
In Situ Observation of ZnO Nanoparticle Formation by a Combination of Time-Resolved X-ray Absorption Spectroscopy and X-ray Diffraction


 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. ZnO Synthesis Procedures
2.2. In Situ Reaction Cell
2.3. X-ray Experiments
3. Results and Discussion
3.1. X-ray Absorption Spectroscopy
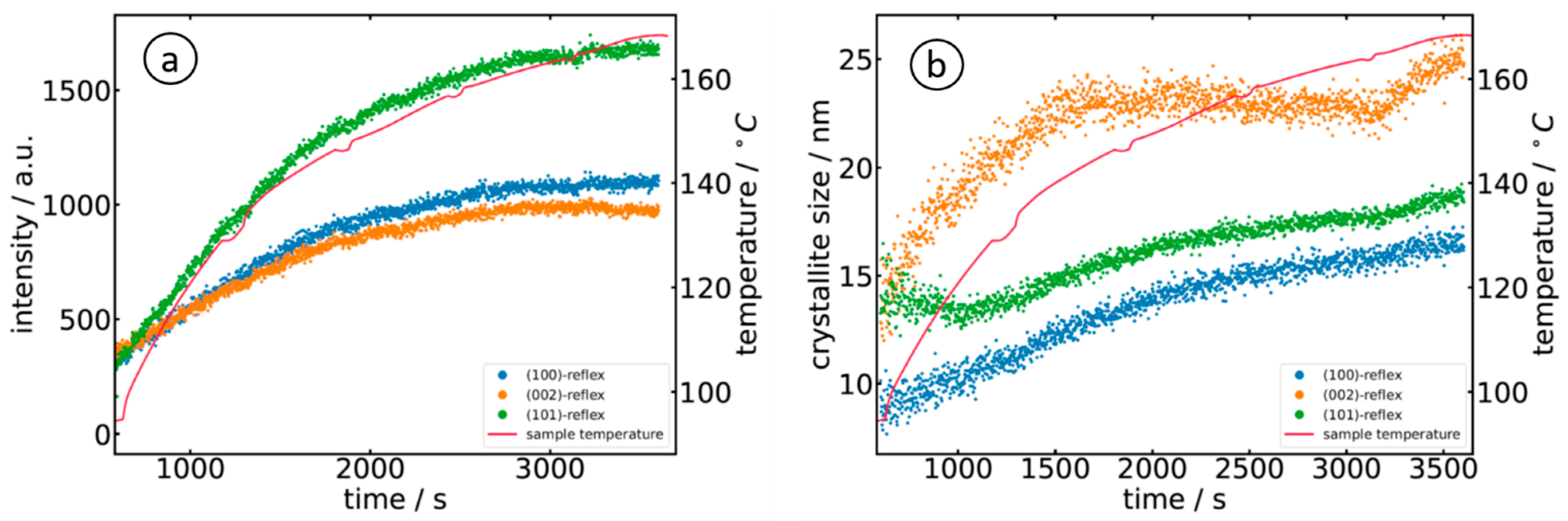
3.2. X-ray Diffraction
3.3. Comprehensive Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Darr, J.A.; Zhang, J.; Makwana, N.M.; Weng, X. Continuous Hydrothermal Synthesis of Inorganic Nanoparticles: Applications and Future Directions. Chem. Rev. 2017, 117, 11125–11238. [Google Scholar] [CrossRef]
- Baruah, S.; Dutta, J. Hydrothermal growth of ZnO nanostructures. Sci. Technol. Adv. Mater. 2009, 10, 013001. [Google Scholar] [CrossRef]
- Demazeau, G. Solvothermal Processes: Definition, Key Factors Governing the Involved Chemical Reactions and New Trends. Z. Naturforsch. 2010, 65, 999–1006. [Google Scholar] [CrossRef]
- Šarić, A.; Štefanić, G.; Dražić, G.; Gotić, M. Solvothermal synthesis of zinc oxide microspheres. J. Alloys Compd. 2015, 652, 91–99. [Google Scholar] [CrossRef]
- Al-Gaashani, R.; Radiman, S.; Daud, A.; Tabet, N.; Al-Douri, Y. XPS and optical studies of different morphologies of ZnO nanostructures prepared by microwave methods. Ceram. Int. 2013, 39, 2283–2292. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, X.; Xu, N.; Xiang, R.; Zhu, Y.; Tang, Z. Morphology and Optical Property of ZnO Nanostructures Grown by Solvothermal Method: Effect of the Solution Pretreatment. J. Nanomater. 2013, 2013, 986157. [Google Scholar] [CrossRef]
- Musić, S.; Šarić, A. Formation of hollow ZnO particles by simple hydrolysis of zinc acetylacetonate. Ceram. Int. 2012, 38, 6047–6052. [Google Scholar] [CrossRef]
- Cho, S.; Jang, J.-W.; Jung, S.-H.; Lee, B.; Oh, E.; Lee, K.-H. Precursor Effects of Citric Acid and Citrates on ZnO Crystal Formation. Langmuir 2009, 25, 3825–3831. [Google Scholar] [CrossRef]
- Razali, R.; Zak, A.K.; Majid, W.A.; Darroudi, M. Solvothermal synthesis of microsphere ZnO nanostructures in DEA media. Ceram. Int. 2011, 37, 3657–3663. [Google Scholar] [CrossRef]
- Šarić, A.; Despotović, I.; Štefanić, G.; Dražić, G. The Influence of Ethanolamines on the Solvothermal Synthesis of Zinc Oxide: A Combined Experimental and Theoretical Study. ChemistrySelect 2017, 2, 10038–10049. [Google Scholar] [CrossRef]
- Šarić, A.; Vrankić, M.; Lützenkirchen-Hecht, D.; Despotović, I.; Petrović, Ž.; Dražić, G.; Eckelt, F. Insight into the Growth Mechanism and Photocatalytic Behavior of Tubular Hierarchical ZnO Structures: An Integrated Experimental and Theoretical Approach. Inorg. Chem. 2022, 61, 2962–2979. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Ke, J.; Hao, J.; Yan, X.; Tian, Y. A new method for synthesis of ZnO flower-like nanostructures and their photocatalytic performance. Phys. B 2022, 624, 413395. [Google Scholar] [CrossRef]
- Wojnarowicz, J.; Chudoba, T.; Lojkowski, W. A Review of Microwave Synthesis of Zinc Oxide Nanomaterials: Reactants, Process Parameters and Morphologies. Nanomaterials 2020, 10, 1086. [Google Scholar] [CrossRef]
- Mohammadi, E.; Aliofkhazraei, M.; Hasanpoor, M.; Chipara, M. Hierarchical and Complex ZnO Nanostructures by Microwave-Assisted Synthesis: Morphologies, Growth Mechanism and Classification. Crit. Rev. Solid State Mater. Sci. 2018, 43, 475–541. [Google Scholar] [CrossRef]
- Sauvik Raha, S.; Ahmaruzzaman, M. ZnO nanostructured materials and their potential applications: Progress, challenges and perspectives. Nanoscale Adv. 2022, 4, 1868–1925. [Google Scholar] [CrossRef]
- Soloviev, A.; Jensen, H.; Søgaard, E.G.; Kanaev, A.V. Aggregation kinetics of sol-gel process based on titanium tetraisopropoxide. J. Mater. Sci. 2003, 38, 3315–3318. [Google Scholar] [CrossRef]
- Kiefer, J.; Grabow, J.; Kurland, H.-D.; Müller, F.A. Characterization of Nanoparticles by Solvent Infrared Spectroscopy. Anal. Chem. 2015, 87, 12313–12317. [Google Scholar] [CrossRef] [PubMed]
- Stötzel, J.; Lützenkirchen-Hecht, D.; Frahm, R.; Santilli, C.V.; Pulcinelli, S.H.; Kaminski, R.; Fonda, E.; Villain, F.; Briois, V. QEXAFS and UV/Vis Simultaneous Monitoring of the TiO2-Nanoparticles Formation by Hydrolytic Sol−Gel Route. J. Phys. Chem. C 2010, 114, 6228–6236. [Google Scholar] [CrossRef]
- Zolotoyabko, E. Basic Concepts of X-ray Diffraction, 1st ed.; Wiley VCH: Weinheim, Germany, 2014. [Google Scholar]
- Koningsberger, D.; Prins, R. X-ray Absorption. Principles, Applications and Techniques of EXAFS, SEXAFS, and XANES; John Wiley & Sons: New York, NY, USA, 1988. [Google Scholar]
- Lützenkirchen-Hecht, D.; Frahm, R. The corrosion of Mo in KOH: Time resolved XAFS investigations. J. Phys. Chem. B 2001, 105, 9988–9993. [Google Scholar] [CrossRef]
- Grote, L.; Zito, C.A.; Frank, K.; Dippel, A.C.; Reisbeck, P.; Pitala, K.; Kvashnina, K.O.; Bauters, S.; Detlefs, B.; Ivashko, O.; et al. X-ray studies bridge the molecular and macro length scales during the emergence of CoO assemblies. Nature Commun. 2021, 12, 4429–4441. [Google Scholar] [CrossRef]
- Karzel, H.; Potzel, W.; Köfferlein, M.; Schiessl, W.; Steiner, M.; Hiller, U.; Kalvius, G.M.; Mitchell, D.W.; Das, T.P.; Blaha, P.; et al. Lattice dynamics and hyperfine interactions in ZnO and ZnSe at high external pressures. Phys. Rev. B 1996, 53, 11425–11438. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, A.; Jagadish, C. Review of zincblende ZnO: Stability of metastable ZnO phases. J. Appl. Phys. 2007, 102, 071101. [Google Scholar] [CrossRef]
- Ashrafi, A.; Ueta, A.; Kumano, H.; Suemune, I. Role of ZnS buffer layers in growth of zincblende ZnO on GaAs substrates by metalorganic molecular-beam epitaxy. J. Cryst. Growth 2000, 221, 435–439. [Google Scholar] [CrossRef]
- Park, S.-H.; Kwak, C.-H.; Seo, S.-Y.; Kim, S.-H.; Kim, B.-H.; Park, C.-I.; Park, Y.-W.; Han, S.-W. A Large Quantity of ZnO Nanorods Grown at Room Temperature. J. Korean Phys. Soc. 2009, 55, 94–97. [Google Scholar] [CrossRef]
- Guo, L.; Yang, S.; Yang, C.; Yu, P.; Ge, W.; Wong, G.K.L. Synthesis and Characterization of Poly(vinylpyrrolidone)-Modified Zinc Oxide Nanoparticles. Chem. Mater. 2000, 12, 2268–2274. [Google Scholar] [CrossRef]
- Zhao, D.; Tian, X.; Doronkin, D.E.; Han, S.; Kondratenko, V.A.; Grunwaldt, J.-D.; Perechodjuk, A.; Vuong, T.H.; Rabeah, J.; Eckelt, R.; et al. In situ formation of ZnOx species for efficient propane dehydrogenation. Nature 2021, 599, 234–241. [Google Scholar] [CrossRef]
- Shaban, M.; Mohamed, F.; Abdallah, S. Production and Characterization of Superhydrophobic and Antibacterial Coated Fabrics Utilizing ZnO Nanocatalyst. Sci. Rep. 2018, 8, 3925. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ye, L.; Liu, S.; Li, Y.; Ji, X. Photocatalytic Reduction of CO2 by ZnO Micro/nanomaterials with Different Morphologies and Ratios of {0001} Facets. Sci. Rep. 2016, 6, 38474. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J. XANES reflects coordination change and underlying surface disorder of zinc adsorbed to silica. J. Synchrotron Radiat. 2021, 28, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Bornmann, B.; Kläs, J.; Müller, O.; Lützenkirchen-Hecht, D.; Frahm, R. The quick EXAFS setup at beamline P64 at PETRA III for up to 200 spectra per second. AIP Conf. Proc. 2019, 2054, 040008. [Google Scholar] [CrossRef]
- Lützenkirchen-Hecht, D.; Bornmann, B.; Frahm, R.; Rothweiler, P.; von Polheim, S.; Eckelt, F.; Müller, O. Simultaneous quick-scanning X-ray absorption spectroscopy and X-ray diffraction. In Proceedings of the 14th International Conference on Synchrotron Radiation Instrumentation, Hamburg, Germany, 28 March–1 April 2022. [Google Scholar]
- Lützenkirchen-Hecht, D.; Wagner, R.; Szillat, S.; Hüsecken, A.K.; Istomin, K.; Pietsch, U.; Frahm, R. The multi-purpose hard X-ray beamline BL10 at the DELTA storage ring. J. Synchrotron Radiat. 2014, 21, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Ravel, B.; Newville, M. ATHENA and ARTEMIS: Interactive Graphical Data Analysis Using IFEFFIT. Phys. Scr. 2005, T115, 1007–1010. [Google Scholar] [CrossRef]
- Castorina, E.; Ingall, E.D.; Morton, P.L.; Tavakoli, D.A.; Lai, B. Zinc K-edge XANES spectroscopy of mineral and organic standards. J. Synchrotron Radiat. 2019, 26, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.A.; Mishra, B.; Myneni, S.C.B. High Energy Resolution-X-ray Absorption Near Edge Structure Spectroscopy Reveals Zn Ligation in Whole Cell Bacteria. J. Phys. Chem. Lett. 2019, 10, 2585–2592. [Google Scholar] [CrossRef]
- Kisi, E.H.; Elcombe, M.M. u parameters for the wurtzite structure of ZnS and ZnO using powder neutron diffraction. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1989, 45, 1867–1870. [Google Scholar] [CrossRef]
- Rehr, J.J.; Kas, J.J.; Vila, F.D.; Prange, M.P.; Jorissen, K. Parameter-free calculations of X-ray spectra with FEFF9. Phys. Chem. Chem. Phys. 2010, 12, 5503–5513. [Google Scholar] [CrossRef]
- Ravel, B. Quantitative EXAFS Analysis. In X-ray Absorption and X-Ray Emission Spectroscopy: Theory and Applications; Van Bokhoven, J.A., Lamberti, C., Eds.; John Wiley & Sons: Chichester, UK, 2016. [Google Scholar]
- Caetano, B.L.; Santilli, C.V.; Meneau, F.; Briois, V.; Pulcinelli, S.H. In Situ and Simultaneous UV−vis/SAXS and UV−vis/XAFS Time-Resolved Monitoring of ZnO Quantum Dots Formation and Growth. J. Phys. Chem. C 2011, 115, 4404–4412. [Google Scholar] [CrossRef]
- Kieffer, J.; Karkoulis, D. PyFAI, a versatile library for azimuthal regrouping. J. Phys. Conf. Ser. 2013, 425, 202012. [Google Scholar] [CrossRef]
- Warren, B.E. X-ray Diffraction; Addison-Wesley Publishing Co.: Boston, MA, USA, 1969. [Google Scholar]
- Wong, E.M.; Bonevich, J.E.; Searson, P.C. Growth Kinetics of Nanocrystalline ZnO Particles from Colloidal Suspensions. J. Phys. Chem. B 1998, 102, 7770–7775. [Google Scholar] [CrossRef]
- Ludi, B.; Niederberger, M. Zinc oxide nanoparticles: Chemical mechanisms and classical and non-classical crystallization. Dalton Trans. 2013, 42, 12554–12568. [Google Scholar] [CrossRef]
- Briois, V.; Giorgetti, C.; Baudelet, F.; Blanchandin, S.; Tokumoto, M.S.; Pulcinelli, S.H.; Santilli, C.V. Dynamical Study of ZnO Nanocrystal and Zn-HDS Layered Basic Zinc Acetate Formation from Sol−Gel Route. J. Phys. Chem. C 2007, 111, 3253–3258. [Google Scholar] [CrossRef]
- Huang, Y.; Shen, Z.; Wu, Y.; Wang, X.; Zhang, S.; Shi, X.; Zeng, H. Amorphous ZnO based resistive random access memory. RSC Adv. 2016, 6, 17867–17872. [Google Scholar] [CrossRef]
- Bruncko, J.; Vincze, A.; Netrvalova, M.; Šutta, P.; Hasko, D.; Michalka, M. Annealing and recrystallization of amorphous ZnO thin films deposited under cryogenic conditions by pulsed laser deposition. Thin Solid Films 2011, 520, 866–870. [Google Scholar] [CrossRef]
- LaMer, V.K.; Dinegar, R.H. Theory, Production and Mechanism of Formation of Monodispersed Hydrosols. J. Am. Chem. Soc. 1950, 72, 4847–4854. [Google Scholar] [CrossRef]
- Briois, V.; Lützenkirchen-Hecht, D.; Villain, F.; Fonda, E.; Belin, S.; Griesebock, B.; Frahm, R. Time-Resolved Study of the Oxidation of Ethanol by Cerium(IV) Using Combined Quick-XANES, UV−Vis, and Raman Spectroscopies. J. Phys. Chem. A 2005, 109, 320–329. [Google Scholar] [CrossRef]
- Bauer, M.; Heusel, G.; Mangold, S.; Bertagnolli, H. Spectroscopic set-up for simultaneous UV-Vis/(Q)EXAFSin situ and in operando studies of homogeneous reactions under laboratory conditions. J. Synchrotron Radiat. 2010, 17, 273–279. [Google Scholar] [CrossRef]
- Tsakoumis, N.; Dehghan, R.; Johnsen, R.E.; Voronov, A.; van Beek, W.; Walmsley, J.C.; Borg, Ø.; Rytter, E.; Chen, D.; Rønning, M.; et al. A combined in situ XAS-XRPD-Raman study of Fischer–Tropsch synthesis over a carbon supported Co catalyst. Catal. Today 2013, 205, 86–93. [Google Scholar] [CrossRef]
- Just, J.; Coughlan, C.; Singh, S.; Ren, H.; Müller, O.; Becker, P.; Unold, T.; Ryan, K.M. Insights into Nucleation and Growth of Colloidal Quaternary Nanocrystals by Multimodal X-ray Analysis. ACS Nano 2021, 15, 6439–6447. [Google Scholar] [CrossRef]
- Rabeah, J.; Briois, V.; Adomeit, S.; La Fontaine, C.; Bentrup, U.; Brückner, A. Multivariate Analysis of Coupled Operando EPR/XANES/EXAFS/UV–Vis/ATR-IR Spectroscopy: A New Dimension for Mechanistic Studies of Catalytic Gas-Liquid Phase Reactions. Chem.–A Eur. J. 2020, 26, 7395–7404. [Google Scholar] [CrossRef]
- Müller, O.; Nachtegaal, M.; Just, J.; Lützenkirchen-Hecht, D.; Frahm, R. Quick-EXAFS setup at the SuperXAS beamline for in situ X-ray absorption spectroscopy with 10 ms time resolution. J. Synchrotron Radiat. 2016, 23, 260–266. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Id. | Zn(acac)2H2O | Solvent | Amount | Temperature | Time |
|---|---|---|---|---|---|
| A | 0.75 g | 10−3 M NaOH | 50 ml | 90 °C | 150 min |
| O | 0.75 g | 1-octanol | 50 ml | 165 °C | 60 min |
| W | 0.75 g | water | 50 ml | 90 °C | 450 min |
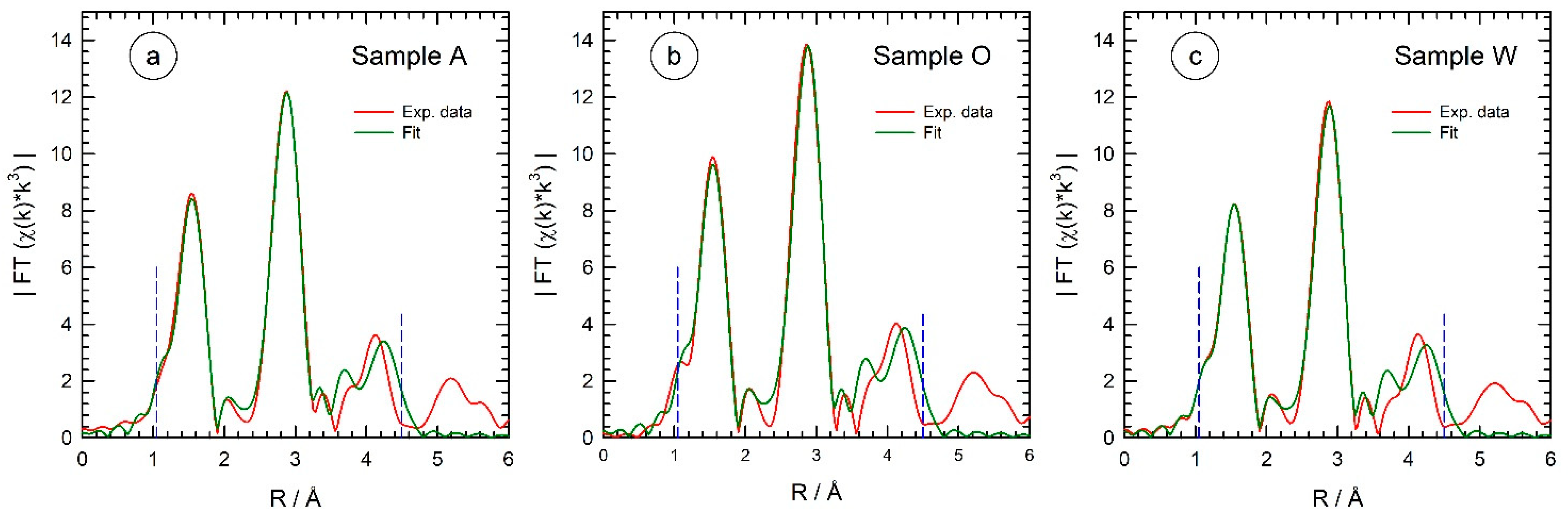
| Sample | Shell | N | S02 | R/Å | σ2/Å2 | E0/eV |
|---|---|---|---|---|---|---|
| Sample A (R-factor: 0.025) | Zn-O | 4 | 0.793 ± 0.097 | 1.978 ± 0.007 | 0.0051 ± 0.0012 | 2.65 ± 0.69 |
| Zn-Zn | 12 | 1.313 ± 0.210 | 3.247 ± 0.006 | 0.0122 ± 0.0012 | 2.65 ± 0.69 | |
| Zn-O | 1 | 0.793 ± 0.097 | 3.255 ± 0.007 | 0.0051 ± 0.0012 | 2.65 ± 0.69 | |
| Zn-O-O | 12 | 0.793 ± 0.097 | 3.600 ± 0.008 | 0.0051 ± 0.0012 | 2.65 ± 0.69 | |
| Zn-O-Zn | 24 | 1.053 ± 0.155 | 3.600 ± 0.008 | 0.0087 ± 0.0012 | 2.65 ± 0.69 | |
| Zn-O | 9 | 0.793 ± 0.097 | 3.808 ± 0.008 | 0.0051 ± 0.0012 | 2.65 ± 0.69 | |
| Zn-O | 4 | 0.793 ± 0.097 | 3.964 ± 0.009 | 0.0051 ± 0.0012 | 2.65 ± 0.69 | |
| Zn-O-Zn-O | 12 | 1.053 ± 0.155 | 3.964 ± 0.009 | 0.0087 ± 0.0012 | 2.65 ± 0.69 | |
| Zn-O-O | 6 | 0.793 ± 0.097 | 4.229 ± 0.009 | 0.0051 ± 0.0012 | 2.65 ± 0.69 | |
| Zn-O-Zn | 36 | 1.053 ± 0.155 | 4.516 ± 0.009 | 0.0087 ± 0.0012 | 2.65 ± 0.69 | |
| Zn-O-O | 36 | 0.793 ± 0.097 | 4.516 ± 0.009 | 0.0051 ± 0.0012 | 2.65 ± 0.69 | |
| Zn-Zn-O | 36 | 1.053 ± 0.155 | 4.516 ± 0.009 | 0.0087 ± 0.0012 | 2.65 ± 0.69 | |
| Zn-Zn | 6 | 1.313 ± 0.210 | 4.577 ± 0.010 | 0.0122 ± 0.0012 | 2.65 ± 0.69 | |
| Zn-O | 6 | 0.793 ± 0.097 | 4.599 ± 0.010 | 0.0051 ± 0.0012 | 2.65 ± 0.69 | |
| Sample O (R-factor: 0.030) | Zn-O | 4 | 0.854 ± 0.114 | 1.971 ± 0.008 | 0.0047 ± 0.0012 | 3.12 ± 0.75 |
| Zn-Zn | 12 | 1.573 ± 0.227 | 3.245 ± 0.006 | 0.0126 ± 0.0013 | 3.12 ± 0.75 | |
| Zn-O | 1 | 0.854 ± 0.114 | 3.250 ± 0.007 | 0.0047 ± 0.0012 | 3.12 ± 0.75 | |
| Zn-O-O | 12 | 0.854 ± 0.114 | 3.595 ± 0.008 | 0.0047 ± 0.0012 | 3.12 ± 0.75 | |
| Zn-O-Zn | 24 | 1.214 ± 0.096 | 3.595 ± 0.008 | 0.0087 ± 0.0013 | 3.12 ± 0.75 | |
| Zn-O | 9 | 0.854 ± 0.114 | 3.802 ± 0.009 | 0.0047 ± 0.0012 | 3.12 ± 0.75 | |
| Zn-O | 4 | 0.854 ± 0.114 | 3.958 ± 0.009 | 0.0047 ± 0.0012 | 3.12 ± 0.75 | |
| Zn-O-Zn-O | 12 | 1.214 ± 0.096 | 3.958 ± 0.009 | 0.0087 ± 0.0013 | 3.12 ± 0.75 | |
| Zn-O-O | 6 | 0.854 ± 0.114 | 4.223 ± 0.009 | 0.0047 ± 0.0012 | 3.12 ± 0.75 | |
| Zn-O-Zn | 36 | 1.214 ± 0.096 | 4.509 ± 0.010 | 0.0087 ± 0.0013 | 3.12 ± 0.75 | |
| Zn-O-O | 36 | 0.854 ± 0.114 | 4.509 ± 0.010 | 0.0047 ± 0.0012 | 3.12 ± 0.75 | |
| Zn-Zn-O | 36 | 1.214 ± 0.096 | 4.509 ± 0.010 | 0.0087 ± 0.0013 | 3.12 ± 0.75 | |
| Zn-Zn | 6 | 1.573 ± 0.277 | 4.570 ± 0.010 | 0.0126 ± 0.0013 | 3.12 ± 0.75 | |
| Zn-O | 6 | 0.854 ± 0.114 | 4.592 ± 0.010 | 0.0047 ± 0.0012 | 3.12 ± 0.75 | |
| Sample W (R-factor: 0.029) | Zn-O | 4 | 0.735 ± 0.096 | 1.972 ± 0.008 | 0.0048 ± 0.0012 | 3.62 ± 0.74 |
| Zn-Zn | 12 | 1.258 ± 0.222 | 3.244 ± 0.006 | 0.0123 ± 0.0013 | 3.62 ± 0.74 | |
| Zn-O | 1 | 0.735 ± 0.096 | 3.248 ± 0.007 | 0.0048 ± 0.0012 | 3.62 ± 0.74 | |
| Zn-O-O | 12 | 0.735 ± 0.096 | 3.593 ± 0.008 | 0.0048 ± 0.0012 | 3.62 ± 0.74 | |
| Zn-O-Zn | 24 | 0.997 ± 0.112 | 3.593 ± 0.008 | 0.0086 ± 0.0013 | 3.62 ± 0.74 | |
| Zn-O | 9 | 0.735 ± 0.096 | 3.800 ± 0.009 | 0.0048 ± 0.0012 | 3.62 ± 0.74 | |
| Zn-O | 4 | 0.735 ± 0.096 | 3.956 ± 0.009 | 0.0048 ± 0.0012 | 3.62 ± 0.74 | |
| Zn-O-Zn-O | 12 | 0.997 ±0.112 | 3.956 ± 0.009 | 0.0086 ± 0.0013 | 3.62 ± 0.74 | |
| Zn-O-O | 6 | 0.735 ± 0.096 | 4.220 ± 0.009 | 0.0048 ± 0.0012 | 3.62 ± 0.74 | |
| Zn-O-Zn | 36 | 0.997 ± 0.112 | 4.506 ± 0.010 | 0.0086 ± 0.0013 | 3.62 ± 0.74 | |
| Zn-O-O | 36 | 0.735 ± 0.096 | 4.506 ± 0.010 | 0.0048 ± 0.0012 | 3.62 ± 0.74 | |
| Zn-Zn-O | 36 | 0.997 ± 0.112 | 4.506 ± 0.010 | 0.0086 ± 0.0013 | 3.62 ± 0.74 | |
| Zn-Zn | 6 | 1.258 ± 0.222 | 4.567 ± 0.010 | 0.0123 ± 0.0013 | 3.62 ± 0.74 | |
| Zn-O | 6 | 0.735 ± 0.096 | 4.589 ± 0.010 | 0.0048 ± 0.0012 | 3.62 ± 0.74 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eckelt, F.; Rothweiler, P.; Braun, F.; Voss, L.; Šarić, A.; Vrankić, M.; Lützenkirchen-Hecht, D. In Situ Observation of ZnO Nanoparticle Formation by a Combination of Time-Resolved X-ray Absorption Spectroscopy and X-ray Diffraction. Materials 2022, 15, 8186. https://doi.org/10.3390/ma15228186
Eckelt F, Rothweiler P, Braun F, Voss L, Šarić A, Vrankić M, Lützenkirchen-Hecht D. In Situ Observation of ZnO Nanoparticle Formation by a Combination of Time-Resolved X-ray Absorption Spectroscopy and X-ray Diffraction. Materials. 2022; 15(22):8186. https://doi.org/10.3390/ma15228186
Chicago/Turabian StyleEckelt, Franz, Patrick Rothweiler, Frederic Braun, Lukas Voss, Ankica Šarić, Martina Vrankić, and Dirk Lützenkirchen-Hecht. 2022. "In Situ Observation of ZnO Nanoparticle Formation by a Combination of Time-Resolved X-ray Absorption Spectroscopy and X-ray Diffraction" Materials 15, no. 22: 8186. https://doi.org/10.3390/ma15228186
APA StyleEckelt, F., Rothweiler, P., Braun, F., Voss, L., Šarić, A., Vrankić, M., & Lützenkirchen-Hecht, D. (2022). In Situ Observation of ZnO Nanoparticle Formation by a Combination of Time-Resolved X-ray Absorption Spectroscopy and X-ray Diffraction. Materials, 15(22), 8186. https://doi.org/10.3390/ma15228186