
Burning Behaviour of Rigid Polyurethane Foams with Histidine and Modified Graphene Oxide

 ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
3. Results
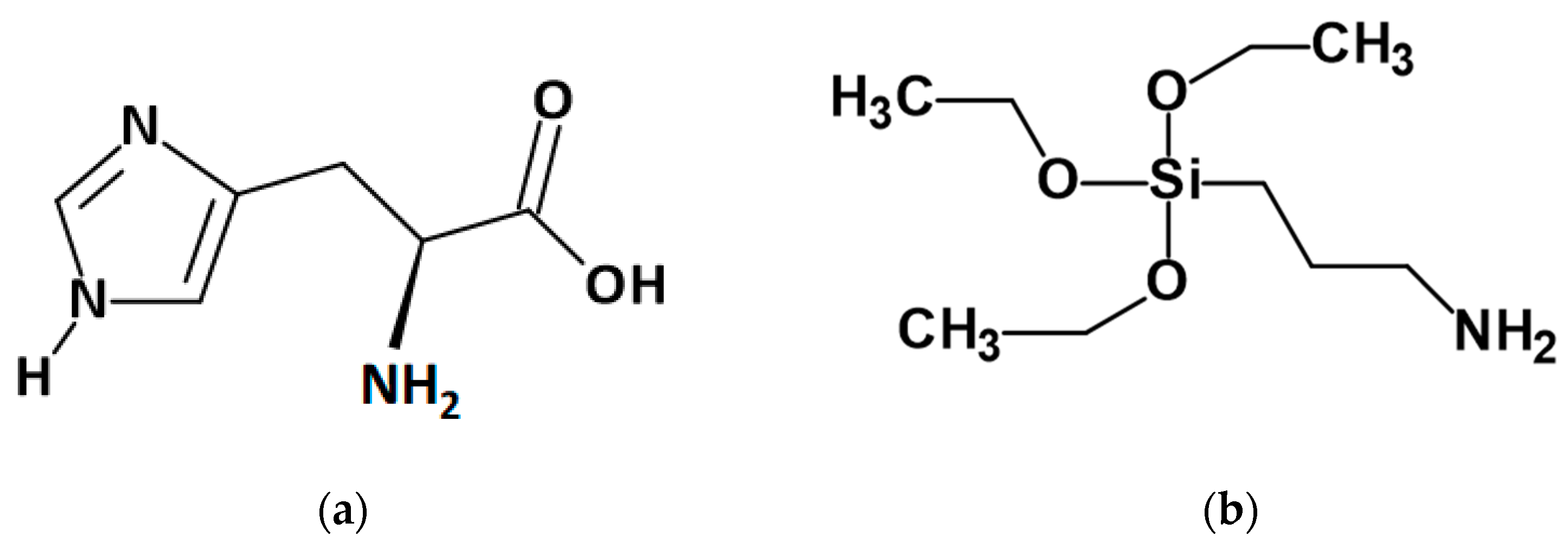
3.1. Fire Retardant’s Characteristic
3.2. Characterization of the Polyurethane Foams
3.3. Physico-Mechanical Properties and Microstructure Analysis of Polyurethane Foams
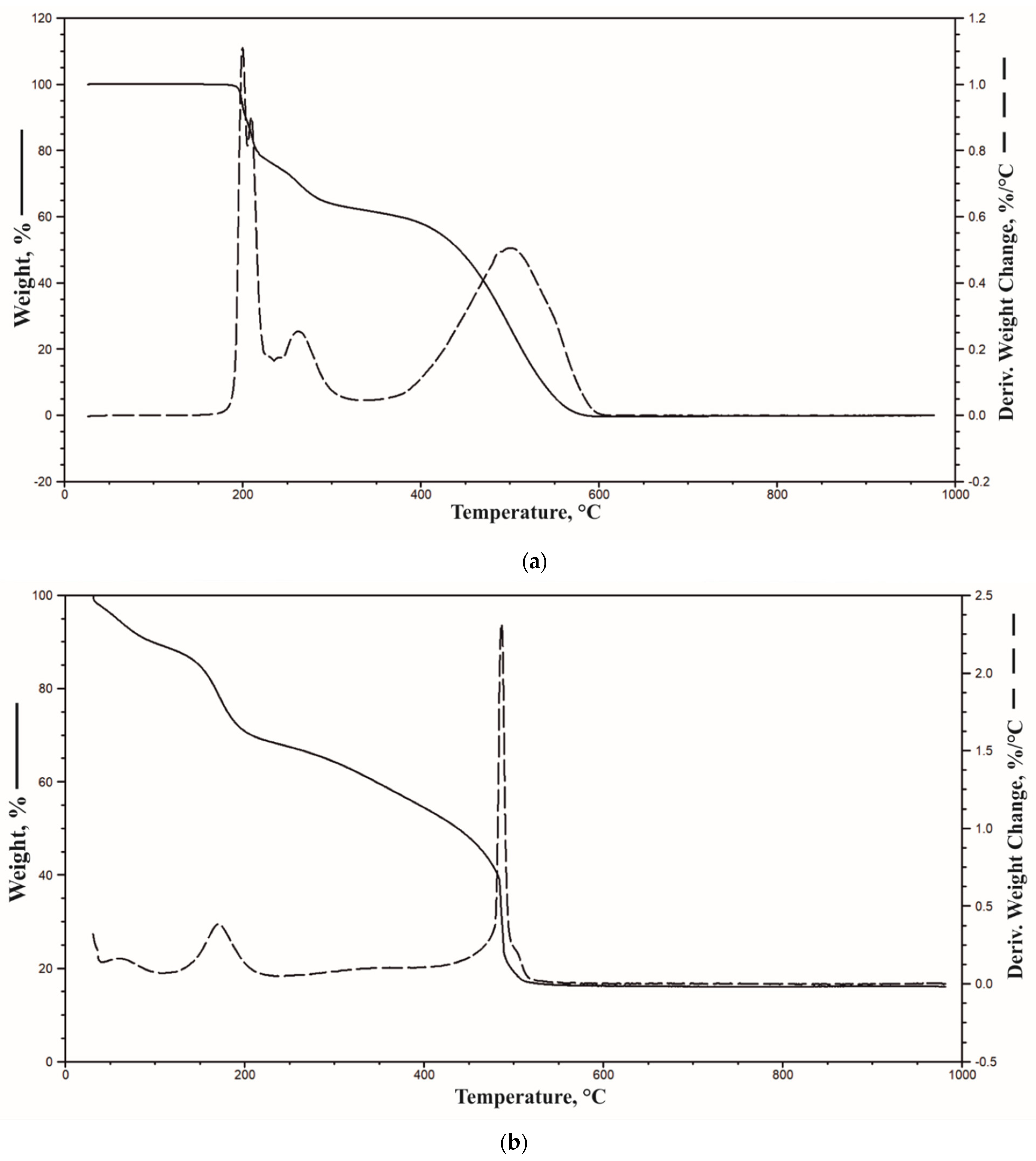
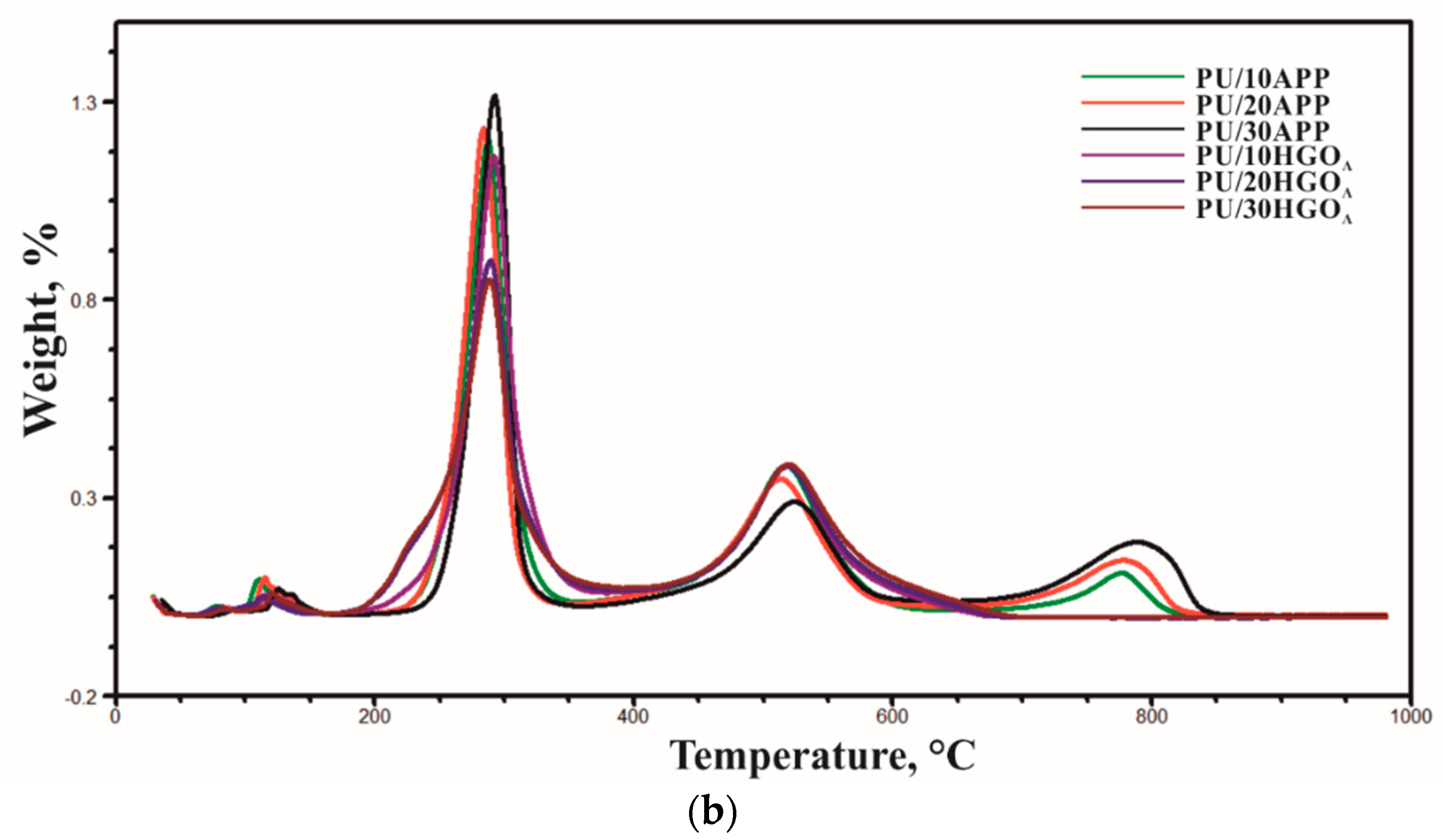
3.4. Thermal Properties
3.5. Fire Behavior
3.6. Smoke Emission
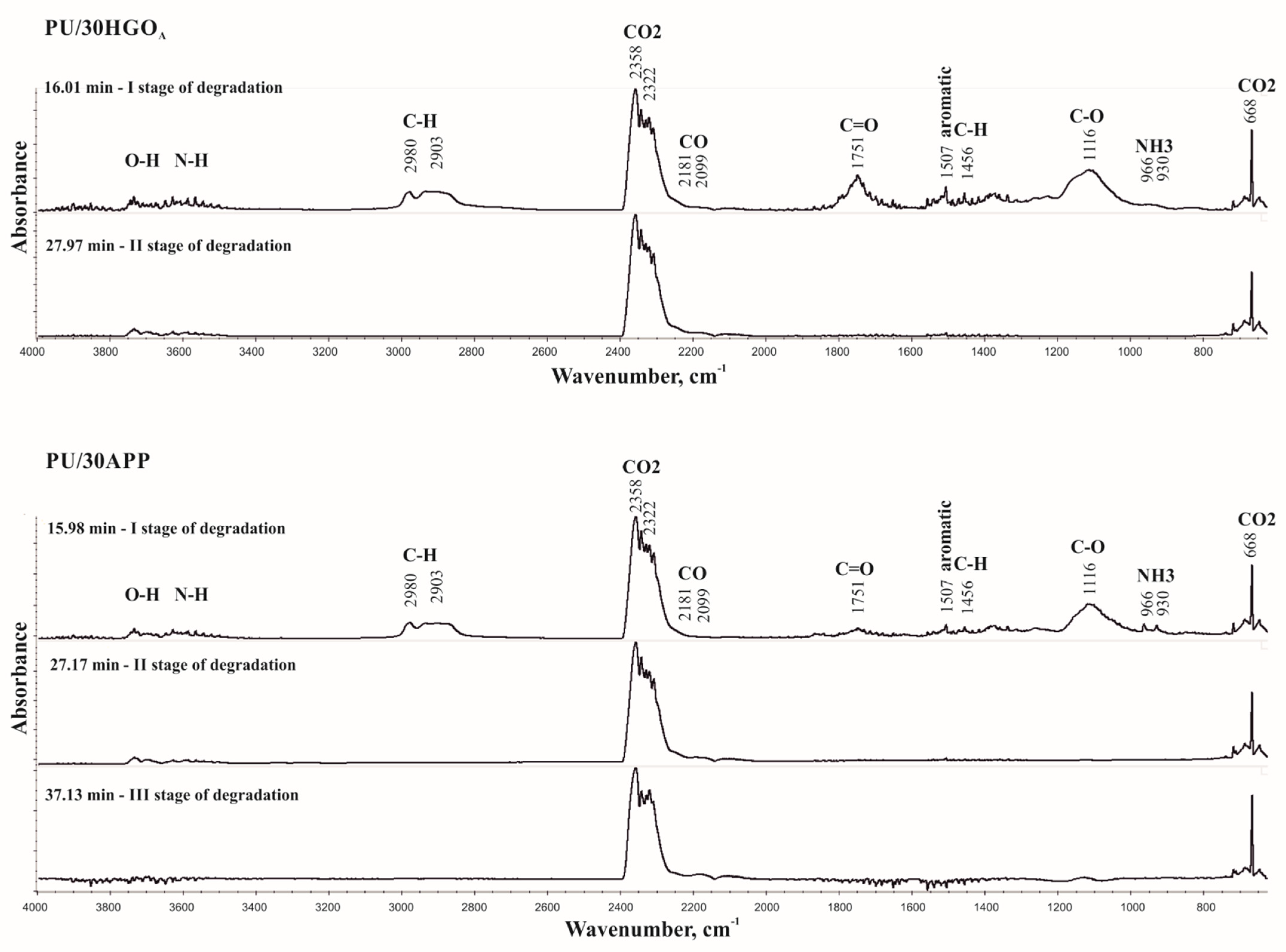
3.7. Characterization of Decomposition Products
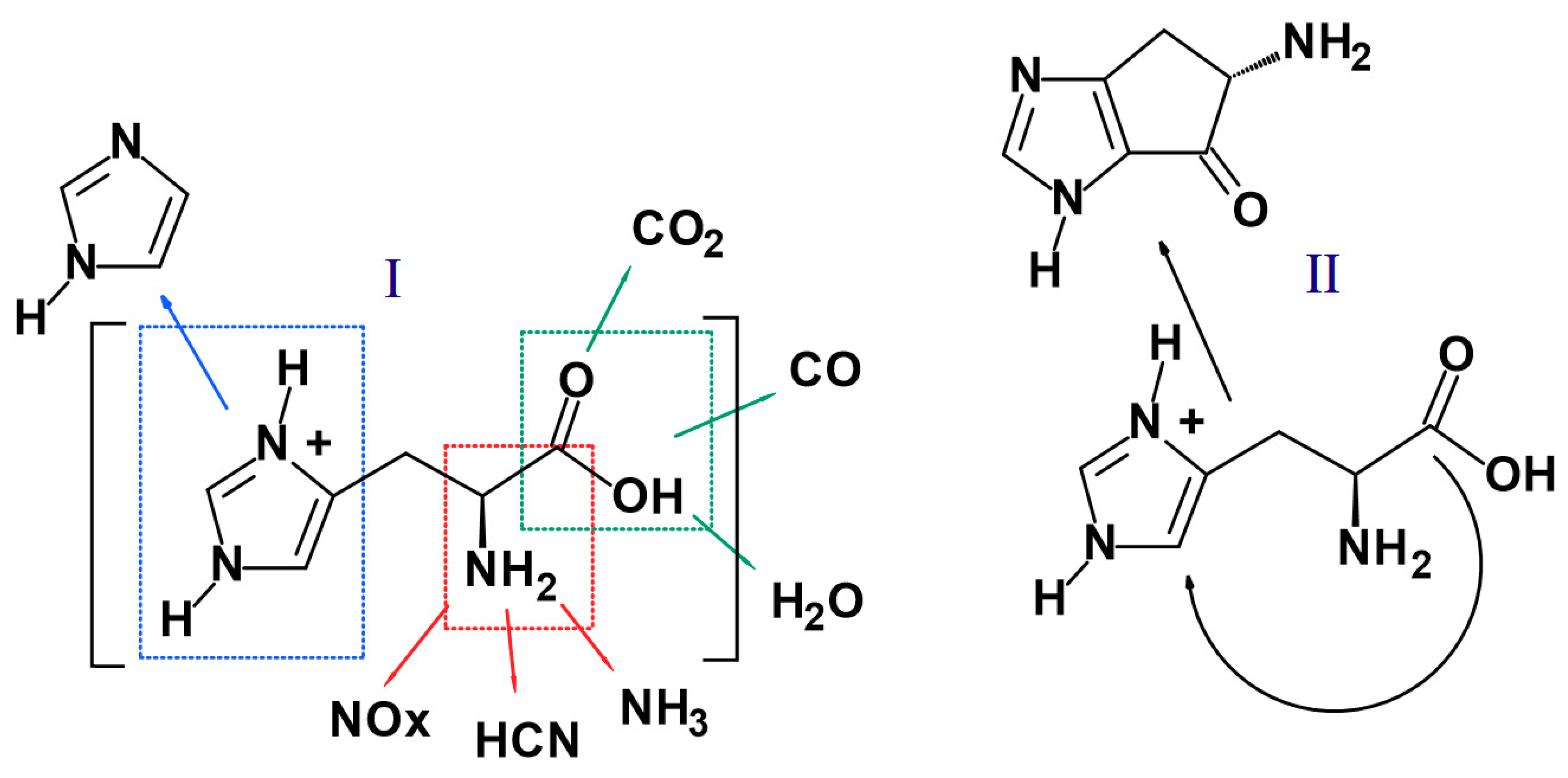
3.8. Fire Retardant Mechanism
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gaidukova, G.; Ivdre, A.; Fridrihsone, A.; Verovkins, A.; Cabulis, U.; Gaidukovs, S. Polyurethane rigid foams obtained from polyols containing bio-based and recycled components and functional additives. Ind. Crops. Prod. 2017, 102, 133–143. [Google Scholar] [CrossRef]
- Ryszkowska, J. The Role of Chemical Structure and Manufacturing Process. Conditions in Shaping the Morphology and Properties of Polyurethane Materials; Publishing House of the Warsaw University of Technology: Warszawa, Poland, 2011. [Google Scholar]
- Somarathna, H.M.C.C.; Raman, S.N.; Mohotti, D.; Mutalib, A.A.; Badri, K.H. The use of polyurethane for structural and infrastructural engineering applications: A state-of-the-art review. Constr. Build. Mater. 2018, 190, 995–1014. [Google Scholar] [CrossRef]
- Członka, S.; Bertino, M.F.; Strzelec, K.; Strąkowska, A.; Masłowski, M. Rigid polyurethane foams reinforced with solid waste generated in leather industry. Polym. Test. 2018, 69, 225–237. [Google Scholar] [CrossRef]
- Prociak, A.; Rokicki, G.; Ryszkowska, J. Polyurethane Materials; Polish Scientific Publishers PWN: Warszawa, Poland, 2014. [Google Scholar]
- Paciorek-Sadowska, J.; Borowicz, M.; Czuprynski, B.; Liszkowska, J. Composites of rigid polyurethane-polyisocyanurate foams with oak bark. Polimery-W. 2017, 62, 666–672. [Google Scholar] [CrossRef]
- Czech-Polak, J.; Oliwa, R.; Oleksy, M.; Budzik, G. Rigid polyurethane foams with increased flame resistance. Polimery-W. 2018, 63, 115–124. [Google Scholar] [CrossRef]
- Piszczyk, Ł. Synthesis and Properties of New Flame Retardant Polyurethane Foams Obtained with the Use of Renewable Raw Materials. Ph.D. Thesis, Gdańsk University of Technology, Gdańsk, Poland, 2011. [Google Scholar]
- Prociak, A. New Generation Polyurethane Thermal Insulation Materials; Publishing House of the Krakaw University of Technology: Warsaw, Poland, 2008. [Google Scholar]
- Wang, J.Q.; Chow, W.K. A brief review on fire retardants for polymeric foams. J. Appl. Polym. Sci. 2005, 97, 366–376. [Google Scholar] [CrossRef]
- Zhan, L.; Zhan, M.; Zhou, Y.; Hu, L. The study of mechanical behavior and flame retardancy of castor oil phosphate-based rigid polyurethane foam composites containing expanded graphite and triethyl phosphate. Polym. Degrad. Stab. 2013, 98, 2784–2794. [Google Scholar] [CrossRef]
- Duquesne, S.; Le Bras, M.; Bourbigot, S.; Delobel, R.; Camino, G.; Eling, B.; Lindsay, C.; Roels, T.; Vezin, H. Mechanism of fire retardancy of polyurethanes using ammonium polyphosphate. J. Appl. Polym. Sci. 2001, 82, 3262–3274. [Google Scholar] [CrossRef]
- Visakh, P.M.; Semkin, A.O.; Rezaev, I.A.; Fateev, A.V. Review on soft polyurethane flame retardant. Constr. Build. Mater. 2019, 227, 116673. [Google Scholar] [CrossRef]
- Ding, H.Y.; Huang, K.; Li, S.H.; Xu, L.N.; Xia, J.L.; Li, M. Flame retardancy and thermal degradation of halogen-free flame-retardant biobased polyurethane composites based on ammonium polyphosphate and aluminium hypophosphite. Polym. Test. 2017, 62, 325–334. [Google Scholar] [CrossRef]
- Weil, E.D.; Levchik, S.V. Commercial Flame Retardancy of Polyurethanes. J. Fire Sci. 2004, 22, 183–210. [Google Scholar] [CrossRef]
- Bastin, B.; Paleja, R.; LeFebvre, J. Behavior of Polyurethane Foams. J. Cel. Plast. 2003, 39, 323–340. [Google Scholar] [CrossRef]
- Frigione, M.; Maffezzoli, A.; Finocchiaro, P.; Failla, S. Cure kinetics and properties of epoxy resins containing a phosphorous-based flame retardant. Adv. Polym. Technol. 2003, 22, 329–342. [Google Scholar] [CrossRef]
- Hummers, W.S.; Offeman, R.E. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- ISO 845 Celluler Plastics and Rubbers. Determination of Apparent Density; BSI: London, UK, 2009. [Google Scholar]
- ASTM C421-08 Standard Test Method for Tumbling Friability of Preformed Block-Type and Preformed Pipe-Covering-Type Thermal Insulation; ASTM International: West Conshohocke, PA, USA, 2014.
- ISO 5660-1 Reaction-to-Fire Tests—Heat Release, Smoke Production and Mass Loss Rate—Part 1: Heat Release Rate (Cone Calorimeter Method) and Smoke Production Rate (Dynamic Measurement); BSI: London, UK, 2015.
- ISO 5659-2, Plastics—Smoke Generation—Part 2: Determination of Optical Density by a Single-Chamber Test; BSI: London, UK, 2017.
- Liu, L.; Wanga, Z.; Zhua, M. Flame retardant, mechanical and thermal insulating properties of rigid polyurethane foam modified by nano zirconium amino-tris-(methylenephosphonate) and expandable graphite. Polym. Degrad. Stab. 2019, 170, 108997. [Google Scholar] [CrossRef]
- Carriço, C.S.; Fraga, T.; Pasa, V.M.D. Production and characterization of polyurethane foams from a simple mixture of castor oil, crude glycerol and untreated lignin as bio-based polyols. Eur. Polym. J. 2016, 85, 53–61. [Google Scholar] [CrossRef]
- Paciorek-Sadowska, J.; Borowicz, M.; Insbrandt, M.; Czupryński, B.; Apiecionek, Ł. The Use of Waste from the Production of Rapeseed Oil for Obtaining of New Polyurethane Composites. Polymers 2019, 11, 1431. [Google Scholar] [CrossRef] [PubMed]
- Chang, L. Improving the Mechanical Performance of Wood Fiber Reinforced Bio-Based Polyurethane Foam. Master’s Thesis, University of Toronto, Toronto, ON, Canada, 2014. [Google Scholar]
- Luo, X.; Mohanty, A.; Misra, M. Lignin as a reactive reinforcing filler for water-blown rigid biofoam composites from soy oil-based polyurethane. Ind. Crop. Prod. 2013, 47, 13–19. [Google Scholar] [CrossRef]
- Sung, G.; Kim, J.H. Influence of filler surface characteristics on morphological, physical, acoustic properties of polyurethane composite foams filled with inorganic fillers. Compos. Sci. Technol. 2017, 146, 147–154. [Google Scholar] [CrossRef]
- Członka, S.; Strąkowska, A.; Kairytė, A.; Kremensas, A. Nutmeg filler as a natural compound for the production of polyurethane composite foams with antibacterial and anti-aging properties. Polym. Test. 2020, 86, 106479. [Google Scholar] [CrossRef]
- Borowicz, M.; Paciorek-Sadowska, J.; Lubczak, J.; Czupryński, B. Biodegradable, Flame-Retardant, and Bio-Based Rigid Polyurethane/Polyisocyanurate Foams for Thermal Insulation Application. Polymers 2019, 11, 1816. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, D.K.; Webster, D.C. Thermal stability and flame retardancy of polyurethanes. Prog. Polym. Sci. 2009, 34, 1068–1133. [Google Scholar] [CrossRef]
- Wang, L.; Tawiah, B.; Shi, Y.; Cai, S.; Rao, X.; Liu, C.; Yang, Y.; Yang, F.; Yu, B.; Liang, Y.; et al. Highly Effective Flame-Retardant Rigid Polyurethane Foams: Fabrication and Applications in Inhibition of Coal Combustion. Polymers 2019, 11, 1776. [Google Scholar] [CrossRef] [PubMed]
- Garrido, M.A.; Font, R. Pyrolysis and combustion study of flexible polyurethane foam. J. Anal. Appl. Pyrol. 2015, 113, 202–215. [Google Scholar] [CrossRef]
- Sałasińska, K.; Mizera, K.; Celiński, M.; Kozikowski, P.; Borucka, M.; Gajek, A. Thermal properties and fire behavior of polyethylene with a mixture of copper phosphate and melamine phosphate as a novel flame retardant. Fire Saf. J. 2020, 115, 103137. [Google Scholar] [CrossRef]
- Krebs, M. The Adjustment of Physical Properties of Viscoelastic Foam—the Role of Different Raw Materials. In Proceedings of the Polyurethanes Technical Conference, Baltimore, MD, USA, 26–28 September2016; Curran Associates, Inc.: New York, NY, USA; pp. 513–532. [Google Scholar]
- Zhang, X.; Shen, Q.; Zhang, X.; Pan, H.; Lu, Y. Graphene oxide-filled multilayer coating to improve flame-retardant and smoke suppression properties of flexible polyurethane foam. J. Mater. Sci. 2016, 51, 10361–10374. [Google Scholar] [CrossRef]
- Gunther, M.; Lorenzetti, A.; Scharel, B. Fire Phenomena of Rigid Polyurethane Foams. Polymers 2018, 10, 1166. [Google Scholar] [CrossRef]
- Gunther, M.; Lechvik, S.V.; Scharel, B. Bubbles and collapses: Fire phenomena of flame-retarded flexible polyurethane foams. Polym. Adv. Technol. 2020, 31, 2185–2198. [Google Scholar] [CrossRef]
- Drysdale, D.D. Fundamentals of the Fire Behaviour of Cellular Polymers. In Fire and Cellular Polymers; Buist, J.M., Grayson, S.J., Woolley, W.D., Eds.; Springer: Dordrecht, The Netherlands, 1986; pp. 61–75. [Google Scholar]
- Levchik, S.V.; Weil, E.D. Thermal decomposition, combustion and fire-retardancy of polyurethanes—A review of the recent literature. Polym. Int. 2004, 53, 1585–1610. [Google Scholar] [CrossRef]
- Feng, C.; Zhang, Y.; Liang, D.; Liu, S.; Chi, Z.; Xu, J. Influence of zinc borate on the flame retardancy and thermal stability of intumescent flame retardant polypropylene composites. J. Anal. Appl. Pyrolysis 2015, 115, 224–232. [Google Scholar] [CrossRef]
- Cervantes, J.M.; Espinosa, J.I.M.; Cauich-Rodríguez, J.V.; Ávila-Ortega, A.; Vázquez-Torres, H.; Marcos-Fernández, A.; San Román, J. TGA/FTIR studies of segmented aliphatic polyurethanes and their nanocomposites prepared with commercial montmorillonites. Polym. Degrad. Stab. 2009, 94, 1666–1677. [Google Scholar] [CrossRef]
- Mizera, K.; Kirpluks, M.; Cabulis, U.; Leszczyńska, M.; Półka, M.; Ryszkowska, J. Characterisation of ureaurethane elastomers containing tall oil based polyols. Ind. Crops. Prod. 2018, 113, 98–110. [Google Scholar] [CrossRef]
- Schaberg, A.; Wroblowski, R.; Goertz, R. Comparative study of the thermal decomposition behaviour of different amino acids and peptides. J. Phys. Conf. Ser. 2018, 1107, 032013. [Google Scholar] [CrossRef]
- Weiss, I.M.; Muth, C.; Drumm, R.; Kirchner, H.O. Thermal decomposition of the amino acids glycine, cysteine, aspartic acid, asparagine, glutamic acid, glutamine, arginine and histidine. BMC Biophys. 2018, 11, 1–15. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Petol 400, g | Petol 480, g | PC CAT TD-33, g | PC41, g | Niax Silicone L-6900, g | Ongronat 2100, g | APP, g | H, g | GOA, g |
|---|---|---|---|---|---|---|---|---|---|
| PU/10APP | 80 | 20 | 1 | 0.5 | 2 | 110 | 10 | 0 | 0 |
| PU/20APP | 80 | 20 | 1 | 0.5 | 2 | 110 | 20 | 0 | 0 |
| PU/30APP | 80 | 20 | 1 | 0.5 | 2 | 110 | 30 | 0 | 0 |
| PU/10HGOA | 80 | 20 | 1 | 0.5 | 2 | 110 | 0 | 9 | 1 |
| PU/20HGOA | 80 | 20 | 1 | 0.5 | 2 | 110 | 0 | 19 | 1 |
| PU/30HGOA | 80 | 20 | 1 | 0.5 | 2 | 110 | 0 | 29 | 1 |
| Samples | Process Parameter | ||
|---|---|---|---|
| Start Time, s | Gel Time, s | Tack-Free Time, s | |
| PU/10APP | 43 | 141 | 181 |
| PU/20APP | 32 | 135 | 182 |
| PU/30APP | 34 | 135 | 197 |
| PU/10HGOA | 22 | 124 | 203 |
| PU/20HGOA | 19 | 165 | 189 |
| PU/30HGOA | 21 | 164 | 193 |
| Component | Water Content, % | T5%, °C | T50%, °C | Residue at 950 °C, % |
|---|---|---|---|---|
| H | 0.1 | 262 | 540 | 0 |
| GOA | 12.5 | 57 | 437 | 16 |
| PU/ 10APP | PU/ 20APP | PU/ 30APP | PU/ 10HGOA | PU/ 20HGOA | PU/ 30HGOA | Bond Type (Vibration) |
|---|---|---|---|---|---|---|
| 3304 | 3304 | 3304 | 3303 | 3303 | 304 | N-H (stretching) |
| 2978, 2925 | 2972, 2925 | 2975, 2927 | 2972, 2927 | 2972, 2927 | 2974, 2924 | C-H (asymmetric stretching) |
| 2870 | 2870 | 2870 | 2871 | 2871 | 2871 | C-H (symmetric stretching) |
| 2275 | 2277 | 2279 | 2277 | 2277 | 2277 | N=C=O (stretching) |
| 1708 | 1708 | 1707 | 1719, 1709 | 1720, 1709 | 1720, 1709 | C=O (stretching) |
| 1595 | 1595 | 1595 | 1595 | 1595 | 1595 | Ar-H (deformation) |
| 1518 | 1517 | 1517 | 1514 | 1514 | 1514 | N-H (bending) |
| 1455 | 1453 | 1455 | 1452 | 1451 | 1452 | C-H (deformation) |
| 1411 | 1411 | 1411 | 1411 | 1411 | 1411 | PIR (deformation) |
| 1219 | 1220 | 1220 | 1220 | 1220 | 1220 | C-N (stretching) |
| 1070 | 1071 | 1070 | 1071 | 1071 | 1071 | C-O (stretching) |
| 600–400 | 600–400 | 600–400 | 623 | 623 | 623 | C-H (bending) |
| Samples | D, µm | Ar | Apparent Density, kg/m3 | Friability, % |
|---|---|---|---|---|
| PU/10APP | 513 | 1.4 | 40.7 ± 2.7 | 6.26 |
| PU/20APP | 558 | 1.2 | 41.5 ± 2.7 | 10.1 |
| PU/30APP | 534 | 1.2 | 44.9 ± 1.7 | 12.23 |
| PU/10HGOA | 567 | 1.5 | 33.1 ± 2.8 | 12.23 |
| PU/20HGOA | 666 | 1.4 | 30.2 ± 1.7 | 26.25 |
| PU/30HGOA | 616 | 1.4 | 36.4 ± 2.0 | 30.38 |
| Samples | T5%, °C | T50%, °C | DTG1, °C; %/°C | DTG2, °C; %/°C | DTG3, °C; %/°C | Residue in 950 °C, % |
|---|---|---|---|---|---|---|
| PU/10APP | 239 | 314 | 288; 12.42 | 517; 3.83 | 776; 1.11 | 1.7 |
| PU/20APP | 240 | 327 | 284; 12.74 | 513; 3.49 | 778; 1.42 | 0.8 |
| PU/30APP | 256 | 396 | 293; 13.6 | 524; 2.93 | 789; 1.89 | 0.9 |
| PU/10HGOA | 232 | 313 | 292; 11.89 | 518; 3.85 | - | 1.4 |
| PU/20HGOA | 221 | 320 | 289; 9.19 | 518; 3.82 | - | 2.7 |
| PU/30HGOA | 220 | 328 | 289; 8.69 | 520; 3.84 | - | 0.9 |
| Samples | TTI, s | pHRR, kW/m2 | MARHE, kW/m2 | THR, MJ/m2 | Residue, % |
|---|---|---|---|---|---|
| PU/10APP | 6 (1 a) | 393 (4) | 292 (20) | 19 (0) | 19.1 (8) |
| PU/20APP | 6 (0) | 419 (25) | 308 (23) | 19 (3) | 19.6 (1) |
| PU/30APP | 6 (1) | 408 (20) | 309 (13) | 19 (2) | 19.4 (4) |
| PU/10HGOA | 5 (1) | 324 (15) | 249 (12) | 19 (1) | 23.0 (5) |
| PU/20HGOA | 6 (1) | 338 (22) | 255 (8) | 16 (1) | 28.3 (10) |
| PU/30HGOA | 6 (0) | 336 (13) | 260 (7) | 17 (2) | 19.0 (5) |
| Samples | TSR, m2/m2 | SEA, m2/kg | Dsmax | VOF4 |
|---|---|---|---|---|
| PU/10APP | 586 (72) | 622 (60 a) | 142 (10) | 308 (15) |
| PU/20APP | 685 (134) | 739 (16) | 158 (29) | 369 (82) |
| PU/30APP | 706 (118) | 757 (52) | 180 (7) | 431 (4) |
| PU/10HGOA | 388 (18) | 466 (12) | 108 (6) | 233 (5) |
| PU/20HGOA | 298 (13) | 359 (79) | 127 (18) | 226 (2) |
| PU/30HGOA | 295 (39) | 381 (27) | 176 (24) | 222 (6) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sałasińska, K.; Leszczyńska, M.; Celiński, M.; Kozikowski, P.; Kowiorski, K.; Lipińska, L. Burning Behaviour of Rigid Polyurethane Foams with Histidine and Modified Graphene Oxide. Materials 2021, 14, 1184. https://doi.org/10.3390/ma14051184
Sałasińska K, Leszczyńska M, Celiński M, Kozikowski P, Kowiorski K, Lipińska L. Burning Behaviour of Rigid Polyurethane Foams with Histidine and Modified Graphene Oxide. Materials. 2021; 14(5):1184. https://doi.org/10.3390/ma14051184
Chicago/Turabian StyleSałasińska, Kamila, Milena Leszczyńska, Maciej Celiński, Paweł Kozikowski, Krystian Kowiorski, and Ludwika Lipińska. 2021. "Burning Behaviour of Rigid Polyurethane Foams with Histidine and Modified Graphene Oxide" Materials 14, no. 5: 1184. https://doi.org/10.3390/ma14051184
APA StyleSałasińska, K., Leszczyńska, M., Celiński, M., Kozikowski, P., Kowiorski, K., & Lipińska, L. (2021). Burning Behaviour of Rigid Polyurethane Foams with Histidine and Modified Graphene Oxide. Materials, 14(5), 1184. https://doi.org/10.3390/ma14051184