
Thermocatalytic CO2 Conversion over a Nickel-Loaded Ceria Nanostructured Catalyst: A NAP-XPS Study

 ,
,  ,
, 
 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Synthesis of the Nickel-Loaded Ceria Nanocubes Catalyst
2.2. Physicochemical Characterisation
2.2.1. Chemical Composition
2.2.2. Structural Characterization
2.2.3. Specific Surface Area
2.2.4. Electron Microscopy Characterization
2.2.5. Reducibility Measurements
2.2.6. Surface Basicity Characterization
2.2.7. XPS and NAP-XPS Measurements
3. Results and Discussion
3.1. Structural and Textural Characterization
3.2. Redox Characterization
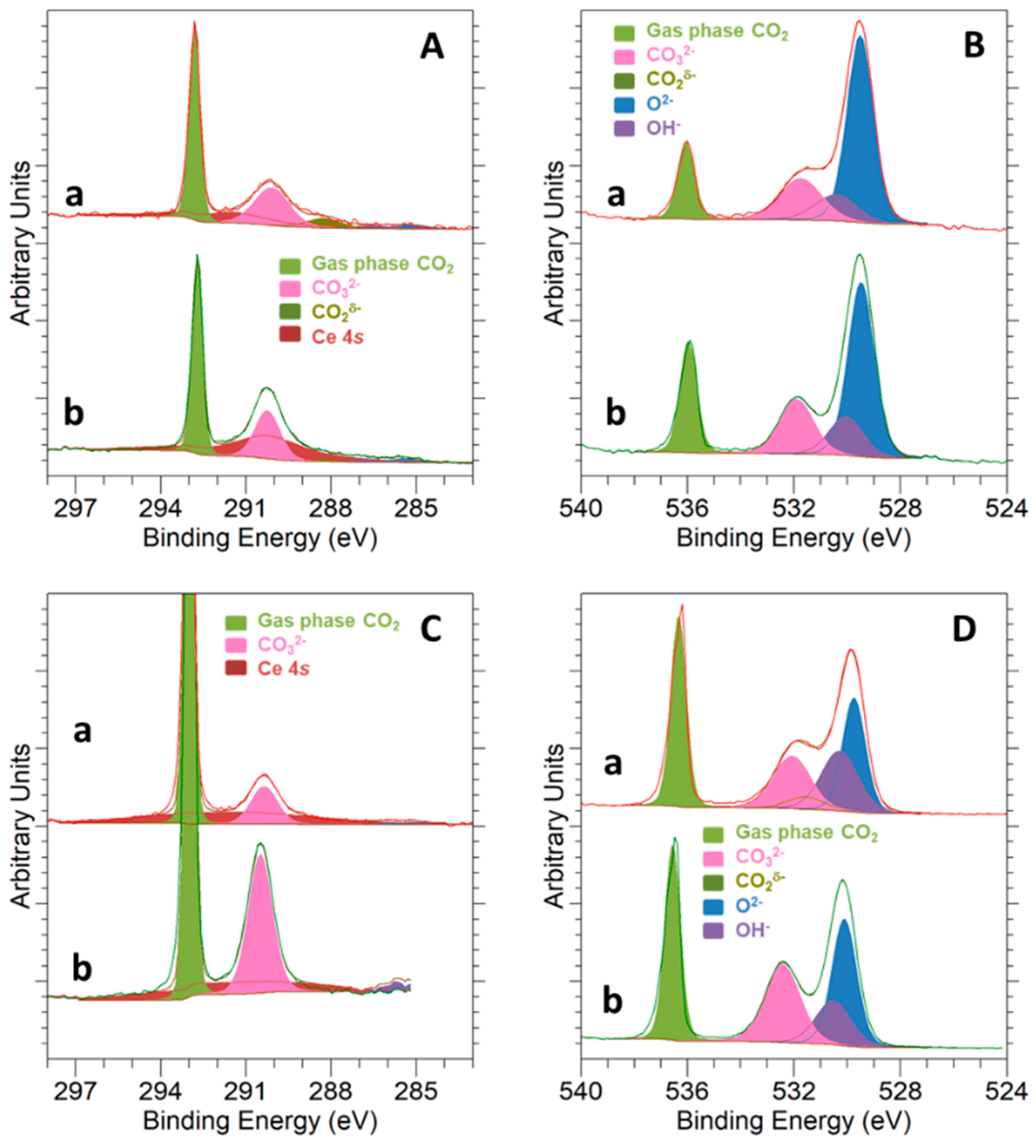
3.3. Surface Basicity and Interaction with the CO2 Molecule
3.4. CO2 Hydrogenation Reaction Followed by NAP-XPS
4. Conclusions
- Below 500 °C, the catalyst behavior was characterized by:
- Well-dispersed nickel particles over ceria surface.
- Both cerium and nickel were in an oxidized state (i.e., Ce4+ and Ni2+), even under reducing conditions.
- CO2 adsorption and reaction with H2 proceeded with high carbonate content on the catalyst surface.
- Only CH4 was detected as hydrogenation product, without evidence of any other reaction, such as reverse water–gas shift (rWGS).
- 2
- Above 500 °C, the catalyst evolution was described by:
- Strong sintering of supported nickel nanoparticles.
- Both nickel and cerium were reduced (100% Ni0 and a large fraction of Ce3+).
- Much lower amounts of carbonate species were detected on the catalyst surface after CO2 adsorption and reaction with H2.
- The appearance of new adsorbed species, which were identified as carboxylates and formates.
- CO2 reduction essentially proceeded via a rWGS mechanism, thus yielding CO as main product and without any evidence of CH4 formation.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kondratenko, E.V.; Mul, G.; Baltrusaitis, J.; Larrazábal, G.O.; Pérez-Ramírez, J. Status and perspectives of CO2 conversion into fuels and chemicals by catalytic, photocatalytic and electrocatalytic processes. Energy Environ. Sci. 2013, 6, 3112–3135. [Google Scholar] [CrossRef]
- U.S. Energy Information Administration. International Energy Outlook 2019 with Projections to 2050; U.S. Energy Information Administration: Washington, DC, USA, 2019.
- Mikkelsen, M.; Jørgensen, M.; Krebs, F.C. The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 2010, 3, 43–81. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Opportunities and prospects in the chemical recycling of carbon dioxide to fuels. Catal. Today 2009, 148, 191–205. [Google Scholar] [CrossRef]
- Perathoner, S.; Centi, G. CO2 Recycling: A key strategy to introduce green energy in the chemical production chain. ChemSusChem 2014, 7, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Winter, L.R.; Chen, R.; Chen, X.; Chang, K.; Liu, Z.; Senanayake, S.D.; Ebrahim, A.M.; Chen, J.G. Elucidating the roles of metallic Ni and oxygen vacancies in CO2 hydrogenation over Ni/CeO2 using isotope exchange and in situ measurements. Appl. Catal. B Environ. 2019, 245, 360–366. [Google Scholar] [CrossRef]
- Graves, C.; Ebbesen, S.D.; Mogensen, M.B.; Lackner, K.S. Sustainable hydrocarbon fuels by recycling CO2 and H2O with renewable or nuclear energy. Renew. Sustain. Energy Rev. 2011, 15, 1–23. [Google Scholar] [CrossRef]
- Costentin, C.; Robert, M.; Savéant, J.-M. Catalysis of the electrochemical reduction of carbon dioxide. Chem. Soc. Rev. 2013, 42, 2423–2436. [Google Scholar] [CrossRef] [PubMed]
- Centi, G.; Quadrelli, E.A.; Perathoner, S. Catalysis for CO2 conversion: A key technology for rapid introduction of renewable energy in the value chain of chemical industries. Energy Environ. Sci. 2013, 6, 1711–1731. [Google Scholar] [CrossRef]
- Appel, A.M.; Bercaw, J.E.; Bocarsly, A.B.; Dobbek, H.; Dubois, D.L.; Dupuis, M.; Ferry, J.G.; Fujita, E.; Hille, R.; Kenis, P.J.A.; et al. Frontiers, opportunities, and challenges in biochemical and chemical catalysis of CO2 fixation. Chem. Rev. 2013, 113, 6621–6658. [Google Scholar] [CrossRef]
- Kattel, S.; Liu, P.; Chen, J.G. Tuning selectivity of CO2 hydrogenation reactions at the metal/oxide interface. J. Am. Chem. Soc. 2017, 139, 9739–9754. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Yan, B.; Chen, J.G. Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: Challenges and opportunities. Energy Environ. Sci. 2016, 9, 62–73. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kuhn, J.N. CO2 conversion by reverse water gas shift catalysis: Comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Chen, C.-S.; Cheng, W.-H.; Lin, S.-S. Study of reverse water gas shift reaction by TPD, TPR and CO2 hydrogenation over potassium-promoted Cu/SiO2 catalyst. Appl. Catal. A Gen. 2003, 238, 55–67. [Google Scholar] [CrossRef]
- Wang, X.; Shi, H.; Kwak, J.H.; Szanyi, J. Mechanism of CO2 hydrogenation on Pd/Al2O3 catalysts: Kinetics and transient DRIFTS-MS studies. ACS Catal. 2015, 5, 6337–6349. [Google Scholar] [CrossRef]
- Chen, C.; Cheng, W.; Lin, S. Mechanism of CO formation in reverse water–gas shift reaction over Cu/Al2O3 catalyst. Catal. Lett. 2000, 68, 45–48. [Google Scholar] [CrossRef]
- Bernal, S.; Blanco, G.; Gatica, J.M.; Perez-Omil, J.A.; Pintado, J.M.; Vidal, H. Chemical reactivity of binary rare earth oxides. In Binary Rare Oxides; Adachi, G., Imanaka, N., Kang, Z.C., Eds.; Springer: Dordrecht, The Netherlands, 2004; pp. 9–55. [Google Scholar]
- Trovarelli, A. Catalytic properties of ceria and CeO2-containing materials. Catal. Rev. 1996, 38, 439–520. [Google Scholar] [CrossRef]
- Du, G.; Lim, S.; Yang, Y.; Wang, C.; Pfefferle, L.; Haller, G.L. Methanation of carbon dioxide on Ni-incorporated MCM-41 catalysts: The influence of catalyst pretreatment and study of steady-state reaction. J. Catal. 2007, 249, 370–379. [Google Scholar] [CrossRef]
- Chang, F.-W.; Kuo, M.-S.; Tsay, M.-T.; Hsieh, M.-C. Hydrogenation of CO2 over nickel catalysts on rice husk ash-alumina prepared by incipient wetness impregnation. Appl. Catal. A Gen. 2003, 247, 309–320. [Google Scholar] [CrossRef]
- Yamasaki, M.; Komori, M.; Akiyama, E.; Habazaki, H.; Kawashima, A.; Asami, K.; Hashimoto, K. CO2 methanation catalysts prepared from amorphous Ni–Zr–Sm and Ni–Zr–misch metal alloy precursors. Mater. Sci. Eng. A 1999, 267, 220–226. [Google Scholar] [CrossRef]
- Yamasaki, M.; Habazaki, H.; Asami, K.; Izumiya, K.; Hashimoto, K. Effect of tetragonal ZrO2 on the catalytic activity of Ni/ZrO2 catalyst prepared from amorphous Ni–Zr alloys. Catal. Commun. 2006, 7, 24–28. [Google Scholar] [CrossRef]
- Lunde, P.J.; Kester, F.L. Carbon dioxide methanation on a ruthenium catalyst. Ind. Eng. Chem. Process. Des. Dev. 1974, 13, 27–33. [Google Scholar] [CrossRef]
- Wang, S.; Lu, G. (Max). Role of CeO2 in Ni/CeO2–Al2O3 catalysts for carbon dioxide reforming of methane. Appl. Catal. B Environ. 1998, 19, 267–277. [Google Scholar] [CrossRef]
- Rahmani, S.; Rezaei, M.; Meshkani, F. Preparation of promoted nickel catalysts supported on mesoporous nanocrystalline gamma alumina for carbon dioxide methanation reaction. J. Ind. Eng. Chem. 2014, 20, 4176–4182. [Google Scholar] [CrossRef]
- Ahmad, W.; Younis, M.N.; Shawabkeh, R.; Ahmed, S. Synthesis of lanthanide series (La, Ce, Pr, Eu & Gd) promoted Ni/γ-Al2O3 catalysts for methanation of CO2 at low temperature under atmospheric pressure. Catal. Commun. 2017, 100, 121–126. [Google Scholar] [CrossRef]
- Schnadt, J.; Knudsen, J.; Johansson, N. Present and new frontiers in materials research by ambient pressure X-ray photoelectron spectroscopy. J. Phys. Condens. Matter 2020, 32, 413003. [Google Scholar] [CrossRef]
- Mai, H.-X.; Sun, L.-D.; Zhang, Y.-W.; Si, R.; Feng, W.; Zhang, H.-P.; Liu, A.H.-C.; Yan, C.-H. Shape-selective synthesis and oxygen storage behavior of ceria nanopolyhedra, nanorods, and nanocubes. J. Phys. Chem. B 2005, 109, 24380–24385. [Google Scholar] [CrossRef]
- Tinoco, M.; Fernandez-Garcia, S.; Lopez-Haro, M.; Hungria, A.B.; Chen, X.; Blanco, G.; Perez-Omil, J.A.; Collins, S.E.; Okuno, H.; Calvino, J.J. Critical influence of nanofaceting on the preparation and performance of supported gold catalysts. ACS Catal. 2015, 5, 3504–3513. [Google Scholar] [CrossRef]
- Fernandez-Garcia, S.; Jiang, L.; Tinoco, M.; Hungria, A.B.; Han, J.; Blanco, G.; Calvino, J.J.; Chen, X. Enhanced hydroxyl radical scavenging activity by doping lanthanum in ceria nanocubes. J. Phys. Chem. C 2015, 120, 1891–1901. [Google Scholar] [CrossRef]
- Collins, S.M.; Fernandez-Garcia, S.; Calvino, J.J.; Midgley, P.A. Sub-nanometer surface chemistry and orbital hybridization in lanthanum-doped ceria nano-catalysts revealed by 3D electron microscopy. Sci. Rep. 2017, 7, 5406. [Google Scholar] [CrossRef]
- Jiang, L.; Fernandez-Garcia, S.; Tinoco, M.; Yan, Z.; Xue, Q.; Blanco, G.; Calvino, J.J.; Hungria, A.B.; Chen, X. Improved oxidase mimetic activity by praseodymium incorporation into ceria nanocubes. ACS Appl. Mater. Interfaces 2017, 9, 18595–18608. [Google Scholar] [CrossRef]
- Cabeza, I.; Souto, L.G.; Pintado, J.M.; Pereira, C.; Freire, C.; Blanco, G. Influence of ceria distribution on the redox behaviour of nanoparticulated CeO2-SiO2 systems with application in catalysis. Surf. Interface Anal. 2014, 46, 712–715. [Google Scholar] [CrossRef]
- Bogeat, A.B.; Núñez-Pérez, B.; Blanco, G.; Pintado, J.M.; Hernández-Garrido, J.C.; Calvino, J.J. Surface and redox characterization of new nanostructured ZrO2@CeO2 systems with potential catalytic applications. Surf. Interface Anal. 2018, 50, 1025–1029. [Google Scholar] [CrossRef]
- Bogeat, A.B.; Raposo, I.D.; Blanco, G.; Pintado, J.M. Tuning the integration rate of Ce(Ln)O2 nanoclusters into nanoparticulated ZrO2 Supports: When the cation size matters. Materials 2020, 13, 2818. [Google Scholar] [CrossRef] [PubMed]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Allen, L.J.; D’Alfonso, A.J.; Freitag, B.; Klenov, D.O. Chemical mapping at atomic resolution using energy-dispersive X-ray spectroscopy. MRS Bull. 2012, 37, 47–52. [Google Scholar] [CrossRef]
- Sánchez, J.J.; López-Haro, M.; Hernández-Garrido, J.C.; Blanco, G.; Cauqui, M.A.; Rodríguez-Izquierdo, J.M.; Pérez-Omil, J.A.; Calvino, J.J.; Yeste, M.P. An atomically efficient, highly stable and redox active Ce0.5Tb0.5Ox (3% mol.)/MgO catalyst for total oxidation of methane. J. Mater. Chem. A 2019, 7, 8993–9003. [Google Scholar] [CrossRef]
- Barr, T.L.; Seal, S. Nature of the use of adventitious carbon as a binding energy standard. J. Vac. Sci. Technol. A 1995, 13, 1239–1246. [Google Scholar] [CrossRef]
- Tanuma, S.; Powell, C.J.; Penn, D.R. Calculations of electron inelastic mean free paths (IMFPS). IV. Evaluation of calculated IMFPs and of the predictive IMFP formula TPP-2 for electron energies between 50 and 2000 eV. Surf. Interface Anal. 1993, 20, 77–89. [Google Scholar] [CrossRef]
- Holgado, J.P.; Alvarez, R.; Munuera, G. Study of CeO2 XPS spectra by factor analysis: Reduction of CeO2. Appl. Surf. Sci. 2000, 161, 301–315. [Google Scholar] [CrossRef]
- Sarma, D.; Rao, C. XPES studies of oxides of second- and third-row transition metals including rare earths. J. Electron. Spectrosc. Relat. Phenom. 1980, 20, 25–45. [Google Scholar] [CrossRef]
- Tanuma, S.; Powell, C.J.; Penn, D.R. Calculations of electron inelastic mean free paths. V. Data for 14 organic compounds over the 50-2000 eV range. Surf. Interface Anal. 1994, 21, 165–176. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Lau, L.W.M.; Gerson, A.; Smart, R.S.C. X-ray photoelectron spectroscopic chemical state quantification of mixed nickel metal, oxide and hydroxide systems. Surf. Interface Anal. 2009, 41, 324–332. [Google Scholar] [CrossRef]
- Boaro, M.; Colussi, S.; Trovarelli, A. Ceria-based materials in hydrogenation and reforming reactions for CO2 valorization. Front. Chem. 2019, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Gao, W.; Zhang, Z.; Zhang, S.; Tian, Z.; Liu, Y.; Ho, J.C.; Qu, Y. Regulating the surface of nanoceria and its applications in heterogeneous catalysis. Surf. Sci. Rep. 2018, 73, 1–36. [Google Scholar] [CrossRef]
- Winter, L.R.; Gomez, E.; Yan, B.; Yao, S.; Chen, J.G. Tuning Ni-catalyzed CO2 hydrogenation selectivity via Ni-ceria support interactions and Ni-Fe bimetallic formation. Appl. Catal. B Environ. 2018, 224, 442–450. [Google Scholar] [CrossRef]
- Yao, H.C.; Yao, Y.F.Y. Ceria in automotive exhaust catalysts: I. Oxygen storage. J. Catal. 1984, 86, 254–265. [Google Scholar] [CrossRef]
- Giordano, F.; Trovarelli, A.; De Leitenburg, C.; Giona, M. A model for the temperature-programmed reduction of low and high surface area ceria. J. Catal. 2000, 193, 273–282. [Google Scholar] [CrossRef]
- Désaunay, T.; Bonura, G.; Chiodo, V.; Freni, S.; Couzinié, J.-P.; Bourgon, J.; Ringuedé, A.; Labat, F.; Adamo, C.; Cassir, M. Surface-dependent oxidation of H2 on CeO2 surfaces. J. Catal. 2013, 297, 193–201. [Google Scholar] [CrossRef]
- Perrichon, V.; Laachir, A.; Bergeret, G.; Fréty, R.; Tournayan, L.; Touret, O. Reduction of cerias with different textures by hydrogen and their reoxidation by oxygen. J. Chem. Soc. Faraday Trans. 1994, 90, 773–781. [Google Scholar] [CrossRef]
- Lechkar, A.; Bogeat, A.B.; Blanco, G.; Pintado, J.M.; El Begrani, M.S. Methanation of carbon dioxide over ceria-praseodymia promoted Ni-alumina catalysts. Influence of metal loading, promoter composition and alumina modifier. Fuel 2018, 234, 1401–1413. [Google Scholar] [CrossRef]
- Romeo, M.; Bak, K.; El Fallah, J.; Le Normand, F.; Hilaire, L. XPS study of the reduction of cerium dioxide. Surf. Interface Anal. 1993, 20, 508–512. [Google Scholar] [CrossRef]
- Rosynek, M.P. Catalytic properties of rare earth oxides. Catal. Rev. 1977, 16, 111–154. [Google Scholar] [CrossRef]
- Bernal, S.; Blanco, G.; Calvino, J.; Omil, J.P.; Pintado, J. Some major aspects of the chemical behavior of rare earth oxides: An overview. J. Alloys Compd. 2006, 408–412, 496–502. [Google Scholar] [CrossRef]
- Sato, S.; Takahashi, R.; Kobune, M.; Gotoh, H. Basic properties of rare earth oxides. Appl. Catal. A Gen. 2009, 356, 57–63. [Google Scholar] [CrossRef]
- Zhong, L.; Chen, D.; Zafeiratos, S. A mini review of in situ near-ambient pressure XPS studies on non-noble, late transition metal catalysts. Catal. Sci. Technol. 2019, 9, 3851–3867. [Google Scholar] [CrossRef]
- Vesselli, E.; Schweicher, J.; Bundhoo, A.; Frennet, A.; Kruse, N. Catalytic CO2 hydrogenation on nickel: Novel insight by chemical transient kinetics. J. Phys. Chem. C 2010, 115, 1255–1260. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Core Level | BE Range | hν (KE = 190 eV) | hν (KE = 550 eV) |
|---|---|---|---|
| Ce 3d+Ni 2p | 850–925 | 1080 | 1440 |
| O 1s | 525–540 | 720 | 1080 |
| C 1s | 281–293 | 475 | 835 |
| Sample | Treatment | %Ce3+ at 3.3 nm | %Ce3+ at 1.8 nm |
|---|---|---|---|
| CeO2 NCs | 1 mbar H2 250 °C | 2 | 2 |
| 1 mbar 1CO2:4H2 250 °C | 1 | 3 | |
| 1 mbar 1CO2:4H2 500 °C | 33 | 52 | |
| 1 mbar 1CO2:4H2 600 °C | 33 | 52 | |
| 1 mbar 1CO2:4H2 640 °C | 37 | 58 | |
| 5Ni-CeO2 NCs | 1 mbar H2 250 °C | 1 | 1 |
| 1 mbar H2 300 °C | 0 | 2 | |
| 1 mbar H2 350 °C | 0 | 3 | |
| 1 mbar H2 500 °C | 36 | 50 | |
| 1 mbar 1CO2:4H2 250 °C | 0 | 3 | |
| 1 mbar 1CO2:4H2 500 °C | 37 | 52 | |
| 1 mbar 1CO2:4H2 600 °C | 42 | 57 |
| Temperature | Ni 2p3/2/Ce 3d at 3.3 nm | Ni 2p3/2/Ce 3d at 1.8 nm |
|---|---|---|
| 250 °C | 0.55 | 0.74 |
| 350 °C | 0.55 | 0.70 |
| 500 °C | 0.20 | 0.23 |
| Sample | 7 mbar CO2 | 1013 mbar CO2 |
|---|---|---|
| CeO2 NCs | 0.048 | 0.119 |
| 5Ni-CeO2 NCs | 0.063 | 0.219 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barroso-Bogeat, A.; Blanco, G.; Pérez-Sagasti, J.J.; Escudero, C.; Pellegrin, E.; Herrera, F.C.; Pintado, J.M. Thermocatalytic CO2 Conversion over a Nickel-Loaded Ceria Nanostructured Catalyst: A NAP-XPS Study. Materials 2021, 14, 711. https://doi.org/10.3390/ma14040711
Barroso-Bogeat A, Blanco G, Pérez-Sagasti JJ, Escudero C, Pellegrin E, Herrera FC, Pintado JM. Thermocatalytic CO2 Conversion over a Nickel-Loaded Ceria Nanostructured Catalyst: A NAP-XPS Study. Materials. 2021; 14(4):711. https://doi.org/10.3390/ma14040711
Chicago/Turabian StyleBarroso-Bogeat, Adrián, Ginesa Blanco, Juan José Pérez-Sagasti, Carlos Escudero, Eric Pellegrin, Facundo C. Herrera, and José María Pintado. 2021. "Thermocatalytic CO2 Conversion over a Nickel-Loaded Ceria Nanostructured Catalyst: A NAP-XPS Study" Materials 14, no. 4: 711. https://doi.org/10.3390/ma14040711
APA StyleBarroso-Bogeat, A., Blanco, G., Pérez-Sagasti, J. J., Escudero, C., Pellegrin, E., Herrera, F. C., & Pintado, J. M. (2021). Thermocatalytic CO2 Conversion over a Nickel-Loaded Ceria Nanostructured Catalyst: A NAP-XPS Study. Materials, 14(4), 711. https://doi.org/10.3390/ma14040711