
Magenta-Blue Electrofluorochromic Device Incorporating Eu(III) Complex, Anthracene Derivative, and Viologen Molecule


Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Synthesis of Red Phosphorescent Eu(III) Complex
- Tris(hexafluoroacetylacetonato)europium(III) bis(triphenylphospine oxide) [Eu(hfa)3(TPPO)2]
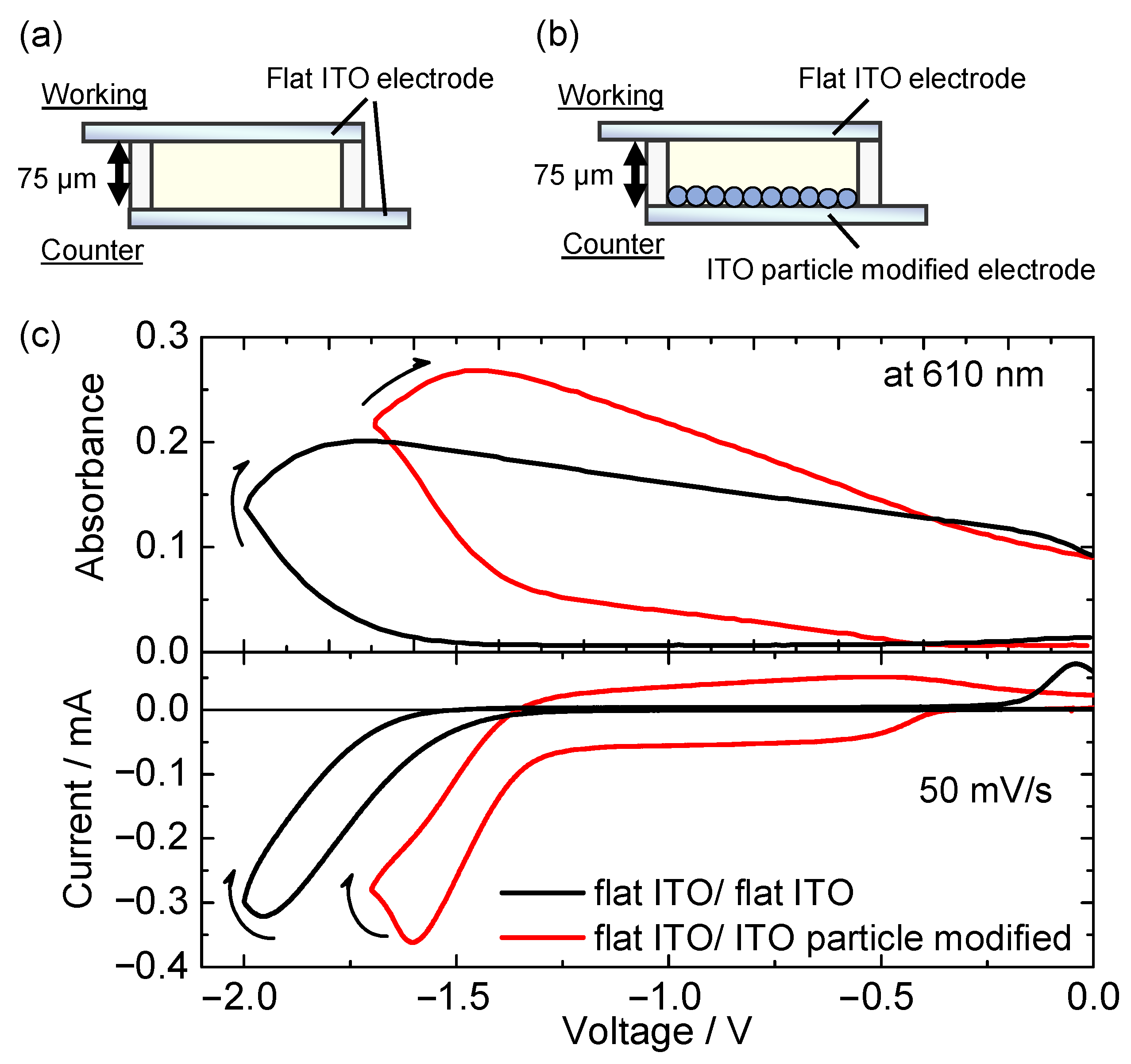
2.3. Preparation of ITO Nanoparticle-Modified Electrodes
2.4. Fabrication of Electrochemical Cell
2.5. Measurements of Electrochemical Properties
2.6. Photophysical Measurements
3. Results and Discussion
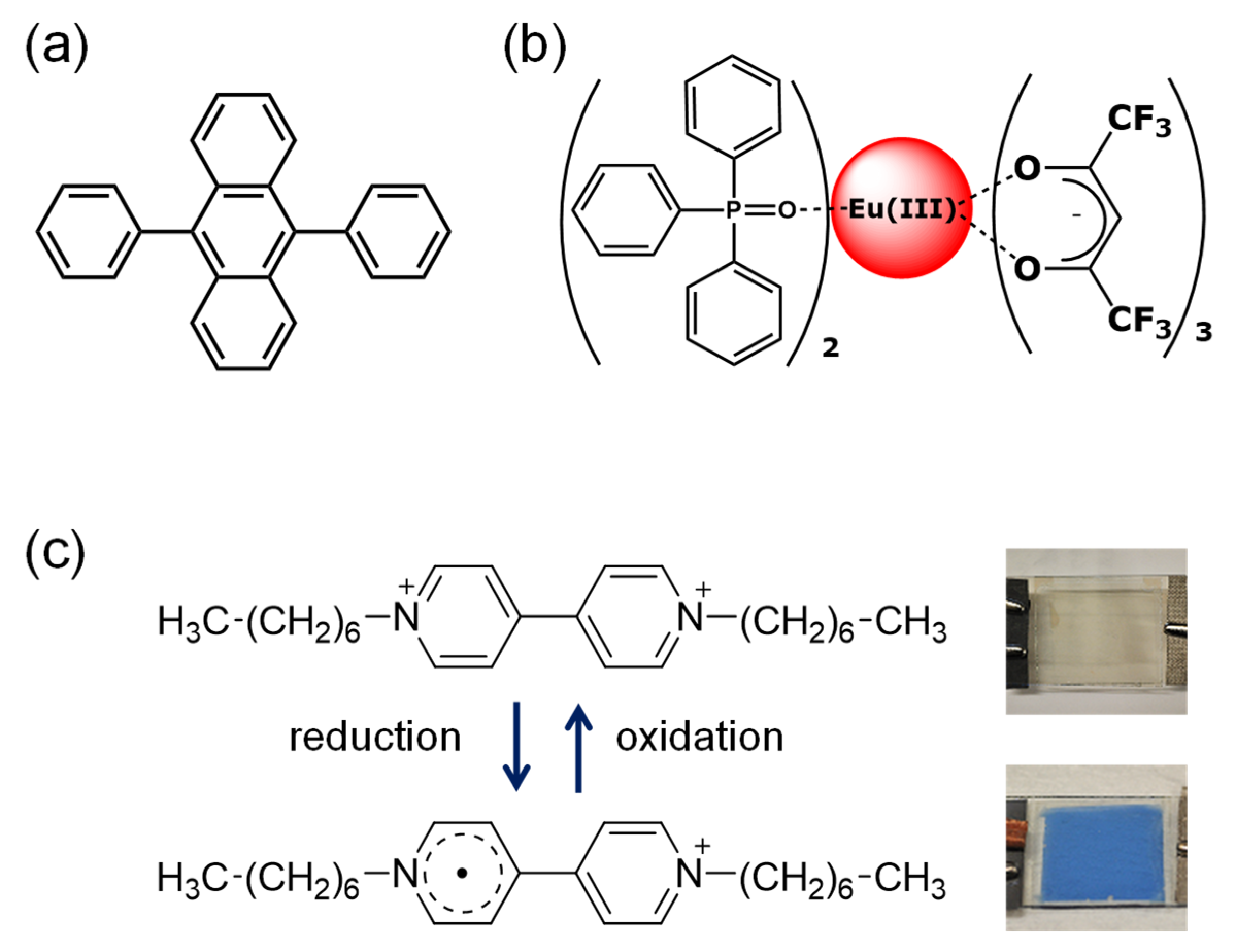
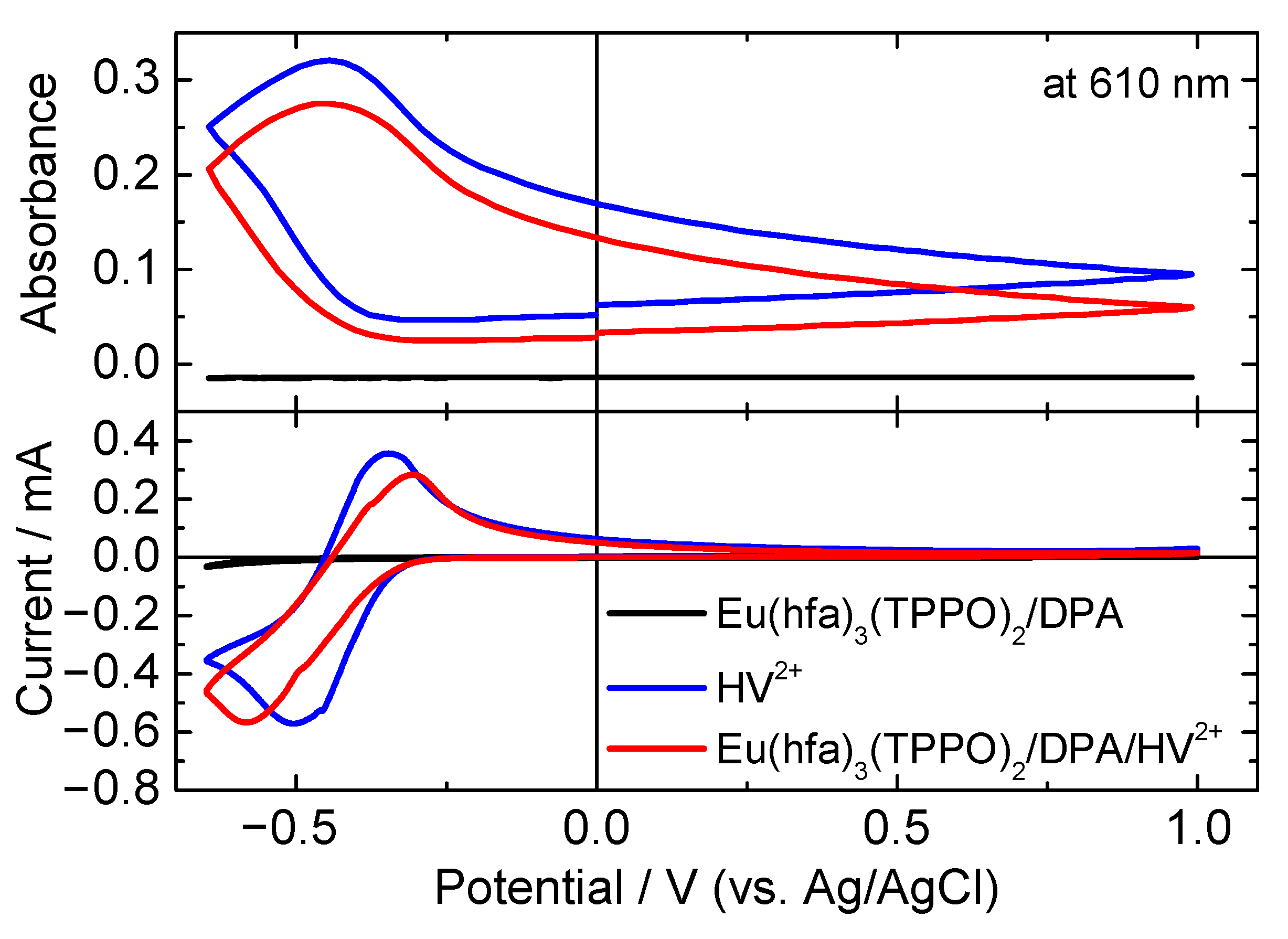
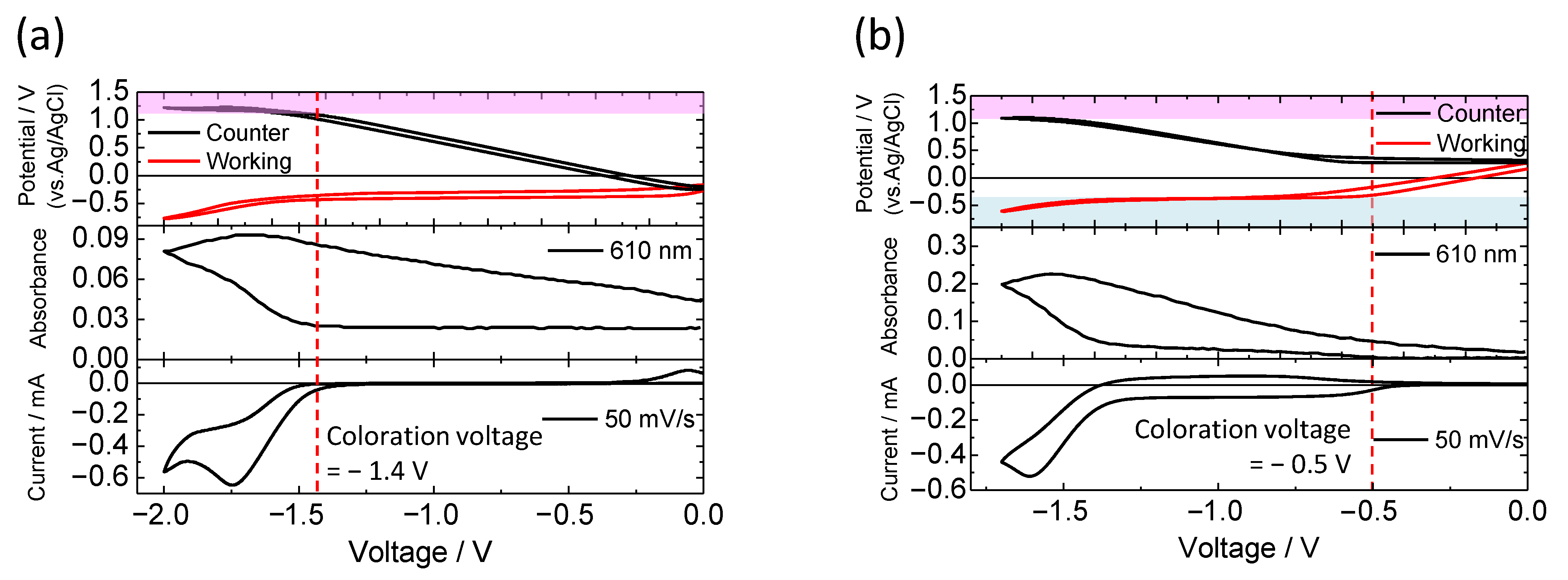
3.1. Electrochemical Properties of the Mixed Solution Containing Luminescent Materials and Electrochromic Molecules
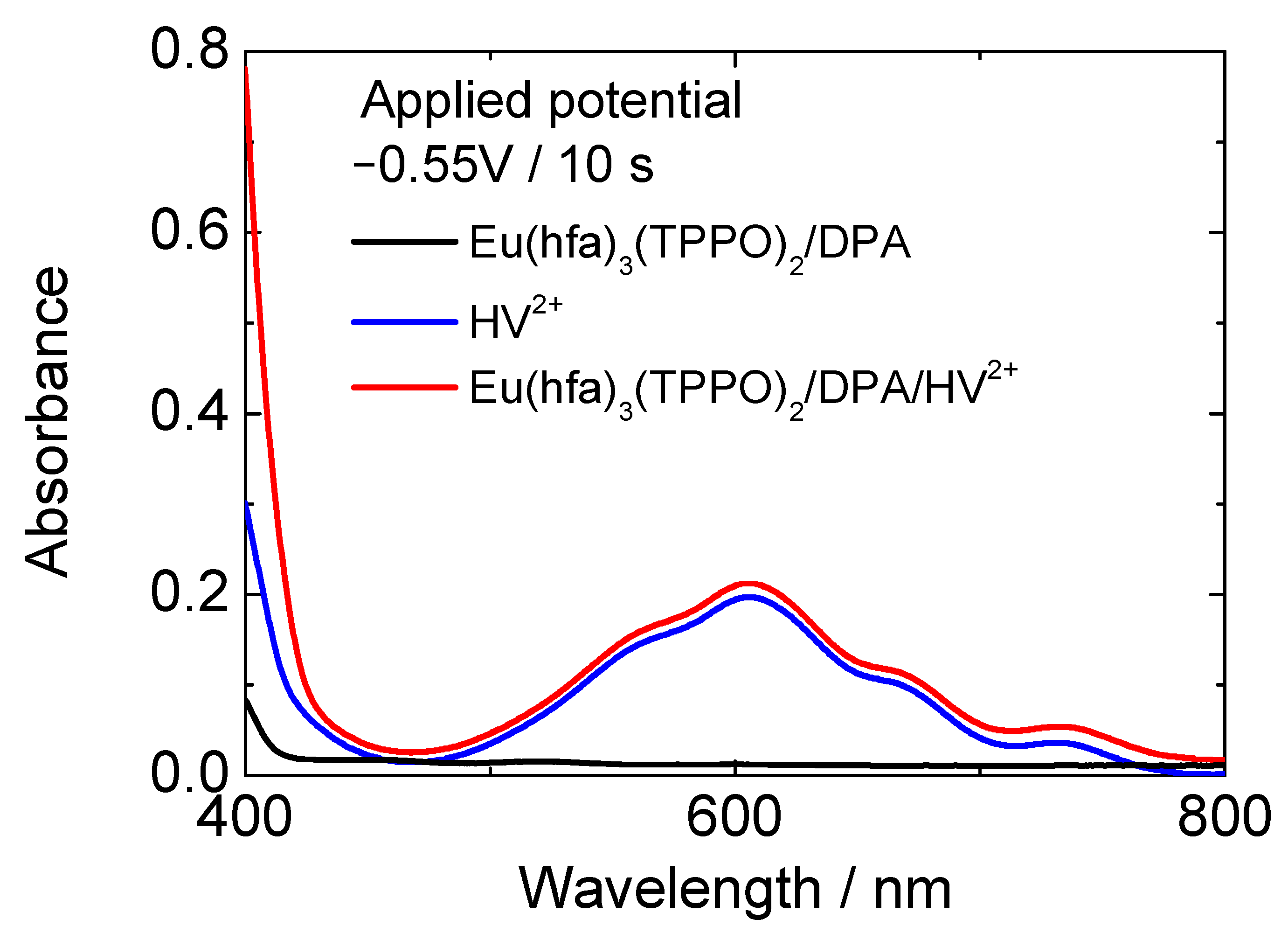
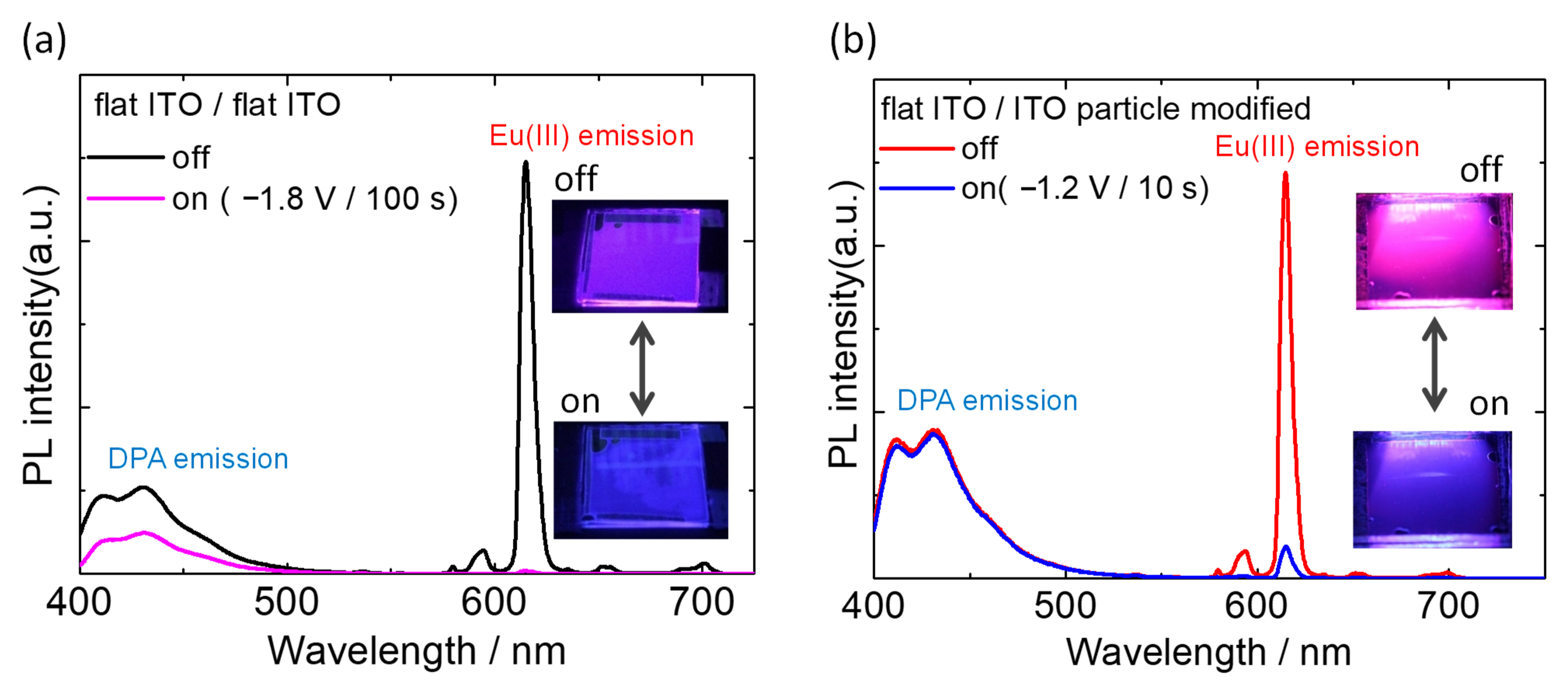
3.2. Electrochemical Control of Luminescence Color
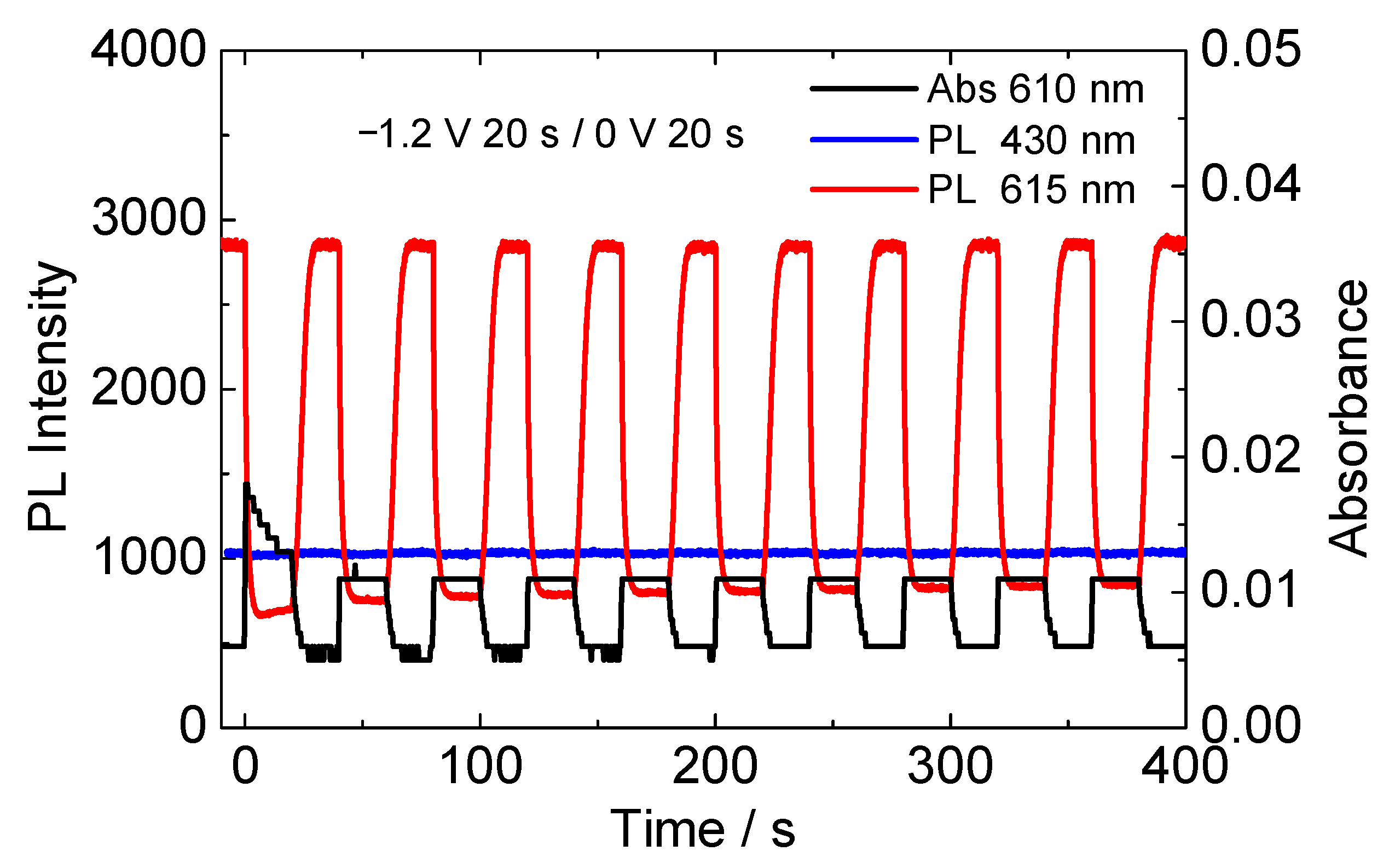
3.3. Photophysical Measurements of the EFC Device
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Matsuda, K.; Irie, M. Diarylethene as a Photoswitching Unit. J. Photochem. Photobiol. C Photochem. Rev. 2004, 5, 169–182. [Google Scholar] [CrossRef]
- de Silva, A.P.; McClenaghan, N.D. Molecular-Scale Logic Gates. Chem.-A Eur. J. 2004, 10, 574–586. [Google Scholar] [CrossRef]
- Granqvist, C.G.; Lansåker, P.C.; Mlyuka, N.R.; Niklasson, G.A.; Avendaño, E. Progress in Chromogenics: New Results for Electrochromic and Thermochromic Materials and Devices. Sol. Energy Mater. Sol. Cells 2009, 93, 2032–2039. [Google Scholar] [CrossRef]
- Kawai, T.; Sasaki, T.; Irie, M. A Photoresponsive Laser Dye Containing Photochromic Dithienylethene Units. Chem. Commun. 2001, 8, 711–712. [Google Scholar] [CrossRef]
- Müller-Buschbaum, K.; Beuerle, F.; Feldmann, C. MOF Based Luminescence Tuning and Chemical/Physical Sensing. Microporous Mesoporous Mater. 2015, 216, 171–199. [Google Scholar] [CrossRef]
- Granqvist, C.G.; Green, S.; Niklasson, G.A.; Mlyuka, N.R.; von Kræmer, S.; Georén, P. Advances in Chromogenic Materials and Devices. Thin Solid Films 2010, 518, 3046–3053. [Google Scholar] [CrossRef]
- Lin, J.; Lai, M.; Dou, L.; Kley, C.S.; Chen, H.; Peng, F.; Sun, J.; Lu, D.; Hawks, S.A.; Xie, C.; et al. Thermochromic Halide Perovskite Solar Cells. Nat. Mater. 2018, 17, 261–267. [Google Scholar] [CrossRef] [Green Version]
- Ohtani, S.; Gon, M.; Tanaka, K.; Chujo, Y. A Flexible, Fused, Azomethine–Boron Complex: Thermochromic Luminescence and Thermosalient Behavior in Structural Transitions between Crystalline Polymorphs. Chem.-A Eur. J. 2017, 23, 11827–11833. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.R.; Sabatino, D.R.; Praisner, T.J. Temperature Sensing with Thermochromic Liquid Crystals. Exp. Fluids 2001, 30, 190–201. [Google Scholar] [CrossRef]
- Tashiro, K.; Ono, K.; Minagawa, Y.; Kobayashi, M.; Kawai, T.; Yoshino, K. Structure and Thermochromic Solid-state Phase Transition of Poly (3-alkylthiophene). J. Polym. Sci. Part B Polym. Phys. 1991, 29, 1223–1233. [Google Scholar] [CrossRef]
- Yoshida, M.; Sääsk, V.; Saito, D.; Yoshimura, N.; Takayama, J.; Hiura, S.; Murayama, A.; Põhako-Esko, K.; Kobayashi, A.; Kato, M.; et al. Thermo-and Mechano-Triggered Luminescence ON/OFF Switching by Supercooled Liquid/Crystal Transition of Platinum(II) Complex Thin Films. Adv. Opt. Mater. 2022, 10, 2102614. [Google Scholar] [CrossRef]
- Kato, M.; Ito, H.; Hasegawa, M.; Ishii, K. Soft Crystals: Flexible Response Systems with High Structural Order. Chem.-A Eur. J. 2019, 25, 5105–5112. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Saito, T.; Oshima, N.; Kitamura, N.; Ishizaka, S.; Hinatsu, Y.; Wakeshima, M.; Kato, M.; Tsuge, K.; Sawamura, M. Reversible Mechanochromic Luminescence of [(C6F5Au)2(μ-1,4-Diisocyanobenzene)]. J. Am. Chem. Soc. 2008, 130, 10044–10045. [Google Scholar] [CrossRef]
- Yagai, S.; Seki, T.; Aonuma, H.; Kawaguchi, K.; Karatsu, T.; Okura, T.; Sakon, A.; Uekusa, H.; Ito, H. Mechanochromic Luminescence Based on Crystal-to-Crystal Transformation Mediated by a Transient Amorphous State. Chem. Mater. 2016, 28, 234–241. [Google Scholar] [CrossRef]
- Sagara, Y.; Kubo, K.; Nakamura, T.; Tamaoki, N.; Weder, C. Temperature-Dependent Mechanochromic Behavior of Mechanoresponsive Luminescent Compounds. Chem. Mater. 2017, 29, 1273–1278. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, M.; Kato, M. Regulation of Metal–Metal Interactions and Chromic Phenomena of Multi-Decker Platinum Complexes Having π-Systems. Coord. Chem. Rev. 2018, 355, 101–115. [Google Scholar] [CrossRef]
- Tsuboi, A.; Nakamura, K.; Kobayashi, N. Multicolor Electrochromism Showing Three Primary Color States (Cyan-Magenta-Yellow) Based on Size- and Shape-Controlled Silver Nanoparticles. Chem. Mater. 2014, 26, 6477–6485. [Google Scholar] [CrossRef]
- Cummins, D.; Boschloo, G.; Ryan, M.; Corr, D.; Nagaraja Rao, S.; Fitzmaurice, D. Ultrafast Electrochromic Windows Based on Redox-Chromophore Modified Nanostructured Semiconducting and Conducting Films. J. Phys. Chem. B 2000, 104, 11449–11459. [Google Scholar] [CrossRef]
- Mortimer, R.J.; Dyer, A.L.; Reynolds, J.R. Electrochromic Organic and Polymeric Materials for Display Applications. Displays 2006, 27, 2–18. [Google Scholar] [CrossRef] [Green Version]
- Somani, P.R.; Radhakrishnan, S. Electrochromic Materials and Devices: Present and Future. Mater. Chem. Phys. 2003, 77, 117–133. [Google Scholar] [CrossRef]
- Mondal, S.; Chandra Santra, D.; Ninomiya, Y.; Yoshida, T.; Higuchi, M. Dual-Redox System of Metallo-Supramolecular Polymers for Visible-to-Near-IR Modulable Electrochromism and Durable Device Fabrication. ACS Appl. Mater. Interfaces 2020, 12, 58277–58286. [Google Scholar] [CrossRef]
- Kobayashi, N.; Watanabe, Y.; Ibata, Y.; Nakamura, K. Reflective and Emissive Dual Mode Display Cell with Electrochromism and Electrochemiluminescence. In Proceedings of the International Conference on Digital Printing Technologies 2013, Seattle, WA, USA, 29 September 2013; pp. 28–31. [Google Scholar]
- Watanabe, Y.; Suemori, K.; Hoshino, S. Electrochromic Response Characteristics of Dye-Modified Porous Electrodes Affected by the Porous Film Structure. Chem. Lett. 2016, 45, 1291–1293. [Google Scholar] [CrossRef]
- Miomandre, F.; Audebert, P. Luminescence in Electrochemistry; Miomandre, F., Audebert, P., Eds.; Springer International Publishing: Cham, Switzerland, 2017; ISBN 978-3-319-49135-6. [Google Scholar]
- Sun, J.; Liang, Z. Swift Electrofluorochromism of Donor-Acceptor Conjugated Polytriphenylamines. ACS Appl. Mater. Interfaces 2016, 8, 18301–18308. [Google Scholar] [CrossRef]
- Audebert, P.; Miomandre, F. Electrofluorochromism: From Molecular Systems to Set-up and Display. Chem. Sci. 2013, 4, 575–584. [Google Scholar] [CrossRef]
- Suzuki, T.; Sato, T.; Zhang, J.; Kanao, M.; Higuchi, M.; Maki, H. Electrochemically Switchable Photoluminescence of an Anionic Dye in a Cationic Metallo-Supramolecular Polymer. J. Mater. Chem. C 2016, 4, 1594–1598. [Google Scholar] [CrossRef]
- Bünzli, J.C.G.; Piguet, C. Taking Advantage of Luminescent Lanthanide Ions. Chem. Soc. Rev. 2005, 34, 1048–1077. [Google Scholar] [CrossRef]
- Eliseeva, S.V.; Bünzli, J.C.G. Lanthanide Luminescence for Functional Materials and Bio-Sciences. Chem. Soc. Rev. 2010, 39, 189–227. [Google Scholar] [CrossRef]
- Wang, C.-M.; Pan, M.-F.; Chen, Y.-C.; Lin, H.-M.; Chung, M.-Y.; Wen, Y.-S.; Lii, K.-H. Two Polymorphs of an Organic−Zincophosphate Incorporating a Terephthalate Bridging Ligand in an Unusual Bonding Mode. Inorg. Chem. 2017, 56, 7602–7605. [Google Scholar] [CrossRef]
- Wang, C.-M.; Pan, M.-F.; Lin, Y.-J.; Chung, M.-Y.; Wen, Y.-S.; Chang, Y.; Lin, H.-M.; Hsu, T. A Series of Organic–Inorganic Hybrid Zinc Phosphites Containing Extra-Large Channels. Inorg. Chem. 2018, 57, 2390–2393. [Google Scholar] [CrossRef] [PubMed]
- Tsukube, H.; Shinoda, S. Lanthanide Complexes in Molecular Recognition and Chirality Sensing of Biological Substrates. Chem. Rev. 2002, 102, 2389–2403. [Google Scholar] [CrossRef]
- Kuriki, K.; Koike, Y.; Okamoto, Y. Plastic Optical Fiber Lasers and Amplifiers Containing Lanthanide Complexes. Chem. Rev. 2002, 102, 2347–2356. [Google Scholar] [CrossRef]
- Hirai, Y.; Nakanishi, T.; Miyata, K.; Fushimi, K.; Hasegawa, Y. Thermo-Sensitive Luminescent Materials Composed of Tb(III) and Eu(III) Complexes. Mater. Lett. 2014, 130, 91–93. [Google Scholar] [CrossRef]
- Hasegawa, M.; Ohmagari, H. Helicate Lanthanide Complexes: The Luminescent Elements. Chem. Lett. 2020, 49, 845–854. [Google Scholar] [CrossRef]
- Kanazawa, K.; Komiya, Y.; Nakamura, K.; Kobayashi, N. Red Luminescence Control of Eu(iii) Complexes by Utilizing the Multi-Colored Electrochromism of Viologen Derivatives. Phys. Chem. Chem. Phys. 2017, 19, 16979–16988. [Google Scholar] [CrossRef]
- Kanazawa, K.; Nakamura, K.; Kobayashi, N. Electrochemical Luminescence Modulation in a Eu(III) Complex-Modified TiO2 Electrode. J. Mater. Chem. C 2015, 3, 7135–7142. [Google Scholar] [CrossRef]
- Nakamura, K.; Kanazawa, K.; Kobayashi, N. Electrochemically Controllable Emission and Coloration by Using Europium(III) Complex and Viologen Derivatives. Chem. Commun. 2011, 47, 10064. [Google Scholar] [CrossRef]
- Kanazawa, K.; Nakamura, K.; Kobayashi, N. A Viologen-Eu(III)-Modified TiO2 Electrode for Electroswitchable Luminescence and Coloration. ChemistrySelect 2018, 3, 9672–9680. [Google Scholar] [CrossRef]
- Tropiano, M.; Kilah, N.L.; Morten, M.; Rahman, H.; Davis, J.J.; Beer, P.D.; Faulkner, S. Reversible Luminescence Switching of a Redox-Active Ferrocene-Europium Dyad. J. Am. Chem. Soc. 2011, 133, 11847–11849. [Google Scholar] [CrossRef] [PubMed]
- Lehr, J.; Tropiano, M.; Beer, P.D.; Faulkner, S.; Davis, J.J. Reversible Redox Modulation of a Lanthanide Emissive Molecular Film. Chem. Commun. 2015, 51, 6515–6517. [Google Scholar] [CrossRef]
- Kim, Y.; Ohmagari, H.; Saso, A.; Tamaoki, N.; Hasegawa, M. Electrofluorochromic Device Based on a Redox-Active Europium(III) Complex. ACS Appl. Mater. Interfaces 2020, 12, 46390–46396. [Google Scholar] [CrossRef]
- Mukaigawa, M.; Ohno, H. Electrochemical Switching of Fluorescence Emission between Red and Blue from Complexed Europium Ions in Poly(Ethylene Oxide). J. Electroanal. Chem. 1998, 452, 141–149. [Google Scholar] [CrossRef]
- Mukaigawa, M.; Ohno, H. Control of Fluorescence Emission Color and Intensity by Electrochemical Redox Reaction of Complexed Europium Ions in PEO. Solid State Ionics 1998, 113–115, 439–442. [Google Scholar] [CrossRef]
- Nakamura, K.; Hasegawa, Y.; Kawai, H.; Yasuda, N.; Kanehisa, N.; Kai, Y.; Nagamura, T.; Yanagida, S.; Wada, Y. Enhanced Lasing Properties of Dissymmetric Eu(III) Complex with Bidentate Phosphine Ligands. J. Phys. Chem. A 2007, 111, 3029–3037. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Yukikawa, M.; Nakamura, K.; Kobayashi, N. A Novel Organic Electrochromic Device with Hybrid Capacitor Architecture towards Multicolour Representation. Phys. Chem. Chem. Phys. 2018, 20, 19892–19899. [Google Scholar] [CrossRef]
- Araki, S.; Nakamura, K.; Kobayashi, K.; Tsuboi, A.; Kobayashi, N. Electrochemical Optical-Modulation Device with Reversible Transformation between Transparent, Mirror, and Black. Adv. Mater. 2012, 24, OP122–OP126. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, R.J. Organic Electrochromic Materials. Electrochim. Acta 1999, 44, 2971–2981. [Google Scholar] [CrossRef]
- Tsuneyasu, S.; Watanabe, Y.; Nakamura, K.; Kobayashi, N. In Situ Measurements of Electrode Potentials of Anode and Cathode in Organic Electrochromic Devices. Sol. Energy Mater. Sol. Cells 2017, 163, 200–203. [Google Scholar] [CrossRef]
- Liang, Z.; Nakamura, K.; Kobayashi, N. A Multicolor Electrochromic Device Having Hybrid Capacitor Architecture with a Porous Carbon Electrode. Sol. Energy Mater. Sol. Cells 2019, 200, 109914. [Google Scholar] [CrossRef]
- Minami, H.; Ichikawa, T.; Nakamura, K.; Kobayashi, N. Electrochemically Triggered Upconverted Luminescence for Light-Emitting Devices. Chem. Commun. 2019, 55, 12611–12614. [Google Scholar] [CrossRef]
- Tsuneyasu, S.; Ichikawa, T.; Nakamura, K.; Kobayashi, N. Electrochemical Stability of Diphenylanthracene and Its Effect on Alternating-Current-Driven Blue-Light Electrochemiluminescence Properties. ChemElectroChem 2017, 4, 1731–1735. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| τ0 (s) | τON (s) | ηENT | Φ0 | kr (s−1) | knr (s−1) | kENT (s−1) | |
|---|---|---|---|---|---|---|---|
| DPA | 7.03 × 10−9 | 7.01 × 10−9 | 0.003 | 0.77 | 1.1 × 108 | 3.3 × 107 | 4.1 × 105 |
| Eu(hfa)3(TPPO)2 | 5.30 × 10−4 | 0.30 × 10−4 | 0.94 | 0.34 | 6.7 × 102 | 1.3 × 103 | 3.1 × 104 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakamura, K.; Yanagawa, N.; Kobayashi, N. Magenta-Blue Electrofluorochromic Device Incorporating Eu(III) Complex, Anthracene Derivative, and Viologen Molecule. Materials 2022, 15, 5202. https://doi.org/10.3390/ma15155202
Nakamura K, Yanagawa N, Kobayashi N. Magenta-Blue Electrofluorochromic Device Incorporating Eu(III) Complex, Anthracene Derivative, and Viologen Molecule. Materials. 2022; 15(15):5202. https://doi.org/10.3390/ma15155202
Chicago/Turabian StyleNakamura, Kazuki, Namiko Yanagawa, and Norihisa Kobayashi. 2022. "Magenta-Blue Electrofluorochromic Device Incorporating Eu(III) Complex, Anthracene Derivative, and Viologen Molecule" Materials 15, no. 15: 5202. https://doi.org/10.3390/ma15155202
APA StyleNakamura, K., Yanagawa, N., & Kobayashi, N. (2022). Magenta-Blue Electrofluorochromic Device Incorporating Eu(III) Complex, Anthracene Derivative, and Viologen Molecule. Materials, 15(15), 5202. https://doi.org/10.3390/ma15155202