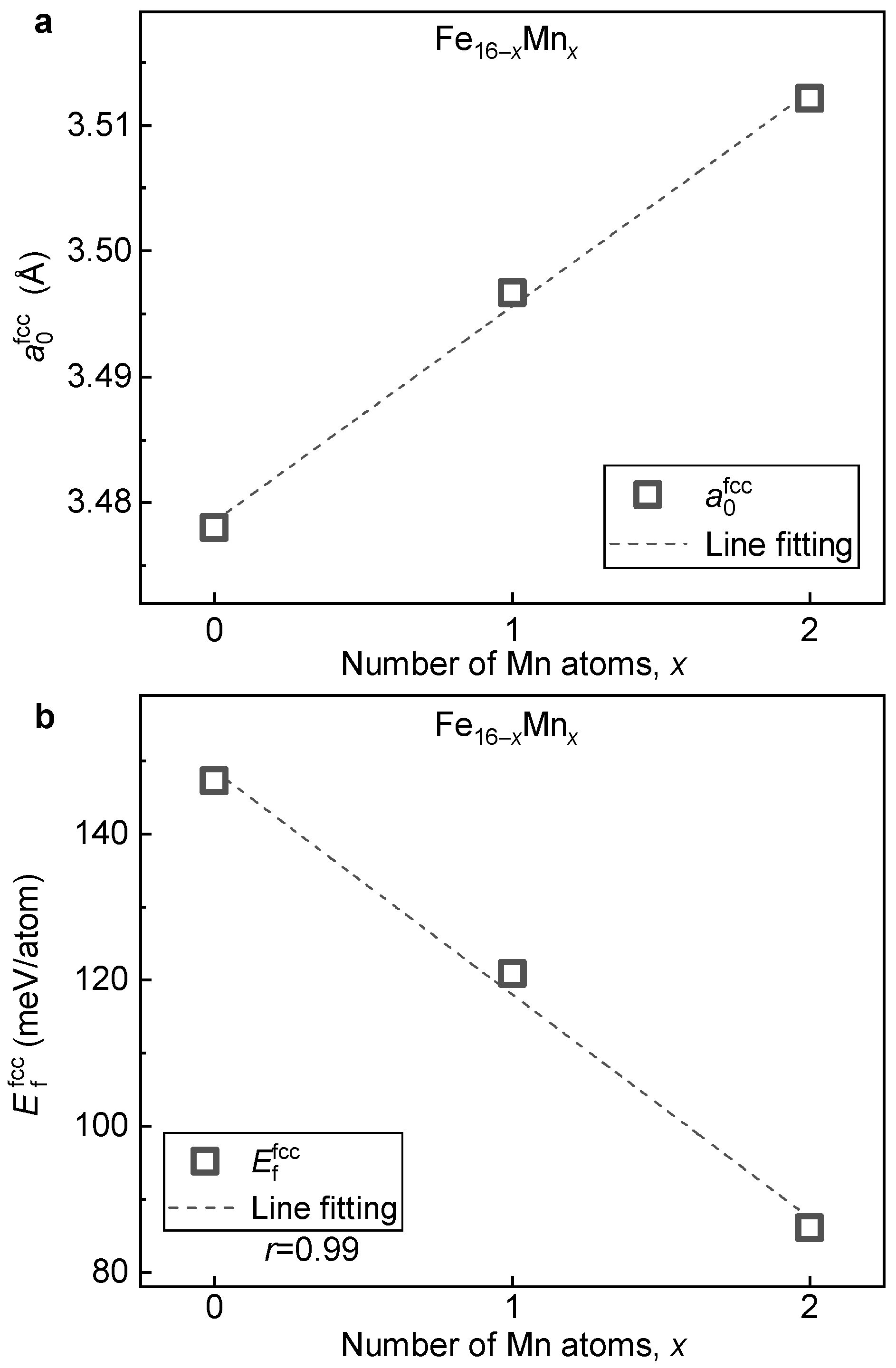
3.2.2. Lattice Stabilities and Magnetic Properties
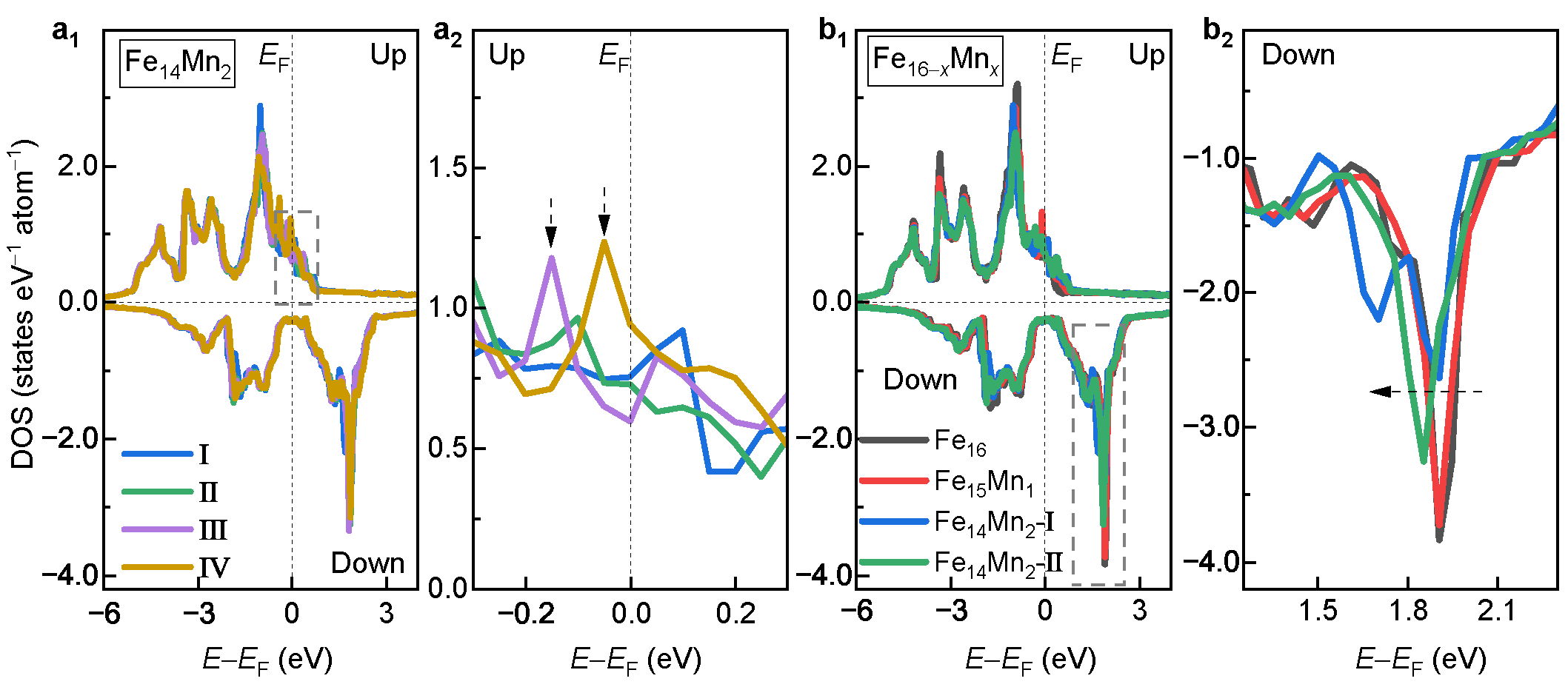
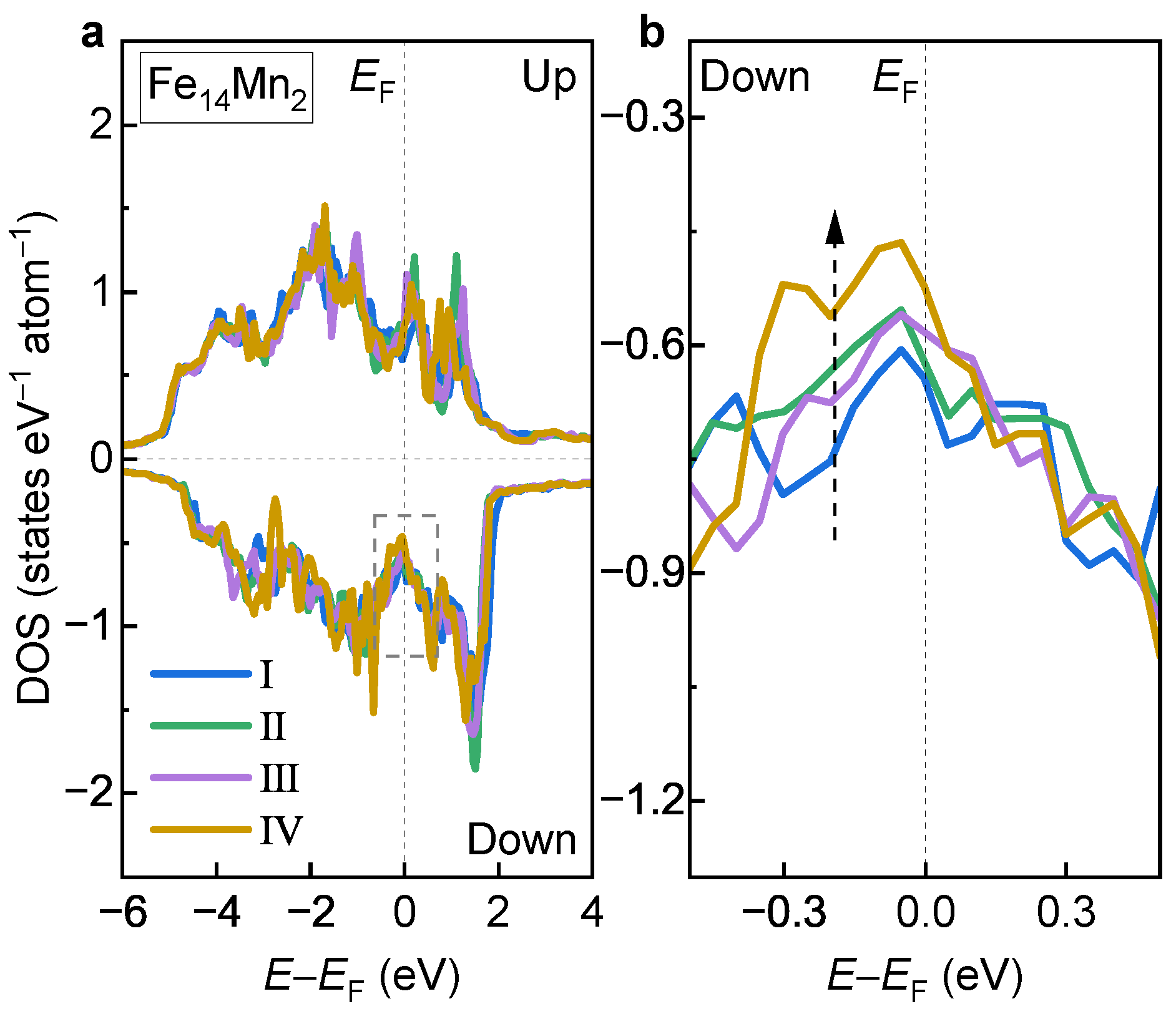
3.2.3. Electronic Structures
3.3. Phase Transition
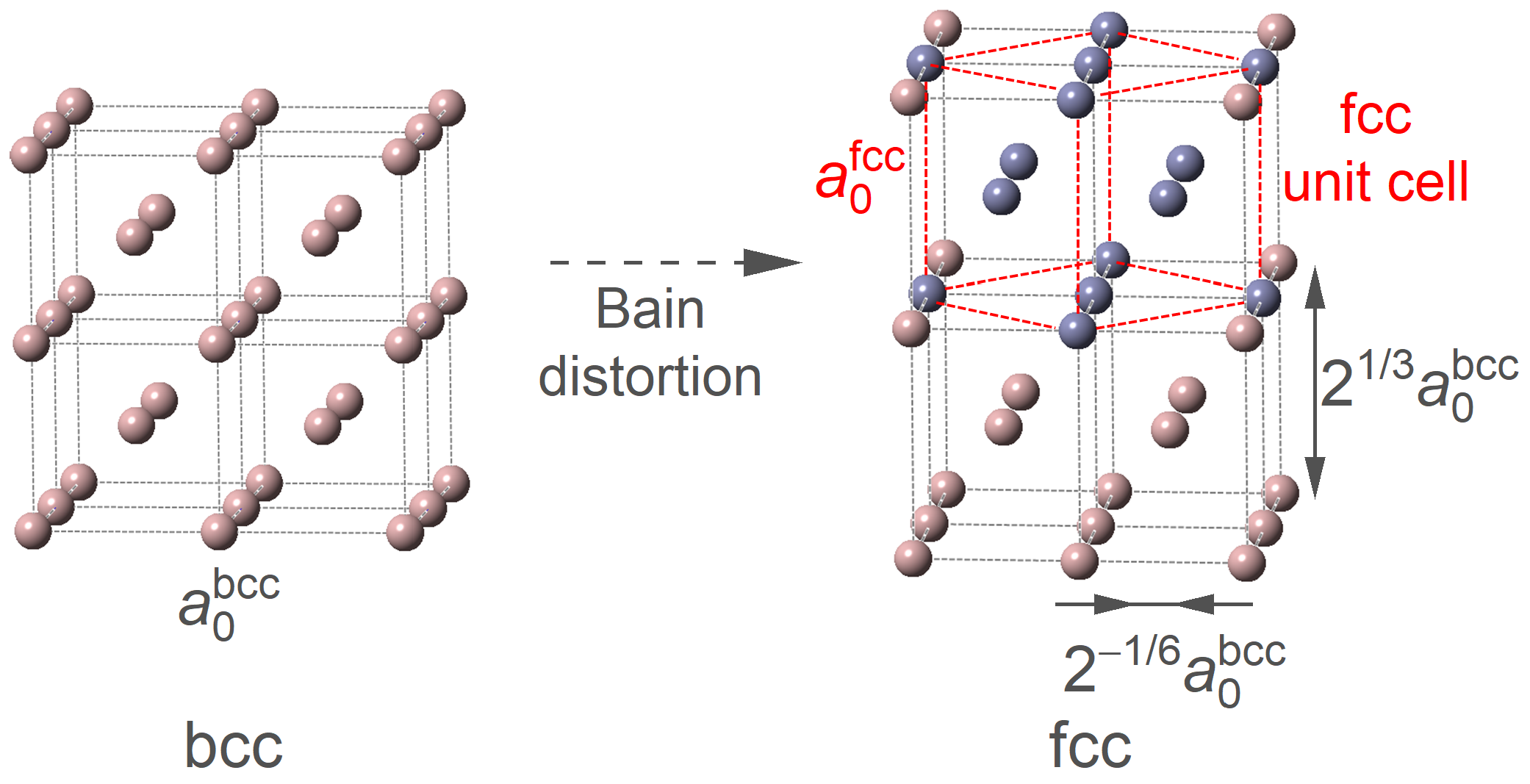
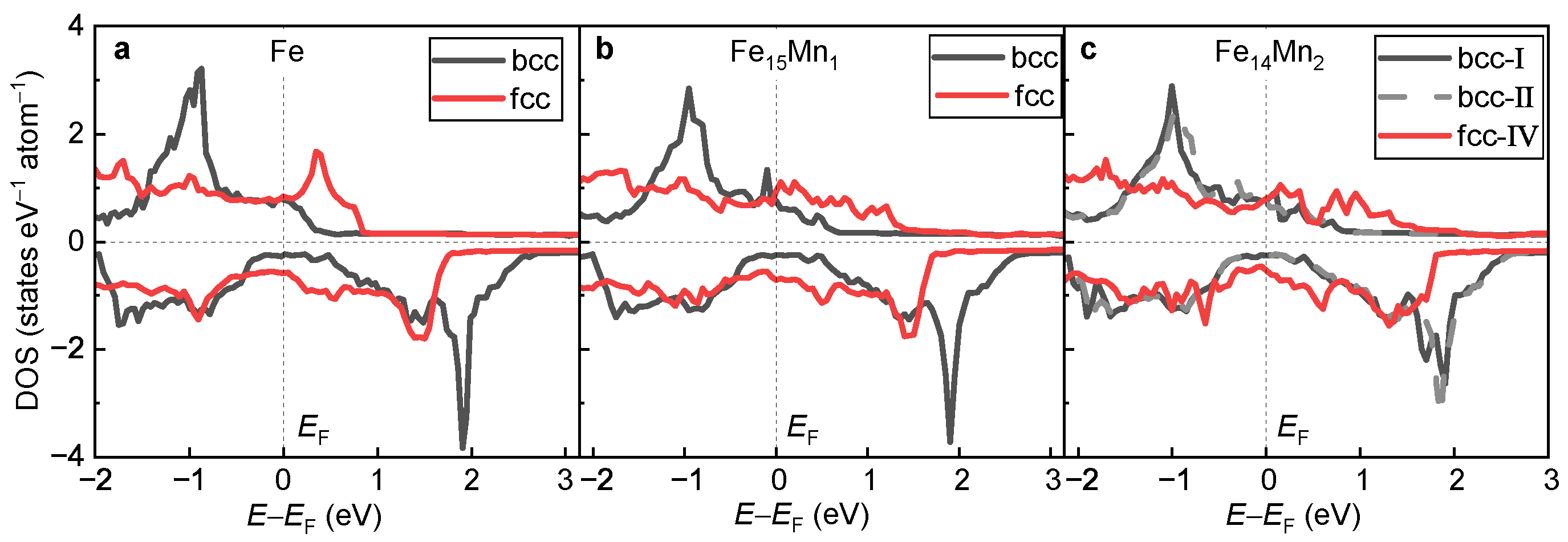
3.3.1. Phase Transition from fcc to bcc
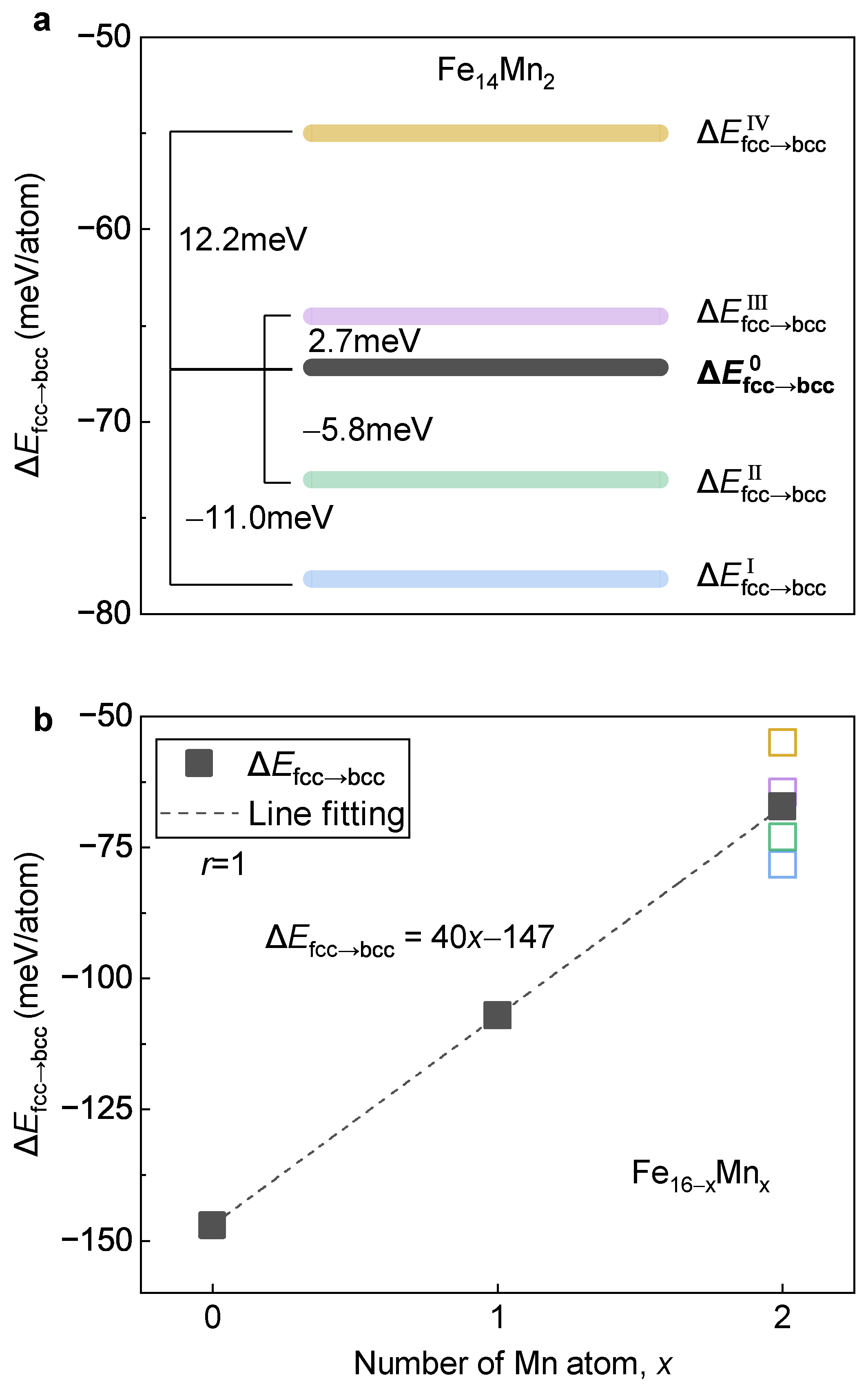
3.3.2. Impact of Mn on Phase Transition
3.3.3. Key Factor Deciding Mn Tailored Phase Transition
4. Conclusions
- (i)
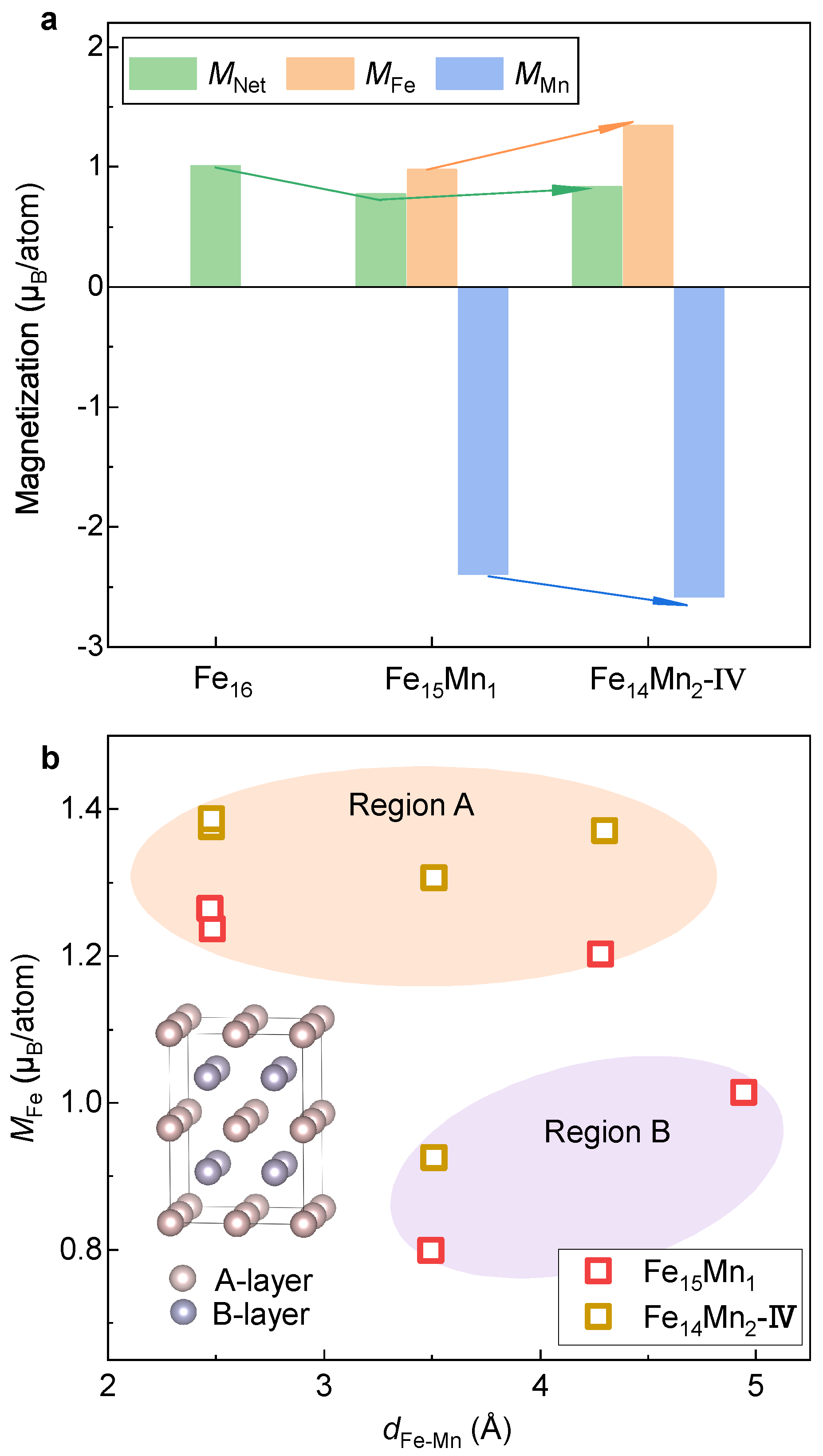
- Mn prefers ferromagnetic and antiferromagnetic interaction with Fe in the bcc and fcc phases, respectively. In these two phases, the magnetic moment of Mn is smaller and larger than Fe, respectively. The local moment of Fe is decided by the Fe-Mn distance in the bcc phase, whereas in the fcc phase, it is determined by spatial orientation with Mn.
- (ii)
- Mn prefers different site occupations in the bcc and fcc phases, which can be understood from the electronic density of states near Fermi energy. This discrepancy implies the possibility of the redistribution of solute Mn in the Fe matrix during the fcc→bcc phase transition, which is favorable for increasing the driving force of transformation.
- (iii)
- Mn alloying tends to destabilize and stabilize the bcc and fcc phases, respectively. With the increase in Mn, the driving force of the fcc→bcc phase transition decreases, in which the stabilization in the fcc phase plays a dominant role. Antiferromagnetism is recognized as the key reason for the enhanced stability of the fcc phase by Mn alloying.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hu, B.; Luo, H.; Yang, F.; Dong, H. Recent progress in medium-Mn steels made with new designing strategies, a review. J. Mater. Sci. Technol. 2017, 33, 1457–1464. [Google Scholar] [CrossRef]
- Lee, Y.K.; Han, J. Current opinion in medium manganese steel. Mater. Sci. Technol. 2015, 31, 843–856. [Google Scholar] [CrossRef]
- Zhang, H.; Cai, M.; Zhu, W.; Sun, S.; Yan, H.; Yao, S.; Luan, Y.; Tang, S.; Hodgson, P. Low-temperature superplastic deformation of cold-rolled Fe–5.6 Mn–1.1 Al–0.2 C steel. Metall. Mater. Trans. A 2022, 53, 3869–3880. [Google Scholar] [CrossRef]
- Mueller, J.; Hu, X.; Sun, X.; Ren, Y.; Choi, K.; Barker, E.; Speer, J.; Matlock, D.; De Moor, E. Austenite formation and cementite dissolution during intercritical annealing of a medium-manganese steel from a martensitic condition. Mater. Des. 2021, 203, 109598. [Google Scholar] [CrossRef]
- Li, C.M.; Sommer, F.; Mittemeijer, E.J. Characteristics of the γ→ α transformation in Fe–Mn alloys. Mater. Sci. Eng. A 2002, 325, 307–319. [Google Scholar] [CrossRef]
- Sahin, O.; Calik, A.; Ekinci, A.; Ucar, N. Work hardening in Fe–Mn binary alloys. Mater. Charact. 2007, 58, 571–574. [Google Scholar] [CrossRef]
- Uenishi, A.; Teodosiu, C. Solid solution softening at high strain rates in Si-and/or Mn-added interstitial free steels. Acta Mater. 2003, 51, 4437–4446. [Google Scholar] [CrossRef]
- Lee, S.; De Cooman, B.C. On the selection of the optimal intercritical annealing temperature for medium Mn TRIP steel. Metall. Mater. Trans. A 2013, 44, 5018–5024. [Google Scholar] [CrossRef]
- Miyamoto, G.; Oh, J.; Hono, K.; Furuhara, T.; Maki, T. Effect of partitioning of Mn and Si on the growth kinetics of cementite in tempered Fe–0.6 mass% C martensite. Acta Mater. 2007, 55, 5027–5038. [Google Scholar] [CrossRef]
- Sun, S.; Cai, M.; Ding, H.; Yan, H.; Tian, Y.; Tang, S.; Hodgson, P. Achieving high ductility in a 1.4 GPa grade medium Mn lightweight TRIP/TWIP steel with hierarchical lamellar structure. Mater. Sci. Eng. A 2022, 858, 144118. [Google Scholar] [CrossRef]
- Li, Z.; Pradeep, K.G.; Deng, Y.; Raabe, D.; Tasan, C.C. Metastable high-entropy dual-phase alloys overcome the strength–ductility trade-off. Nature 2016, 534, 227–230. [Google Scholar] [CrossRef] [PubMed]
- La Roca, P.; Baruj, A.; Sade, M. Shape-memory effect and pseudoelasticity in Fe–Mn-based alloys. Shape Mem. Superelasticity 2017, 3, 37–48. [Google Scholar] [CrossRef]
- Hu, G.; Cai, X.; Rong, Y. Fundamentals of Materials Science; Shanghai Jiao Tong University Press: Shanghai, China, 2010; pp. 19–57. [Google Scholar]
- Yan, H.L.; Liu, H.X.; Zhao, Y.; Jia, N.; Bai, J.; Yang, B.; Li, Z.; Zhang, Y.; Esling, C.; Zhao, X.; et al. Unraveling the abnormal dependence of phase stability on valence electron concentration in Ni-Mn-based metamagnetic shape memory alloys. J. Appl. Phys. 2020, 128, 045104. [Google Scholar] [CrossRef]
- Zayak, A.; Adeagbo, W.; Entel, P.; Rabe, K. e/a dependence of the lattice instability of cubic Heusler alloys from first principles. Appl. Phys. Lett. 2006, 88, 111903. [Google Scholar] [CrossRef]
- Kuang, W.; Wang, H.; Li, X.; Zhang, J.; Zhou, Q.; Zhao, Y. Application of the thermodynamic extremal principle to diffusion-controlled phase transformations in Fe-C-X alloys: Modeling and applications. Acta Mater. 2018, 159, 16–30. [Google Scholar] [CrossRef]
- Wang, D.; Wang, X.X.; Jin, M.L.; He, P.; Zhang, S. Molecular level manipulation of charge density for solid-liquid TENG system by proton irradiation. Nano Energy 2022, 103, 107819. [Google Scholar] [CrossRef]
- Zhang, B.; Hao, M.; Yao, Y.; Xiong, J.; Li, X.; Murphy, A.B.; Sinha, N.; Antony, B.; Ambalampitiya, H.B. Determination and assessment of a complete and self-consistent electron-neutral collision cross-section set for the C4F7N molecule. J. Phys. D 2023, 56, 134001. [Google Scholar] [CrossRef]
- Zhang, B.; Hao, M.; Xiong, J.; Li, X.; Koopman, J. Ab initio molecular dynamics calculations on electron ionization induced fragmentations of C4F7N and C5F10O for understanding their decompositions under discharge conditions. Phys. Chem. Chem. Phys 2023, 25, 7540–7549. [Google Scholar] [CrossRef]
- Akai, H.; Akai, M.; Kanamori, J. Electronic structure of impurities in ferromagnetic iron. II. 3d and 4d impurities. J. Phys. Soc. Japan 1985, 54, 4257–4264. [Google Scholar] [CrossRef]
- Anisimov, V.I.; Antropov, V.P.; Liechtenstein, A.I.; Gubanov, V.A.; Postnikov, A.V. Electronic structure and magnetic properties of 3d impurities in ferromagnetic metals. Phys. Rev. B 1988, 37, 5598–5602. [Google Scholar] [CrossRef]
- Drittler, B.; Stefanou, N.; Blügel, S.; Zeller, R.; Dederichs, P.H. Electronic structure and magnetic properties of dilute Fe alloys with transition-metal impurities. Phys. Rev. B 1989, 40, 8203–8212. [Google Scholar] [CrossRef] [PubMed]
- Kulikov, N.I.; Demangeat, C. Spin polarization of disordered Fe-Cr and Fe-Mn alloys. Phys. Rev. B 1997, 55, 3533–3542. [Google Scholar] [CrossRef]
- Medvedeva, N.; Van Aken, D.; Medvedeva, J.E. Magnetism in bcc and fcc Fe with carbon and manganese. J. Phys. Condens Mat. 2010, 22, 316002. [Google Scholar] [CrossRef] [PubMed]
- Medvedeva, N.; Van Aken, D.C.; Medvedeva, J.E. The effect of carbon distribution on the manganese magnetic moment in bcc Fe–Mn alloy. J. Phys. Condens. Matter 2011, 23, 326003. [Google Scholar] [CrossRef] [PubMed]
- Rahman, G.; Kim, I.G.; Bhadeshia, H.K.D.H.; Freeman, A.J. First-principles investigation of magnetism and electronic structures of substitutional 3d transition-metal impurities in bcc Fe. Phys. Rev. B 2010, 81, 184423. [Google Scholar] [CrossRef]
- Comtesse, D.; Herper, H.C.; Hucht, A.; Entel, P. A first-principles study aided with Monte Carlo simulations of carbon doped iron-manganese alloys. Eur. Phys. J. B 2012, 85, 1–13. [Google Scholar] [CrossRef]
- King, D.J.; Middleburgh, S.C.; Burr, P.A.; Whiting, T.; Fossati, P.; Wenman, M.R. Density functional theory study of the magnetic moment of solute Mn in bcc Fe. Phys. Rev. B 2018, 98, 024418. [Google Scholar] [CrossRef]
- Schneider, A.; Fu, C.C.; Barreteau, C. Local environment dependence of Mn magnetism in bcc iron-manganese alloys: A first-principles study. Phys. Rev. B 2018, 98, 094426. [Google Scholar] [CrossRef]
- Schneider, A.; Fu, C.C.; Waseda, O.; Barreteau, C.; Hickel, T. Ab initio based models for temperature-dependent magnetochemical interplay in bcc Fe-Mn alloys. Phys. Rev. B 2021, 103, 024421. [Google Scholar] [CrossRef]
- Kou, B.; Kou, Z.; Liu, Y.; Han, P. First principles study on interstitial C and substitutional Mn in bcc iron: Structural stability and elastic properties. Mater. Res. Innov. 2014, 18, S2-775–S2-779. [Google Scholar] [CrossRef]
- Music, D.; Takahashi, T.; Vitos, L.; Asker, C.; Abrikosov, I.A.; Schneider, J.M. Elastic properties of Fe–Mn random alloys studied by ab initio calculations. Appl. Phys. Lett. 2007, 91, 191904. [Google Scholar] [CrossRef]
- Zhang, H.; Lu, S.; Zhou, M.; Punkkinen, M.P.; Johansson, B.; Vitos, L. Ab initio determination of the elastic properties of ferromagnetic body-centered cubic Fe-Mn-Al alloys. J. Appl. Phys. 2015, 118, 103904. [Google Scholar] [CrossRef]
- Gebhardt, T.; Music, D.; Hallstedt, B.; Ekholm, M.; Abrikosov, I.A.; Vitos, L.; Schneider, J. Ab initio lattice stability of fcc and hcp Fe–Mn random alloys. J. Phys. Condens. Matter 2010, 22, 295402. [Google Scholar] [CrossRef] [PubMed]
- Acciarri, M.; La Roca, P.; Guerrero, L.; Baruj, A.; Curiale, J.; Sade, M. Effect of FCC anti-ferromagnetic ordering on the stability of phases in Fe60-xMn30Cr10Co10 high entropy alloys. J. Alloys Compd. 2020, 823, 153845. [Google Scholar] [CrossRef]
- Choi, Y.; Dong, Z.; Li, W.; Lizárraga, R.; Kwon, S.K.; Vitos, L. Density functional theory description of paramagnetic hexagonal close-packed iron. Materials 2022, 15, 1276. [Google Scholar] [CrossRef]
- Nakano, J.; Jacques, P.J. Effects of the thermodynamic parameters of the hcp phase on the stacking fault energy calculations in the Fe–Mn and Fe–Mn–C systems. Calphad 2010, 34, 167–175. [Google Scholar] [CrossRef]
- Wang, C.; Zu, W.; Wang, H.; Wang, Y. First-principles study on stacking fault energy of γ-Fe–Mn alloys. Met. Mater. Int. 2021, 27, 3205–3213. [Google Scholar] [CrossRef]
- Okatov, S.V.; Kuznetsov, A.R.; Gornostyrev, Y.N.; Urtsev, V.N.; Katsnelson, M.I. Effect of magnetic state on the γ-α transition in iron: First-principles calculations of the Bain transformation path. Phys. Rev. B 2009, 79, 094111. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223–16233. [Google Scholar] [CrossRef] [PubMed]
- Herper, H.; Hoffmann, E.; Entel, P. Ab initio investigations of iron-based martensitic systems. J. Phys. 1997, 7, C5-71–C5-76. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to solid state physics, 6th ed. Phys. Today 1957, 10, 43–44. [Google Scholar] [CrossRef]
- Wang, C.S.; Klein, B.M.; Krakauer, H. Theory of magnetic and structural ordering in Iron. Phys. Rev. Lett. 1985, 54, 1852–1855. [Google Scholar] [CrossRef]
- Clementi, E.; Raimondi, D.L.; Reinhardt, W.P. Atomic screening constants from SCF functions. II. Atoms with 37 to 86 electrons. J. Chem. Phys. 2004, 47, 1300–1307. [Google Scholar] [CrossRef]
- Schilfgaarde, M.; Abrikosov, I.; Johansson, B. Origin of the Invar effect in iron–nickel alloys. Nature 1999, 400, 46–49. [Google Scholar] [CrossRef]
- Yan, H.L.; Liu, H.X.; Zhao, Y.; Jia, N.; Bai, J.; Yang, B.; Li, Z.; Zhang, Y.; Esling, C.; Zhao, X.; et al. Impact of B alloying on ductility and phase transition in the Ni-Mn-based magnetic shape memory alloys: Insights from first-principles calculation. J. Mater. Sci. Technol. 2021, 74, 27–34. [Google Scholar] [CrossRef]
- Abrahams, S.C.; Guttman, L.; Kasper, J.S. Neutron diffraction determination of antiferromagnetism in Face-centered cubic (γ) iron. Phys. Rev. 1962, 127, 2052–2055. [Google Scholar] [CrossRef]
- Lintzen, S.; von Appen, J.; Hallstedt, B.; Dronskowski, R. The Fe–Mn enthalpy phase diagram from first principles. J. Alloys Compd. 2013, 577, 370–375. [Google Scholar] [CrossRef]
- Krauss, G.; Thompson, S.W. Ferritic microstructures in continuously cooled low-and ultralow-carbon steels. ISIJ Int. 1995, 35, 937–945. [Google Scholar] [CrossRef]
- Calcagnotto, M.; Ponge, D.; Raabe, D. On the effect of manganese on grain size stability and hardenability in ultrafine-grained ferrite/martensite dual-phase steels. Metall. Mater. Trans. A 2012, 43, 37–46. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alloy | Model | |||||
|---|---|---|---|---|---|---|
| Å | µ/at. | µ/at. | µ/at. | eV/at. | ||
| Fe | − | 2.831 | 2.19 | 2.19 | − | −8.243 |
| − | 2.84 | 2.17 | ||||
| − | 2.83 | 2.22 | ||||
| − | 2.824 | 2.11 | 2.18 | 1.05 | −8.266 | |
| I | 2.829 | 2.02 | 2.23 | 0.46 | −8.298 | |
| II | 2.820 | 2.02 | 2.17 | 1.02 | −8.298 | |
| III | 2.819 | 2.01 | 2.13 | 1.06 | −8.293 | |
| IV | 2.820 | 2.03 | 2.07 | 1.70 | −8.286 |
| Alloy | Model | |||||
|---|---|---|---|---|---|---|
| Å | µ/at. | µ/at. | µ/at. | eV/at. | ||
| Fe | − | 3.478 | 1.02 | 1.02 | − | −8.095 |
| − | 3.48 | 1.23 | ||||
| − | 3.588 | 0.75 | ||||
| − | 3.497 | 0.77 | 0.98 | −2.39 | −8.159 | |
| I | 3.522 | 0.93 | 1.4 | −2.33 | −8.220 | |
| II | 3.507 | 0.79 | 1.26 | −2.48 | −8.225 | |
| III | 3.502 | 0.75 | 1.23 | −2.37 | −8.229 | |
| IV | 3.512 | 0.83 | 1.34 | −2.58 | −8.231 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Yang, J.-H.; Zhao, Y.; Ma, H.; Tian, Y.; Cai, M.; Tang, S.; Liu, Y.; Zhao, X.; Yan, H.-L.; et al. Impact of Mn Alloying on Phase Stabilities, Magnetic Properties and Electronic Structures in Fe. Materials 2023, 16, 6679. https://doi.org/10.3390/ma16206679
Yang H, Yang J-H, Zhao Y, Ma H, Tian Y, Cai M, Tang S, Liu Y, Zhao X, Yan H-L, et al. Impact of Mn Alloying on Phase Stabilities, Magnetic Properties and Electronic Structures in Fe. Materials. 2023; 16(20):6679. https://doi.org/10.3390/ma16206679
Chicago/Turabian StyleYang, Hao, Jin-Han Yang, Ying Zhao, Han Ma, Yanzhong Tian, Minghui Cai, Shuai Tang, Yandong Liu, Xiang Zhao, Hai-Le Yan, and et al. 2023. "Impact of Mn Alloying on Phase Stabilities, Magnetic Properties and Electronic Structures in Fe" Materials 16, no. 20: 6679. https://doi.org/10.3390/ma16206679
APA StyleYang, H., Yang, J.-H., Zhao, Y., Ma, H., Tian, Y., Cai, M., Tang, S., Liu, Y., Zhao, X., Yan, H.-L., & Zuo, L. (2023). Impact of Mn Alloying on Phase Stabilities, Magnetic Properties and Electronic Structures in Fe. Materials, 16(20), 6679. https://doi.org/10.3390/ma16206679