
Effects of Mono-Vacancies of Oxygen and Manganese on the Properties of the MnO2/Graphene Heterostructure


Abstract
:1. Introduction
2. Computational Method
3. Results and Discussions
3.1. Strucural Parameters
3.2. Electronic Properties
3.2.1. Bader Charge Transfer
3.2.2. Electronic Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, W.Q.; Wu, L.; Zhu, Y.; Watanabe, K.; Taniguchi, T. Structure of Chemically Derived Mono- and Few-Atomic-Layer Boron Nitride Sheets. Appl. Phys. Lett. 2008, 93, 223103. [Google Scholar] [CrossRef]
- Chhowalla, M.; Shin, H.S.; Eda, G.; Li, L.J.; Loh, K.P.; Zhang, H. The Chemistry of Two-Dimensional Layered Transition Metal Dichalcogenide Nanosheets. Nat. Chem. 2013, 5, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Humanez-Tobar, Á.; Murillo, J.F.; Ortega-López, C.; Rodríguez-Mateínez, J.A.; Espitia-Rico, M.J. Study of the structural and electronic properties of three- and two-dimensional transition-metal dioxides using first-principles calculations. Comput. Condens. Matter 2020, 25, e00498. [Google Scholar] [CrossRef]
- Berrio, G.; Murillo, J.F.; Ortega, C.; Rodriguez, J.A.; Espitia, M.J. Adsorption effect of a chromium atom on the structure and electronic, properties of a single ZnO monolayer. Phys. B Condens. Matter 2019, 565, 44–47. [Google Scholar] [CrossRef]
- Wang, Q.H.; Kalantar-Zadeh, K.; Kis, A.; Coleman, J.N.; Strano, M.S. Electronics and Optoelectronics of Two-Dimensional Transition Metal Dichalcogenides. Nat. Nanotechnol. 2012, 7, 699–712. [Google Scholar] [CrossRef]
- Song, L.; Ci, L.; Lu, H.; Sorokin, P.B.; Jin, C.; Ni, J.; Kvashnin, A.G.; Kvashnin, D.G.; Lou, J.; Yakobson, B.I.; et al. Large Scale Growth and Characterization of Atomic Hexagonal Boron Nitride Layers. Nano Lett. 2010, 10, 3209–3215. [Google Scholar] [CrossRef]
- Tang, Q.; Zhou, Z. Graphene-Analogous Low-Dimensional Materials. Prog. Mater. Sci. 2013, 58, 1244–1315. [Google Scholar] [CrossRef]
- Ataca, C.; Sahin, H.; Ciraci, S. Stable, single-layer MX2 transition-metal oxides and dichalcogenides in a honeycomb-like structure. J. Phys. Chem. C 2012, 116, 8983–8999. [Google Scholar] [CrossRef]
- Rasmussen, F.A.; Thygesen, K.S. Computational 2D Materials Database: Electronic Structure of Transition-Metal Dichalcogenides and Oxides. J. Phys. Chem. C 2015, 119, 13169–13183. [Google Scholar] [CrossRef]
- Omomo, Y.; Sasaki, T.; Watanabe, M. Redoxable Nanosheet Crystallites of MnO2 Derived via Delamination of a Layered Manganese Oxide. J. Am. Chem. Soc. 2003, 125, 3568–3575. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, F.; Izumi, N.; Yasuo, E.; Masahiko, T.; Takeharu, M.; Takayoshi, S. Structure analysis of exfoliated unilamellar crystallites of manganese oxide nanosheets. J. Phys. Chem. B 2006, 110, 17070–17075. [Google Scholar] [CrossRef] [PubMed]
- Kai, K.; Yoshida, Y.; Kageyama, H.; Saito, G.; Ishigaki, T.; Furukawa, Y.; Kawamata, J. Room-temperature synthesis of manganese oxide monosheets. J. Am. Chem. Soc. 2008, 130, 15938–15943. [Google Scholar] [CrossRef] [PubMed]
- Oaki, Y.; Imai, H. One-pot synthesis of manganese oxide nanosheets in aqueous solution: Chelation-mediated parallel control of reaction and morphology. Angew. Chem. Int. Ed. Engl. 2007, 46, 4951–4955. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xu, K.; Sun, H.; Yin, S. One-step synthesis of single-layer MnO2 nanosheets with multi-role sodium dodecyl sulfate for high-performance pseudocapacitors. Small 2015, 11, 2182–2191. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Hou, P.; Dong, L.; Cai, L.; Chen, Z.; Zhao, M.; Li, J. Manganese dioxide nanosheets: From preparation to biomedical applications. Int. J. Nanomed. 2019, 14, 4781–4800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kan, M.; Zhou, J.; Sun, Q.; Kawazoe, Y.; Jena, P. The intrinsic ferromagnetism in a MnO2 monolayer. J. Phys. Chem. Lett. 2013, 4, 3382–3386. [Google Scholar] [CrossRef]
- Sun, C.; Wang, Y.; Zoub, J.; Smith, S.C. A formation mechanism of oxygen vacancies in a MnO2 monolayer: A DFT + U study. Phys. Chem. Chem. Phys. 2011, 13, 11325–11328. [Google Scholar] [CrossRef]
- Li, L.; Feng, X.; Nie, Y.; Chen, S.; Shi, F.; Xiong, K.; Ding, W.; Qi, X.; Hu, J.; Wei, Z.; et al. Insight into the Effect of Oxygen Vacancy Concentration on the Catalytic Performance of MnO2. ACS Catal. 2015, 5, 4825–4832. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, C.; Zou, J.; Wang, L.; Smith, S.; Lu, G.Q.; Cockayne, D.J.H. Oxygen vacancy induced structural Variations of exfoliated monolayer MnO2 sheets. Phys. Rev. B 2010, 81, 081401. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Ma, J.; Yang, L.; He, G.; Zhang, C.; Zhang, R.; He, H. Oxygen Vacancies Induced by Transition Metal Doping in γ-MnO2 for Highly Efficient Ozone Decomposition. Environ. Sci. Technol. 2018, 52, 12685–12696. [Google Scholar] [CrossRef] [PubMed]
- Kabongo, G.L.; Khawula, T.; Thokozani, T.; Nyongombe, E.G.; Ozoemena, K.; Dhlamini, S. Microwave Irradiation Induces Oxygen Vacancy in Metal Oxides based Materials and Devices: A Review. J. Nanosci. Curr. Res. 2018, 3, 1000125. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, J.; Hang, X.; Zhang, X.; Xie, J.; Pan, B.; Xie, Y. Half-Metallicity in Single-Layered Manganese Dioxide Nanosheets by Defect Engineering. Angew. Chem. Int. Ed. 2015, 54, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Ebina, Y.; Takada, K.; Sasaki, T. Photocurrent Generation from Semiconducting Manganese Oxide Nanosheets in Response to Visible Light. J. Phys. Chem. B 2005, 109, 9651–9655. [Google Scholar] [CrossRef]
- Ostovari, F.; Hasanpoori, M.; Abbasnejad, M.; Salehi, M.A. DFT calculations of graphene monolayer in presence of Fe dopant and vacancy. Phys. B Condens. Matter 2018, 541, 6–13. [Google Scholar] [CrossRef]
- Withers, F.; Del Pozo-Zamudio, O.; Mishchenko, A.; Rooney, A.; Gholinia, A.; Watanabe, K.; Taniguchi, T.; Haigh, S.J.; Geim, A.K.; Tartakovskii, A.I.; et al. Light-Emitting Diodes by Band-Structure Engineering in van der Waals Heterostructures. Nat. Mater. 2015, 14, 301–306. [Google Scholar] [CrossRef]
- Britnell, L.; Ribeiro, R.; Eckmann, A.; Jalil, R.; Belle, B.; Mishchenko, A.; Kim, Y.; Gorbachev, R.; Georgiou, T.; Morozov, S.; et al. Strong Light-Matter Interactions in Heterostructures of Atomically Thin Films. Science 2013, 340, 1311–1314. [Google Scholar] [CrossRef] [Green Version]
- Geim, A.K.; Grigorieva, I.V. Van der Waals Heterostructures. Nature 2013, 499, 419–425. [Google Scholar] [CrossRef]
- Gong, Y.; Lin, J.; Wang, X.; Shi, G.; Lei, S.; Lin, Z.; Zou, X.; Ye, G.; Vajtai, R.; Yakobson, B.I.; et al. Vertical and In-Plane Heterostructures from WS2/MoS2 Monolayers. Nat. Mater. 2014, 13, 1135–1142. [Google Scholar] [CrossRef] [Green Version]
- Bonaccorso, F.; Colombo, L.; Yu, G.; Stoller, M.; Tozzini, V.; Ferrari, A.C.; Ruoff, R.S.; Pellegrini, V. Graphene, Related Two-Dimensional Crystals, and Hybrid Systems for Energy Conversion and Storage. Science 2015, 347, 1246501. [Google Scholar] [CrossRef]
- Wang, X.; Xia, F. Stacked 2D Materials Shed Light. Nat. Mater. 2015, 14, 264–265. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Kumar, N.; Bellus, M.; Chiu, H.; He, D.; Wang, Y.; Zhao, H. Electron Transfer and Coupling in Graphene-Tungsten Disulfide van der Waals Heterostructures. Nat. Commun. 2014, 5, 5622. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Zhou, Z.; Chen, Z. Graphene-Related Nanomaterials: Tuning Properties by Functionalization. Nanoscale 2013, 5, 4541–4583. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Tang, Q.; Zhou, Z. Structural and Electronic Properties of Graphene−ZnO Interfaces: Dispersion-Corrected Density Functional Theory Investigations. Nanotechnology 2013, 24, 305401. [Google Scholar] [CrossRef]
- Padilha, J.; Fazzio, A.; da Silva, A. van der Waals Heterostructure of Phosphorene and Graphene: Tuning the Schottky Barrier and Doping by Electrostatic Gating. Phys. Rev. Lett. 2015, 114, 066803. [Google Scholar] [CrossRef]
- Huang, X.; Tan, C.; Yin, Z.; Zhang, H. 25th Anniversary Article: Hybrid Nanostructures Based on Two Dimensional Nanomaterials. Adv. Mater. 2014, 26, 2185–2204. [Google Scholar] [CrossRef]
- Zhang, H.; Du, X.; Ding, S.; Wang, Q.; Chang, L.; Ma, X.; Xiaogang, M.; Pen, C. DFT calculations of the synergistic effect of λ-MnO2/graphene composites for electrochemical adsorption of lithium ions. Phys. Chem. Chem. Phys. 2019, 21, 8133–8140. [Google Scholar] [CrossRef]
- Wu, S.; Fan, K.; Wu, M.; Yin, G. Two-dimensional MnO2/graphene hybrid nanostructures as anode for lithium ion batteries. Int. J. Mod. Phys. B 2016, 30, 1650208. [Google Scholar] [CrossRef]
- Deng, J.; Wang, X.; Duan, X.; Liu, P. Facile Preparation of MnO2/Graphene Nanocomposites with Spent Battery Powder for Electrochemical Energy Storage. ACS Sustain. Chem. Eng. 2015, 3, 1330–1338. [Google Scholar] [CrossRef]
- Peng, L.; Peng, X.; Liu, B.; Wu, C.; Xie, Y.; Yu, G. Ultrathin two-dimensional MnO2/graphene hybrid nanostructures for high-performance, flexible planar supercapacitors. Nano Lett. 2013, 13, 2151–2157. [Google Scholar] [CrossRef]
- Lee, H.; Kang, J.; Cho, M.S.; Choi, J.; Lee, Y. MnO2/graphene composite electrodes for supercapacitors: The effect of graphene intercalation on capacitance. J. Mater. Chem. 2011, 21, 18215. [Google Scholar] [CrossRef]
- Mao, L.; Zhang, K.; Chan, H.S.; Wu, J. Nanostructured MnO2/graphene composites for supercapacitor electrodes: The effect of morphology, crystallinity and composition. J. Mater. Chem. 2012, 22, 1845. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X.; He, D.; Ma, F.; Fu, Q.; Hu, Y. An amperometric glucose biosensor based on a MnO2/graphene composite modified electrode. RSC Adv. 2016, 6, 18654. [Google Scholar] [CrossRef]
- Gan, L.; Zhang, Q.; Guo, C.; Schwingenschlögl, U.; Zhao, Y. Two-Dimensional MnO2/Graphene Interface: Half-Metallicity and Quantum Anomalous Hall State. J. Phys. Chem. C 2016, 120, 2119–2125. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Tian, H.; He, J.; Yang, Q. Graphene–MnO2 Hybrid Nanostructure as a New Catalyst for Formaldehyde Oxidation. J. Phys. Chem. C 2016, 120, 23660–23668. [Google Scholar] [CrossRef]
- Song, Z.; Ma, Y.; Li, C. The residual tetracycline in pharmaceutical wastewater was effectively removed by using MnO2/graphene nanocomposite. Sci. Total Environ. 2019, 651, 580–590. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. B 1964, 36, 864. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. A 1965, 140, 1133. [Google Scholar] [CrossRef] [Green Version]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.; Calandra, M.; Baroni, S. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [Green Version]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM Espresso: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, S. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monkhorst, H.; Pack, J. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.F.W. Atoms in molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Espitia-Rico, M.; Rodríguez-Martínez, J.A.; Moreno-Armenta, M.G.; Takeichi, N. Graphene monolayers on GaN(0001). Appl. Surf. Sci. 2015, 326, 7–11. [Google Scholar] [CrossRef]
- Fan, Y.; Ma, X.; Liu, X.; Wang, J.; Ai, H.; Zhao, M. Theoretical Design of an InSe/GaTe vdW Heterobilayer: A Potential Visible-Light Photocatalyst for Water Splitting. J. Phys. Chem. C 2018, 122, 27803–27810. [Google Scholar] [CrossRef]
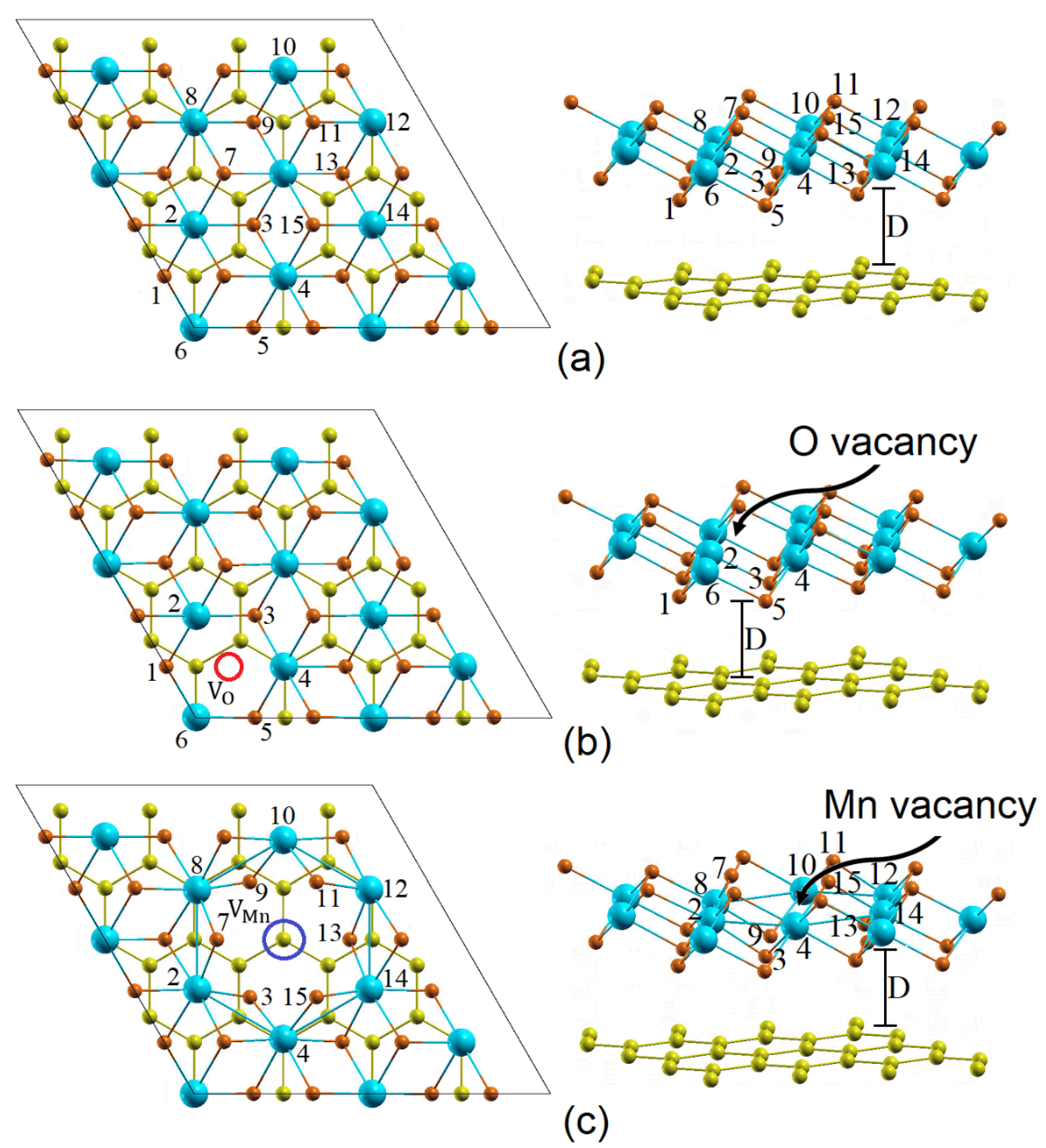
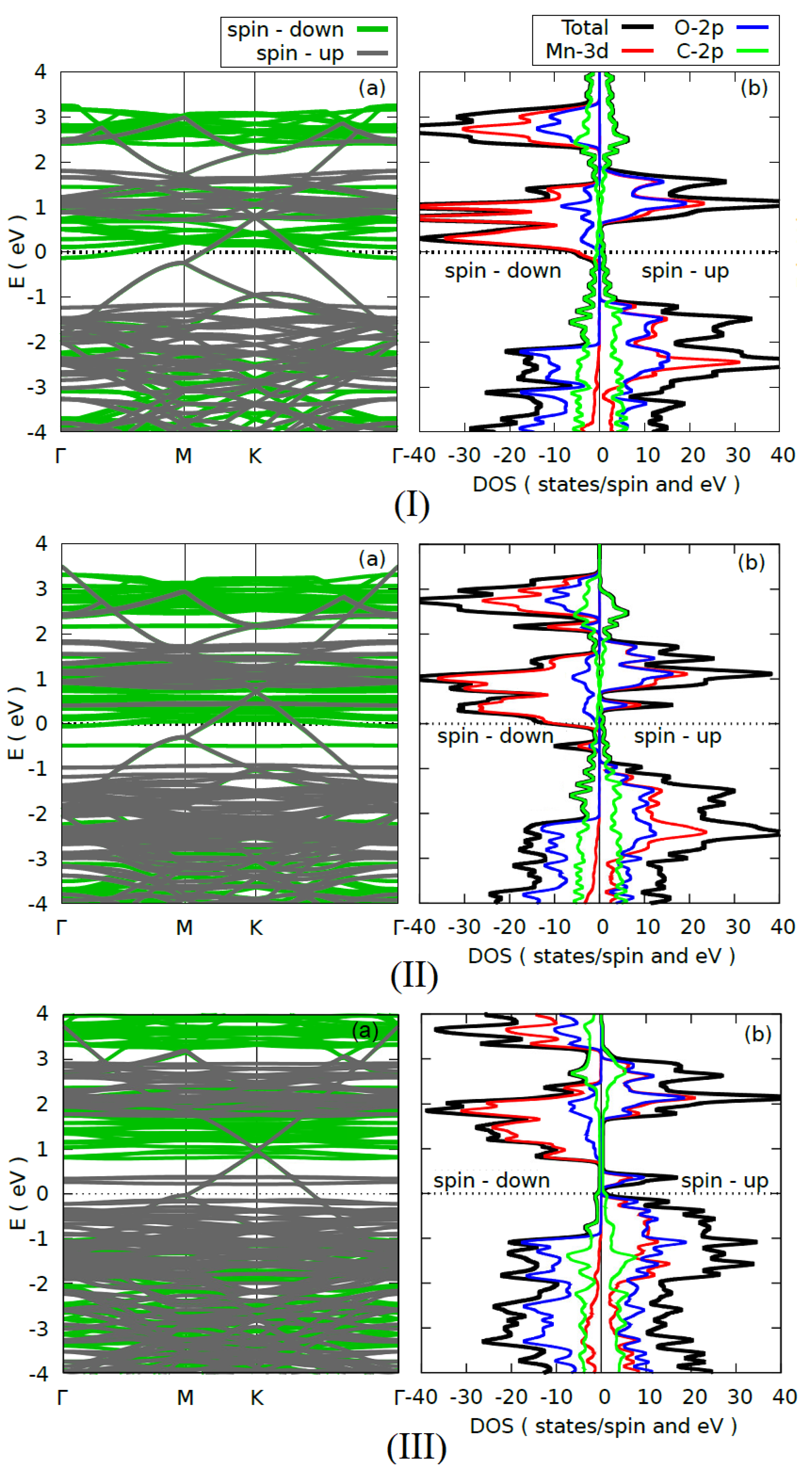

{kind=link}
{kind=link}
| Heterostructure | l1-2 | l2-3 | l3-4 | l4-5 | l5-6 | D |
|---|---|---|---|---|---|---|
| Without vacancies | 1.917 | 1.917 | 1.916 | 1.916 | 1.916 | 2.981 |
| With VO | 1.875 | 1.875 | 1.875 | 1.875 | 1.874 | 2.977 |
| Heterostructure | l7-8 | l8-9 | l9-10 | l10-11 | l11-12 | l12-13 | l13-14 | l14-15 | l15-4 | l2-7 | l3-4 | D |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Without vacancies | 1.910 | 1.916 | 1.917 | 1.910 | 1.910 | 1.916 | 1.917 | 1.910 | 1.910 | 1.910 | 1.916 | 2.981 |
| With VMn | 1.788 | 1.802 | 1.799 | 1.786 | 1.788 | 1.802 | 1.799 | 1.787 | 1.788 | 1.787 | 1.802 | 2.939 |
| Neighbors | Atoms i | |
|---|---|---|
| First | 2 | +0.214 |
| 4 | +0.213 | |
| 6 | +0.205 | |
| Second | 1 | +0.053 |
| 3 | +0.047 | |
| 5 | +0.048 |
| Neighbors | Atoms i | |
|---|---|---|
| First | 3 | −0.157 |
| 7 | −0.188 | |
| 9 | −0.156 | |
| 11 | −0.189 | |
| 13 | −0.150 | |
| 15 | −0.189 | |
| Second | 2 | −0.064 |
| 4 | −0.067 | |
| 8 | −0.064 | |
| 10 | −0.067 | |
| 12 | −0.063 | |
| 14 | −0.061 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morinson-Negrete, J.D.; Ortega-López, C.; Espitia-Rico, M.J. Effects of Mono-Vacancies of Oxygen and Manganese on the Properties of the MnO2/Graphene Heterostructure. Materials 2022, 15, 2731. https://doi.org/10.3390/ma15082731
Morinson-Negrete JD, Ortega-López C, Espitia-Rico MJ. Effects of Mono-Vacancies of Oxygen and Manganese on the Properties of the MnO2/Graphene Heterostructure. Materials. 2022; 15(8):2731. https://doi.org/10.3390/ma15082731
Chicago/Turabian StyleMorinson-Negrete, Juan David, César Ortega-López, and Miguel J. Espitia-Rico. 2022. "Effects of Mono-Vacancies of Oxygen and Manganese on the Properties of the MnO2/Graphene Heterostructure" Materials 15, no. 8: 2731. https://doi.org/10.3390/ma15082731
APA StyleMorinson-Negrete, J. D., Ortega-López, C., & Espitia-Rico, M. J. (2022). Effects of Mono-Vacancies of Oxygen and Manganese on the Properties of the MnO2/Graphene Heterostructure. Materials, 15(8), 2731. https://doi.org/10.3390/ma15082731