
Self-Discharge Processes in Symmetrical Supercapacitors with Activated Carbon Electrodes


 , ,
, , 
Abstract
:1. Introduction
- 1.
- 2.
- 3.
- (a)
- The stage of polarization of electrodes due to reactions on their surfaces, which depends on the surface catalytic capability;
- (b)
- The barrier stage in the formation of the of electric double layer (EDL);
- (c)
- The stages of diffusion and ohmic transfer of ions between electrodes.
- 4.
2. Experimental Section
2.1. Electrode Fabrication
2.2. Electrochemical Measurements
2.3. Contact Angle Measurement
2.4. IR Spectroscopy
2.5. X-ray Photoelectron Spectroscopy
3. Results and Discussion
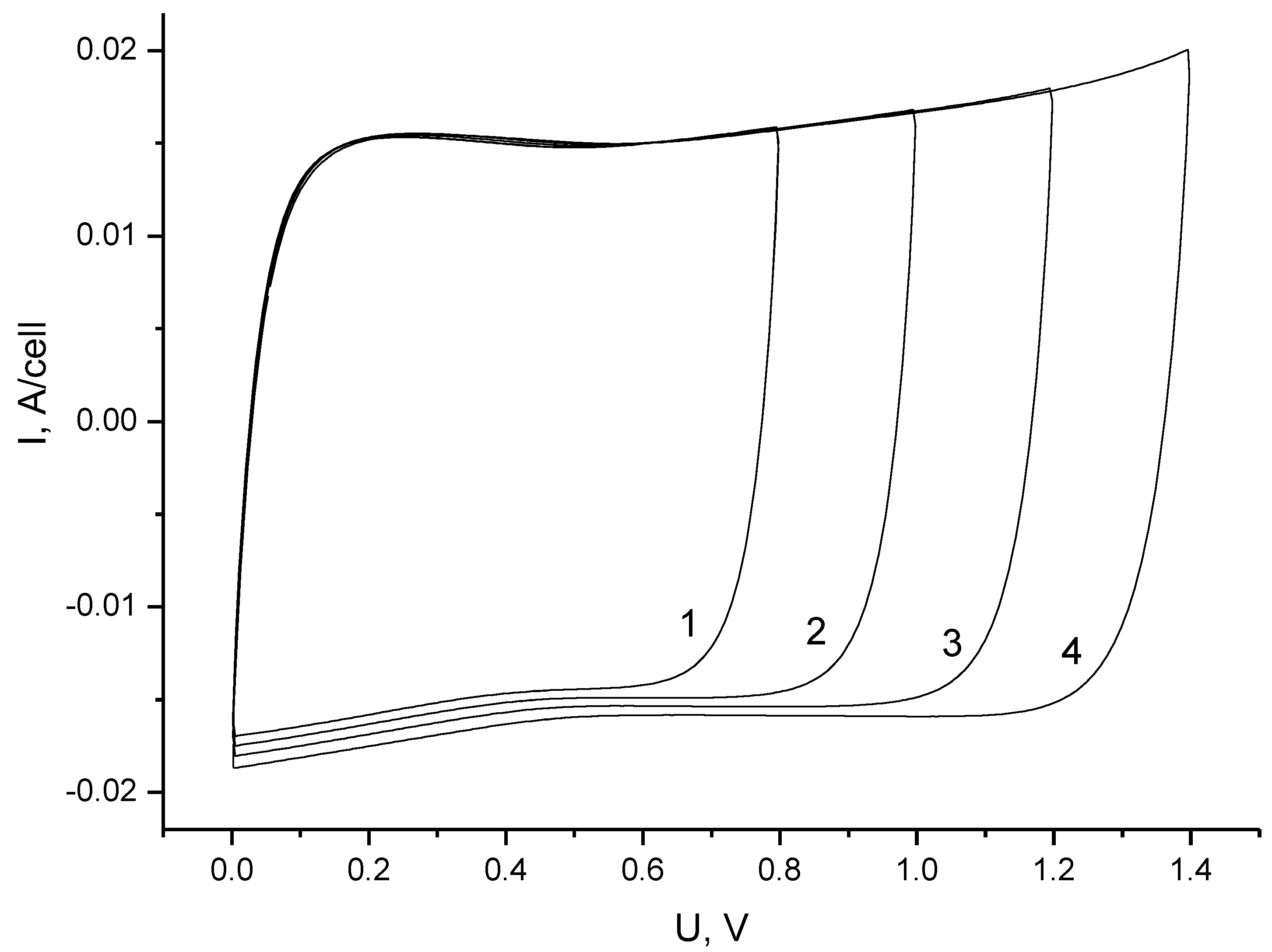
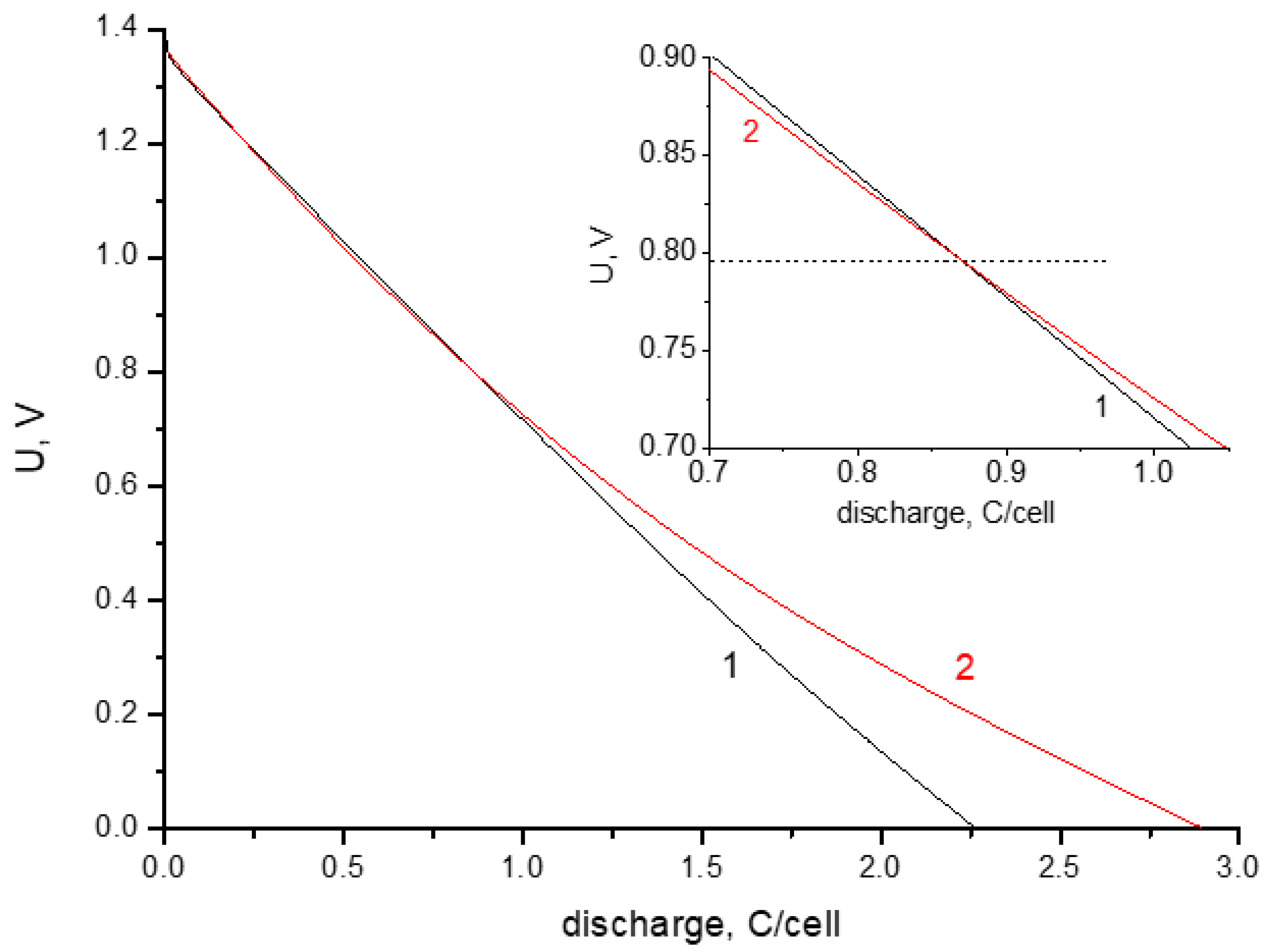
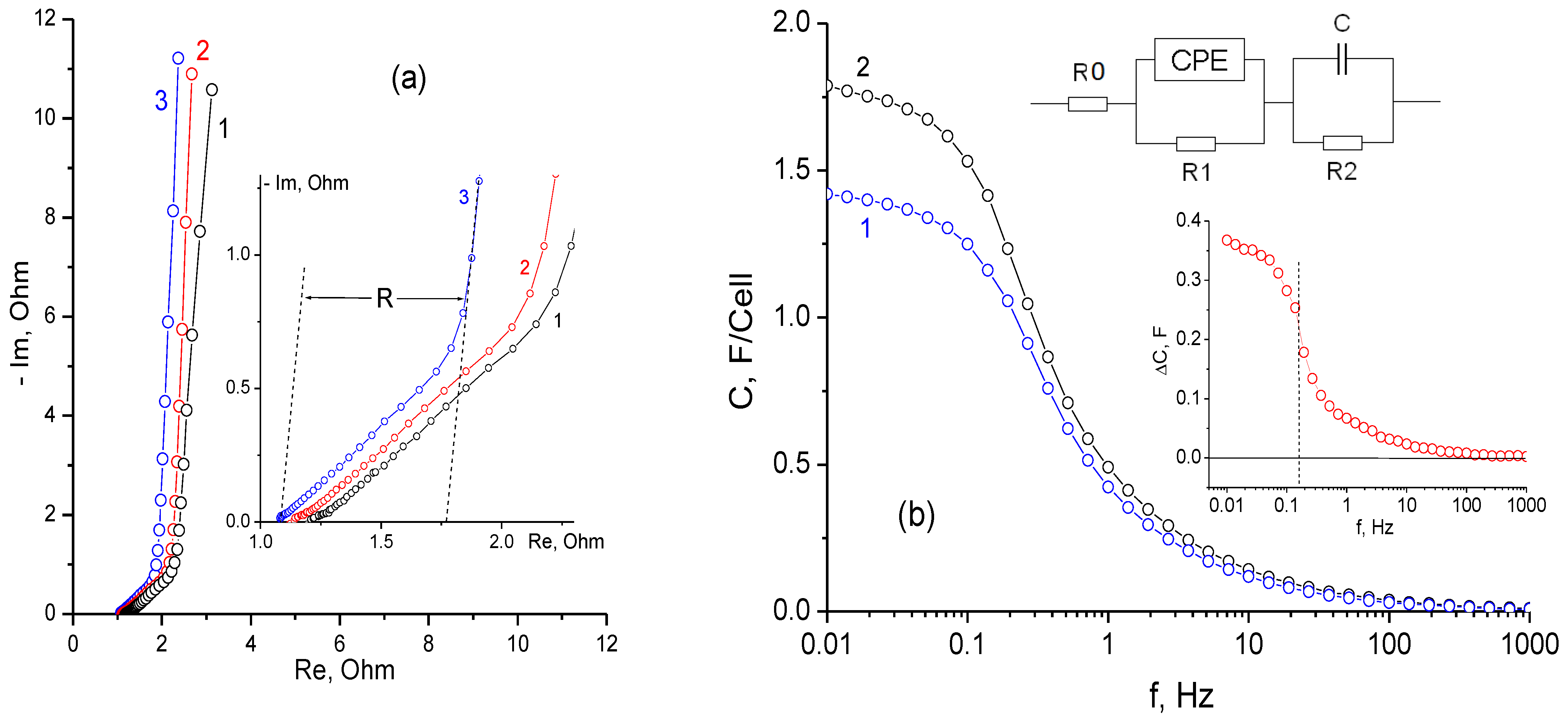
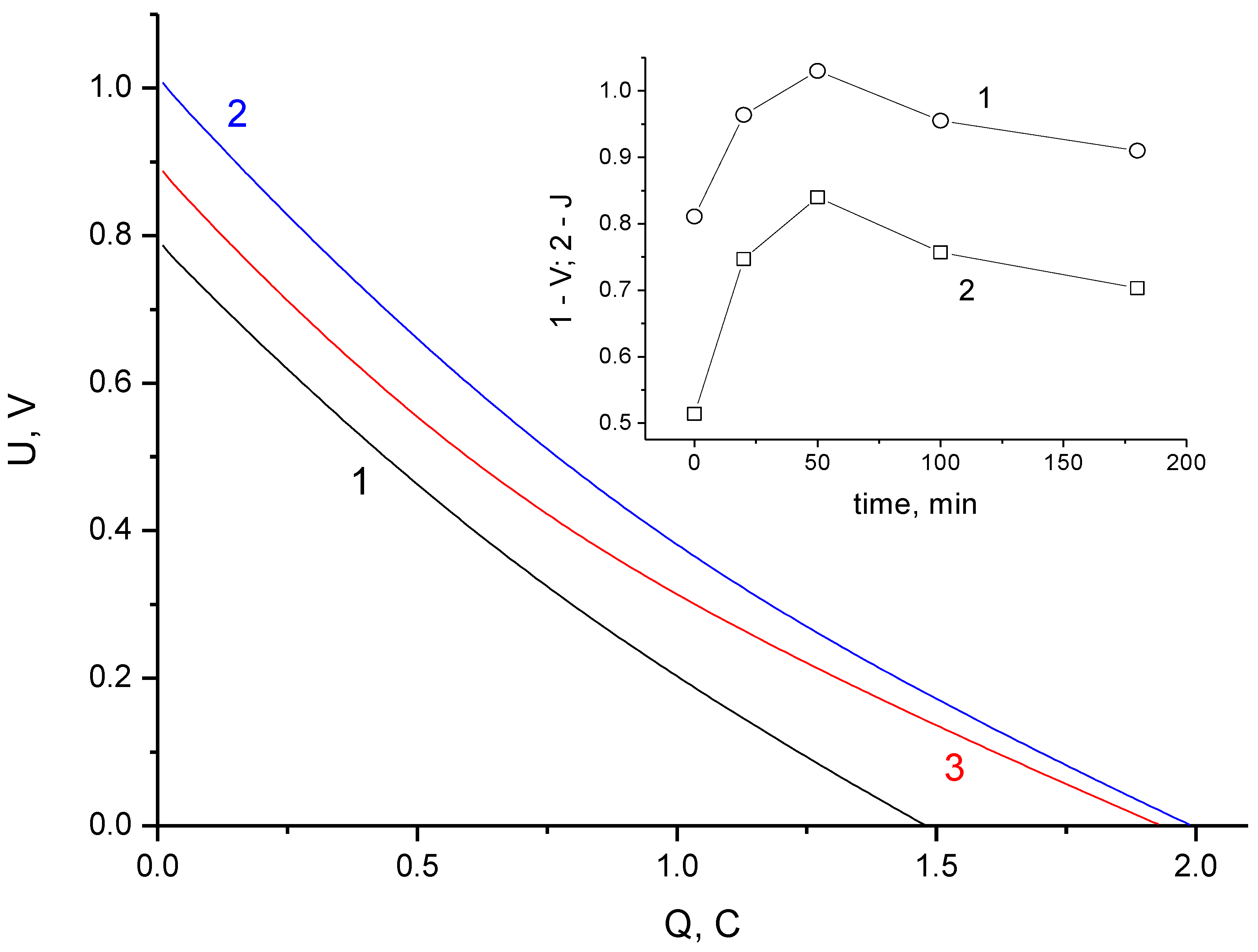
3.1. Electrochemical Measurements
3.2. Contact Angle Measurements
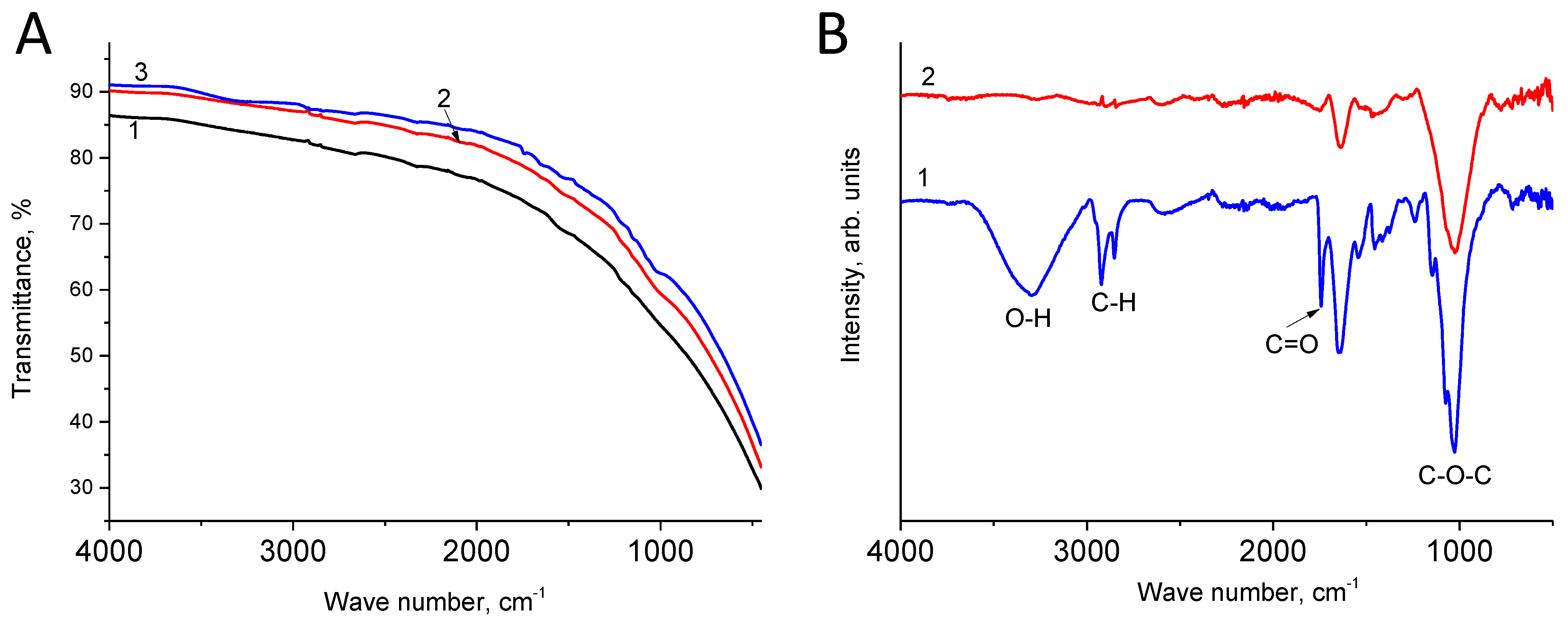
3.3. IR Spectroscop
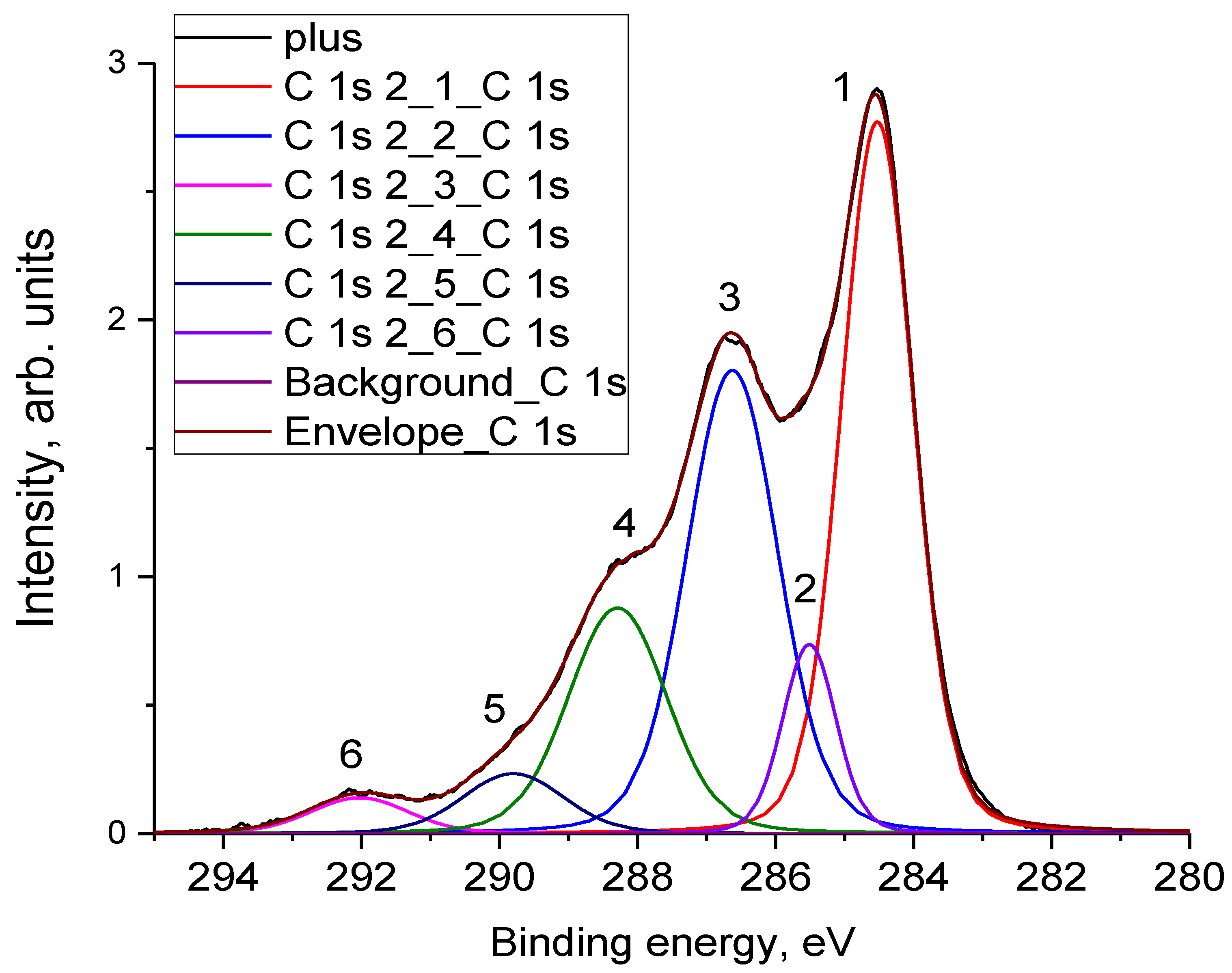
3.4. X-ray Photoelectron Spectroscopy
4. Discussion
5. Conclusions
- (a)
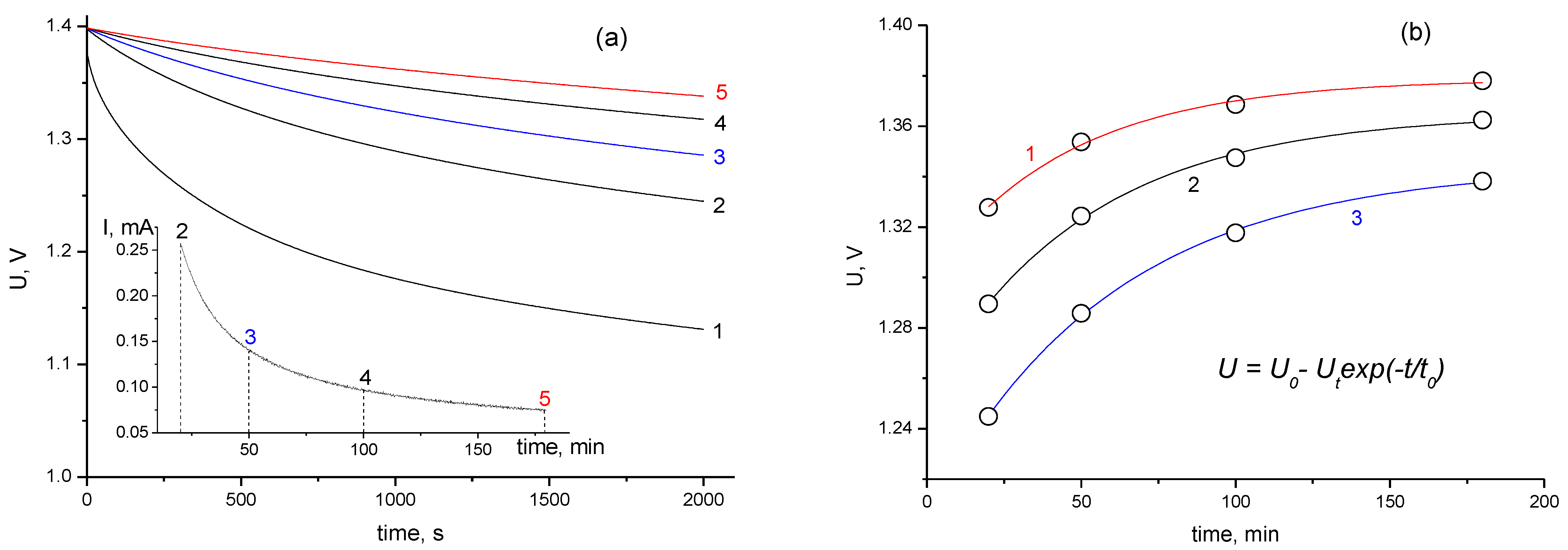
- During the long-term stabilization (the potentiostatic charge), a decrease in the discharge capacity is observed at voltages exceeding 0.8 V; this is associated with the blocking of the surface by chemisorbed particles.
- (b)
- The process of self-discharge can be conditionally divided into two stages: initial and main. For the initial stage, with a duration of about an hour, the voltage drop in the charged cell is determined by processes of charge redistribution inside the porous structure of the electrodes. For the main stage with a duration more than 2–3 h, the cell voltage drop is due to the processes of charge transfer between the electrodes.
- (c)
- The self-discharge rate at the initial stage decreases with increasing stabilization time and is well-described by a simple first-order exponential equation over a wide time interval.
- (d)
- The self-discharge rate of the main stage (~20 h) has a pronounced minimum. Under our experimental conditions, the optimal stabilization time of 50 min was obtained; this represented an increase of 60% in the residual energy compared to a cell charged without stabilization.
- (e)
- According to the measurement of the contact (wetting) angle, hydrophilization of the negative electrode occurs during the long-term cell polarization process. In this case, the difference in the IR spectra of the negative electrode reveals the maximum corresponds to vibrations of the epoxy functional groups. Based on these data and considering that long-term stabilization leads to an increase in the average self-discharge current, an assumption was made about the participation of particles with a high oxidizing ability in the process of the main self-discharge.
- (f)
- The oxidation of the electrodes that were in a charged state for a long time was established using XPS measurements. The oxidation state of the positive electrode significantly exceeds the oxidation state of the negative electrode, mainly due to the formation of hydroxyl and epoxy groups. The number of carboxyl groups on both electrodes increases to the same extent. The XPS data are consistent with the self-discharge mechanism 2 presented in the introduction.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Conway, B.E. Electrochemical Supercapacitors: Scientific Fundamentals and Technological Applications; Springer Science & Business Media: Berlin, Germany, 2013. [Google Scholar]
- Bagotsky, V.S.; Skundin, A.M.; Volfkovich, Y.M. Electrochemical Power Sources: Batteries, Fuel Cells, and Supercapacitors; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Volfkovich, Y.M.; Bograchev, D.A.; Rychagov, A.Y.; Sosenkin, V.E.; Chaika, M.Y. Supercapacitors with Carbon Electrodes. Energy Efficiency: Modeling and Experimental Verification. J. Solid State Electrochem. 2015, 19, 2771–2779. [Google Scholar] [CrossRef]
- Oren, Y. Capacitive Deionization (CDI) for Desalination and Water Treatment—Past, Present and Future (a Review). Desalination 2008, 228, 10–29. [Google Scholar] [CrossRef]
- Volfkovich, Y.M. Electrochemical Supercapacitors (a Review). Russ. J. Electrochem. 2021, 57, 311–347. [Google Scholar] [CrossRef]
- Kowal, J.; Avaroglu, E.; Chamekh, F.; Šenfelds, A.; Thien, T.; Wijaya, D.; Sauer, D.U. Detailed Analysis of the Self-Discharge of Supercapacitors. J. Power Sources 2011, 196, 573–579. [Google Scholar] [CrossRef]
- Diab, Y.; Venet, P.; Gualous, H.; Rojat, G. Self-Discharge Characterization and Modeling of Electrochemical Capacitor Used for Power Electronics Applications. IEEE Trans. Power Electron. 2009, 24, 510–517. [Google Scholar] [CrossRef]
- Kurzweil, P.; Shamonin, M. State-of-Charge Monitoring by Impedance Spectroscopy during Long-Term Self-Discharge of Supercapacitors and Lithium-Ion Batteries. Batteries 2018, 4, 35. [Google Scholar] [CrossRef]
- Liu, K.; Yu, C.; Guo, W.; Ni, L.; Yu, J.; Xie, Y.; Wang, Z.; Ren, Y.; Qiu, J. Recent Research Advances of Self-Discharge in Supercapacitors: Mechanisms and Suppressing Strategies. J. Mater. Chem. A Mater. Energy Sustain. 2021, 58, 94–109. [Google Scholar] [CrossRef]
- Tevi, T.; Yaghoubi, H.; Wang, J.; Takshi, A. Application of Poly (p-Phenylene Oxide) as Blocking Layer to Reduce Self-Discharge in Supercapacitors. J. Power Sources 2013, 241, 589–596. [Google Scholar] [CrossRef]
- Shen, J.-F.; He, Y.-J.; Ma, Z.-F. A Systematical Evaluation of Polynomial Based Equivalent Circuit Model for Charge Redistribution Dominated Self-Discharge Process in Supercapacitors. J. Power Sources 2016, 303, 294–304. [Google Scholar] [CrossRef]
- Saha, P.; Khanra, M. Equivalent Circuit Model of Supercapacitor for Self-Discharge Analysis—A Comparative Study. In Proceedings of the 2016 International Conference on Signal Processing, Communication, Power and Embedded System (SCOPES), Paralakhemundi, India, 3–5 October 2016; pp. 1381–1386. [Google Scholar]
- El Brouji, H.; Vinassa, J.-M.; Briat, O.; Bertrand, N.; Woirgard, E. Ultracapacitors Self Discharge Modelling Using a Physical Description of Porous Electrode Impedance. In Proceedings of the 2008 IEEE Vehicle Power and Propulsion Conference, Harbin, China, 3–5 September 2008; pp. 1–6. [Google Scholar]
- Bamgbopa, M.O.; Belaineh, D.; Mengistie, D.A.; Edberg, J.; Engquist, I.; Berggren, M.; Tybrandt, K. Modelling of Heterogeneous Ion Transport in Conducting Polymer Supercapacitors. J. Mater. Chem. A Mater. Energy Sustain. 2021, 9, 2184–2194. [Google Scholar] [CrossRef]
- Volfkovich, Y.M.; Rychagov, A.Y.; Mikhalin, A.A.; Sosenkin, V.E.; Kabachkov, E.N.; Shulga, Y.M.; Michtchenko, A. Self-Discharge of a Supercapacitor with Electrodes Based on Activated Carbon Cloth. J. Electroanal. Chem. 2022, 910, 116198. [Google Scholar] [CrossRef]
- Wu, F.; Liu, M.; Li, Y.; Feng, X.; Zhang, K.; Bai, Y.; Wang, X.; Wu, C. High-Mass-Loading Electrodes for Advanced Secondary Batteries and Supercapacitors. Electrochem. Energy Rev. 2021, 4, 382–446. [Google Scholar] [CrossRef]
- Xu, Z.; Deng, W.; Wang, X. 3D Hierarchical Carbon-Rich Micro-/Nanomaterials for Energy Storage and Catalysis. Electrochem. Energy Rev. 2021, 4, 269–335. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, S.; Zhou, Y.-N.; Tang, W.; Yang, J.; Peng, C.; Guo, Z. Biomass-Derived Carbon Materials for High-Performance Supercapacitors: Current Status and Perspective. Electrochem. Energy Rev. 2021, 4, 219–248. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Fan, H.; Ma, L. Solid Electrolyte Interface in Zn-Based Battery Systems. Nano-Micro Lett. 2022, 14, 205. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, X.; Ma, L.; Huang, W. Solvation Structures in Aqueous Metal-Ion Batteries. Adv. Energy Mater. 2022, 12, 2202068. [Google Scholar] [CrossRef]
- Ke, Q.; Zhang, Y.; Fu, Y.; Yang, C.; Wu, F.; Li, Z.; Wei, Y.; Zhang, K. Study on Electrochemical Performance of MnO@rGO/Carbon Fabric-Based Wearable Supercapacitors. Materials 2023, 16, 4687. [Google Scholar] [CrossRef]
- Kumar, S.; Ahmed, F.; Shaalan, N.M.; Arshi, N.; Dalela, S.; Chae, K.H. Investigations of Structural, Magnetic, and Electrochemical Properties of NiFe2O4 Nanoparticles as Electrode Materials for Supercapacitor Applications. Materials 2023, 16, 4328. [Google Scholar] [CrossRef]
- Yang, X.; Fan, H.; Hu, F.; Chen, S.; Yan, K.; Ma, L. Aqueous Zinc Batteries with Ultra-Fast Redox Kinetics and High Iodine Utilization Enabled by Iron Single Atom Catalysts. Nano-Micro Lett. 2023, 15, 126. [Google Scholar] [CrossRef]
- Auer, A.; Ding, X.; Bandarenka, A.S.; Kunze-Liebhäuser, J. The Potential of Zero Charge and the Electrochemical Interface Structure of Cu(111) in Alkaline Solutions. J. Phys. Chem. C Nanomater. Interfaces 2021, 125, 5020–5028. [Google Scholar] [CrossRef]
- Trasatti, S.; Lust, E. The Potential of Zero Charge. In Modern Aspects of Electrochemistry; White, R.E., Bockris, J.O., Conway, B.E., Eds.; Springer: Boston, MA, USA, 1999; pp. 1–215. ISBN 9780306469176. [Google Scholar]
- Gajewska, K.; Moyseowicz, A.; Minta, D.; Gryglewicz, G. Effect of Electrolyte and Carbon Material on the Electrochemical Performance of High-Voltage Aqueous Symmetric Supercapacitors. J. Mater. Sci. 2023, 58, 1721–1738. [Google Scholar] [CrossRef]
- Klementov, A.D.; Litvinenko, S.V.; Stepanov, A.B.; Varakin, I.N. Internal Losses and Features of Asymmetric Capacitor Operation. In Proceedings of the 11th International Seminar on Double Lauer Capacitors and Similar Energy Storage Devices, Deerfield Beach, FL, USA, 3–5 December 2001. [Google Scholar]
- Shulga, Y.M.; Baskakov, S.A.; Baskakova, Y.V.; Volfkovich, Y.M.; Shulga, N.Y.; Skryleva, E.A.; Parkhomenko, Y.N.; Belay, K.G.; Gutsev, G.L.; Rychagov, A.Y.; et al. Supercapacitors with Graphene Oxide Separators and Reduced Graphite Oxide Electrodes. J. Power Sources 2015, 279, 722–730. [Google Scholar] [CrossRef]
- Si, Y.; Samulski, E.T. Synthesis of Water Soluble Graphene. Nano Lett. 2008, 8, 1679–1682. [Google Scholar] [CrossRef]
- Jeong, H.-K.; Lee, Y.P.; Jin, M.H.; Kim, E.S.; Bae, J.J.; Lee, Y.H. Thermal Stability of Graphite Oxide. Chem. Phys. Lett. 2009, 470, 255–258. [Google Scholar] [CrossRef]
- Cote, L.J.; Cruz− Silva, R.; Huang, J. Flash Reduction and Patterning of Graphite Oxide and Its Polymer Composite. J. Am. Chem. Soc. 2009, 131, 11027–11032. [Google Scholar] [CrossRef]
- Karthika, P.; Rajalakshmi, N.; Dhathathreyan, K.S. Functionalized Graphene Oxide Based on Biomass Waste: Synthesis and Applications. Soft Nanosci. Lett. 2012, 2, 59–66. [Google Scholar] [CrossRef]
- Xue, Y.; Liu, J.; Chen, H.; Wang, R.; Li, D.; Qu, J.; Dai, L. Nitrogen-Doped Graphene Foams as Metal-Free Counter Electrodes in High-Performance Dye-Sensitized Solar Cells. Angew. Chem. Int. Ed Engl. 2012, 51, 12124–12127. [Google Scholar] [CrossRef] [PubMed]
- Niu, H.; Zhang, S.; Wang, R.; Guo, Z.; Shang, X.; Gan, W.; Qin, S.; Wan, L.; Xu, J. Dye-Sensitized Solar Cells Employing a Multifunctionalized Hierarchical SnO2 Nanoflower Structure Passivated by TiO2 Nanogranulum. J. Phys. Chem. C 2014, 118, 3504–3513. [Google Scholar] [CrossRef]
- Iamprasertkun, P.; Krittayavathananon, A.; Sawangphruk, M. N-Doped Reduced Graphene Oxide Aerogel Coated on Carboxyl-Modified Carbon Fiber Paper for High-Performance Ionic-Liquid Supercapacitors. Carbon 2016, 102, 455–461. [Google Scholar] [CrossRef]
- Du, C.; Pan, N. High Power Density Supercapacitor Electrodes of Carbon Nanotube Films by Electrophoretic Deposition. Nanotechnology 2006, 17, 5314. [Google Scholar] [CrossRef]
- Arulepp, M.; Leis, J.; Lätt, M.; Miller, F.; Rumma, K.; Lust, E.; Burke, A.F. The Advanced Carbide-Derived Carbon Based Supercapacitor. J. Power Sources 2006, 162, 1460–1466. [Google Scholar] [CrossRef]
- Park, S.; An, J.; Potts, J.R.; Velamakanni, A.; Murali, S.; Ruoff, R.S. Hydrazine-Reduction of Graphite- and Graphene Oxide. Carbon 2011, 49, 3019–3023. [Google Scholar] [CrossRef]
- Shulga, Y.M.; Baskakov, S.A.; Knerelman, E.I.; Davidova, G.I.; Badamshina, E.R.; Shulga, N.Y.; Skryleva, E.A.; Agapov, A.L.; Voylov, D.N.; Sokolov, A.P.; et al. Carbon Nanomaterial Produced by Microwave Exfoliation of Graphite Oxide: New Insights. RSC Adv. 2013, 4, 587–592. [Google Scholar] [CrossRef]
- Voylov, D.N.; Agapov, A.L.; Sokolov, A.P.; Shulga, Y.M.; Arbuzov, A.A. Room Temperature Reduction of Multilayer Graphene Oxide Film on a Copper Substrate: Penetration and Participation of Copper Phase in Redox Reactions. Carbon 2014, 69, 563–570. [Google Scholar] [CrossRef]
- Shulga, Y.M.; Baskakov, S.A.; Baskakova, Y.V.; Lobach, A.S.; Kabachkov, E.N.; Volfkovich, Y.M.; Sosenkin, V.E.; Shulga, N.Y.; Nefedkin, S.I.; Kumar, Y.; et al. Preparation of Graphene Oxide-Humic Acid Composite-Based Ink for Printing Thin Film Electrodes for Micro-Supercapacitors. J. Alloys Compd. 2018, 730, 88–95. [Google Scholar] [CrossRef]
- Pohjalainen, E.; Rauhala, T.; Valkeapää, M.; Kallioinen, J.; Kallio, T. Effect of Li4Ti5O12 particle size on the performance of lithium ionbattery electrodes at high rates and low temperatures. Phys. Chem. C 2015, 119, 2277–2283. [Google Scholar] [CrossRef]
- Chaban, M.O.; Rozhdestvenska, L.M.; Palchyk, O.V.; Dzyazko, Y.S.; Dzyazko, O.G. Structural characteristics and sorption properties of lithium-selective composite materials based on TiO2 and MnO2. Appl. Nanosci. 2019, 9, 1037–1045. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode | С | О | N | F | S |
|---|---|---|---|---|---|
| initial | 81.27 | 11.98 | 2.54 | 4.05 | - |
| negative | 76.13 | 15.66 | 4 | 3.66 | 0.2 |
| positive | 74.81 | 16.83 | 4.25 | 3.75 | 0.19 |
| Electrode | Peak | Eb, eV | I, % | Assignment |
|---|---|---|---|---|
| Initial (0) | 1 | 284.6 | 48.75 | C–C (sp2) |
| 2 | 285.5 | 7.02 | C–C (sp3) | |
| 3 | 286.6 | 25.55 | C–OH, C–O–C | |
| 4 | 288.3 | 11.25 | C(O)OH | |
| 5 | 289.8 | 4.13 | π→π* | |
| 6 | 292.1 | 3.3 | C–F2 | |
| Negative (−) | 1 | 284.6 | 41.91 | C–C (sp2) |
| 2 | 285.5 | 7.51 | C–C (sp3) | |
| 3 | 286.6 | 28.4 | C–OH, C–O–C | |
| 4 | 288.3 | 17.38 | C(O)OH | |
| 5 | 289.8 | 2.33 | π→π* | |
| 6 | 292.1 | 2.45 | C–F2 | |
| 1 | 284.6 | 37.83 | C–C (sp2) | |
| 2 | 285.5 | 7.16 | C–C (sp3) | |
| Positive (+) | 3 | 286.6 | 31.27 | C–OH, C–O–C |
| 4 | 288.3 | 16.39 | C(O)OH | |
| 5 | 289.8 | 4.46 | π→π* | |
| 6 | 292.1 | 2.59 | C–F2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rychagov, A.Y.; Sosenkin, V.E.; Izmailova, M.Y.; Kabachkov, E.N.; Shulga, Y.M.; Volfkovich, Y.M.; Gutsev, G.L. Self-Discharge Processes in Symmetrical Supercapacitors with Activated Carbon Electrodes. Materials 2023, 16, 6415. https://doi.org/10.3390/ma16196415
Rychagov AY, Sosenkin VE, Izmailova MY, Kabachkov EN, Shulga YM, Volfkovich YM, Gutsev GL. Self-Discharge Processes in Symmetrical Supercapacitors with Activated Carbon Electrodes. Materials. 2023; 16(19):6415. https://doi.org/10.3390/ma16196415
Chicago/Turabian StyleRychagov, Alexey Yu., Valentin E. Sosenkin, Marianna Yu. Izmailova, Evgeny N. Kabachkov, Yury M. Shulga, Yury M. Volfkovich, and Gennady L. Gutsev. 2023. "Self-Discharge Processes in Symmetrical Supercapacitors with Activated Carbon Electrodes" Materials 16, no. 19: 6415. https://doi.org/10.3390/ma16196415
APA StyleRychagov, A. Y., Sosenkin, V. E., Izmailova, M. Y., Kabachkov, E. N., Shulga, Y. M., Volfkovich, Y. M., & Gutsev, G. L. (2023). Self-Discharge Processes in Symmetrical Supercapacitors with Activated Carbon Electrodes. Materials, 16(19), 6415. https://doi.org/10.3390/ma16196415