
DFT/TD-DFT Framework of Mixed-Metal Complexes with Symmetrical and Unsymmetrical Bridging Ligands—Step-By-Step Investigations: Mononuclear, Dinuclear Homometallic, and Heterometallic for Optoelectronic Applications


Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
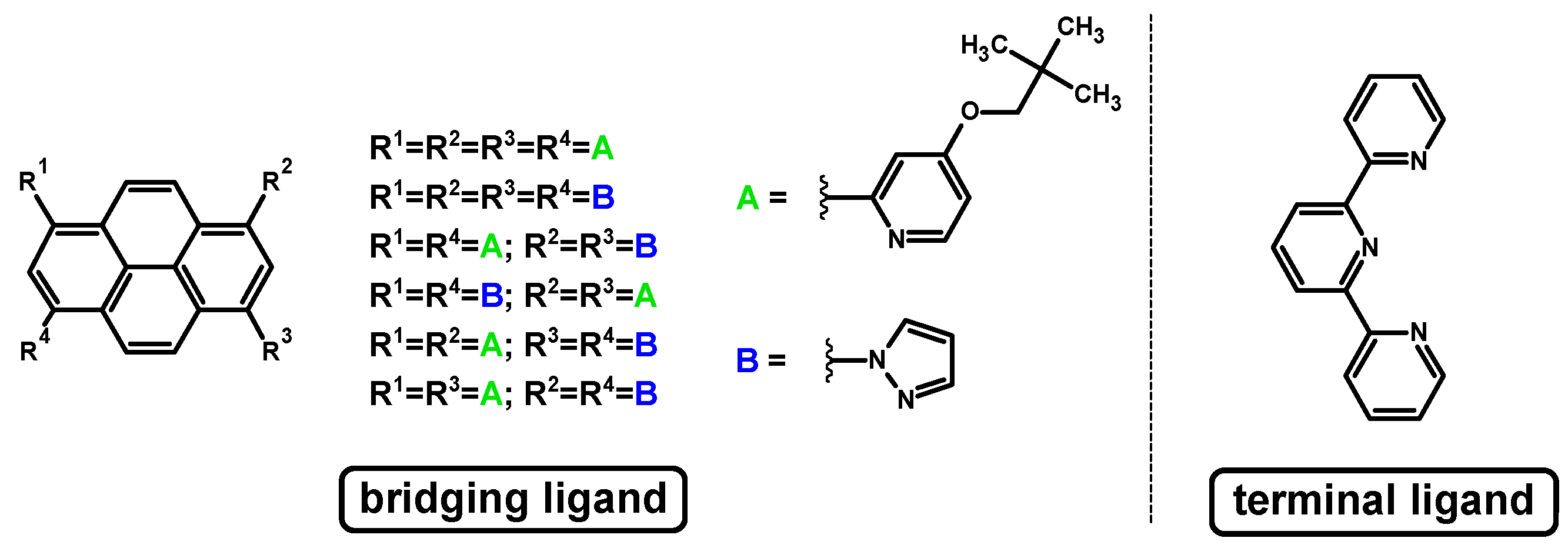
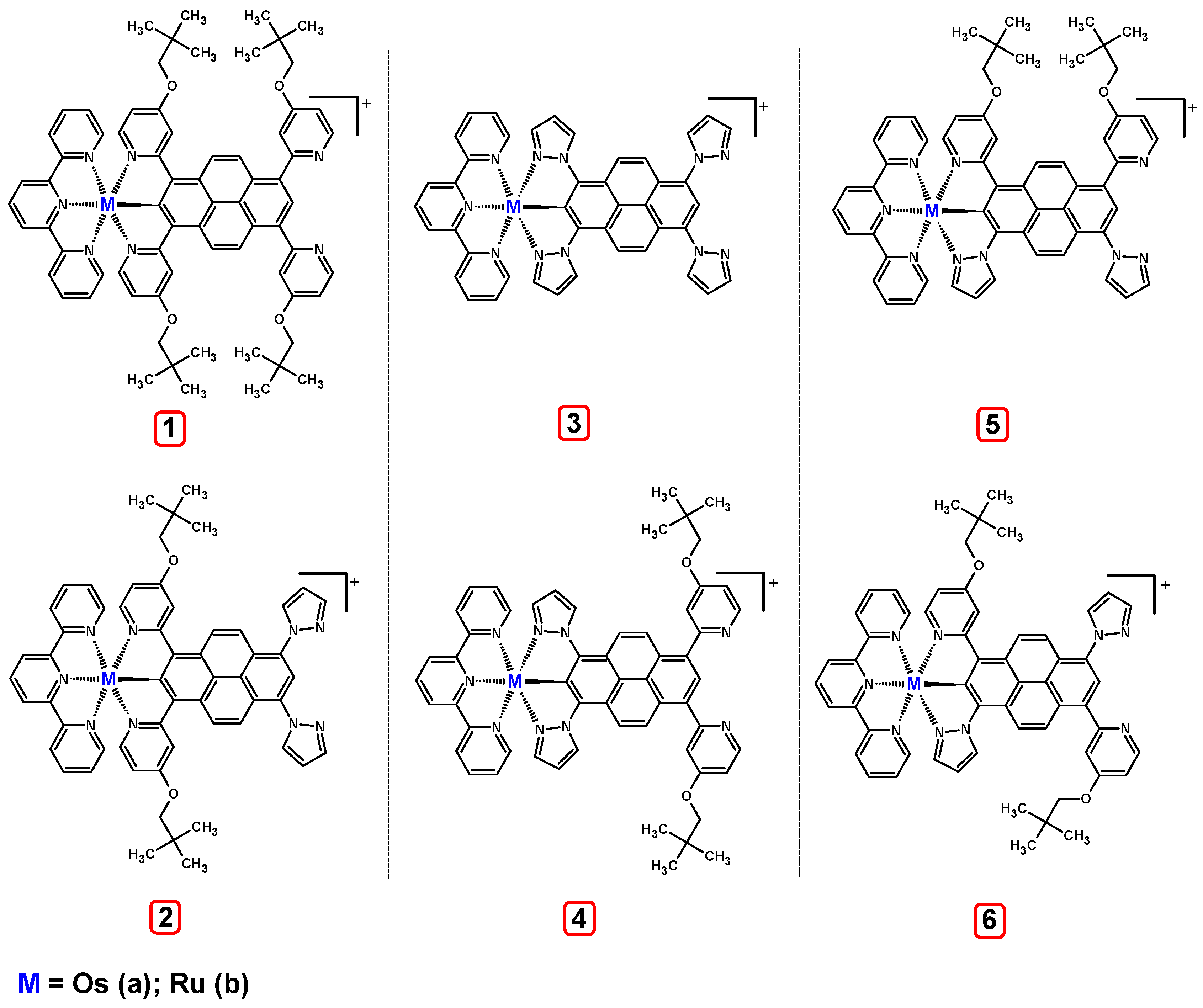
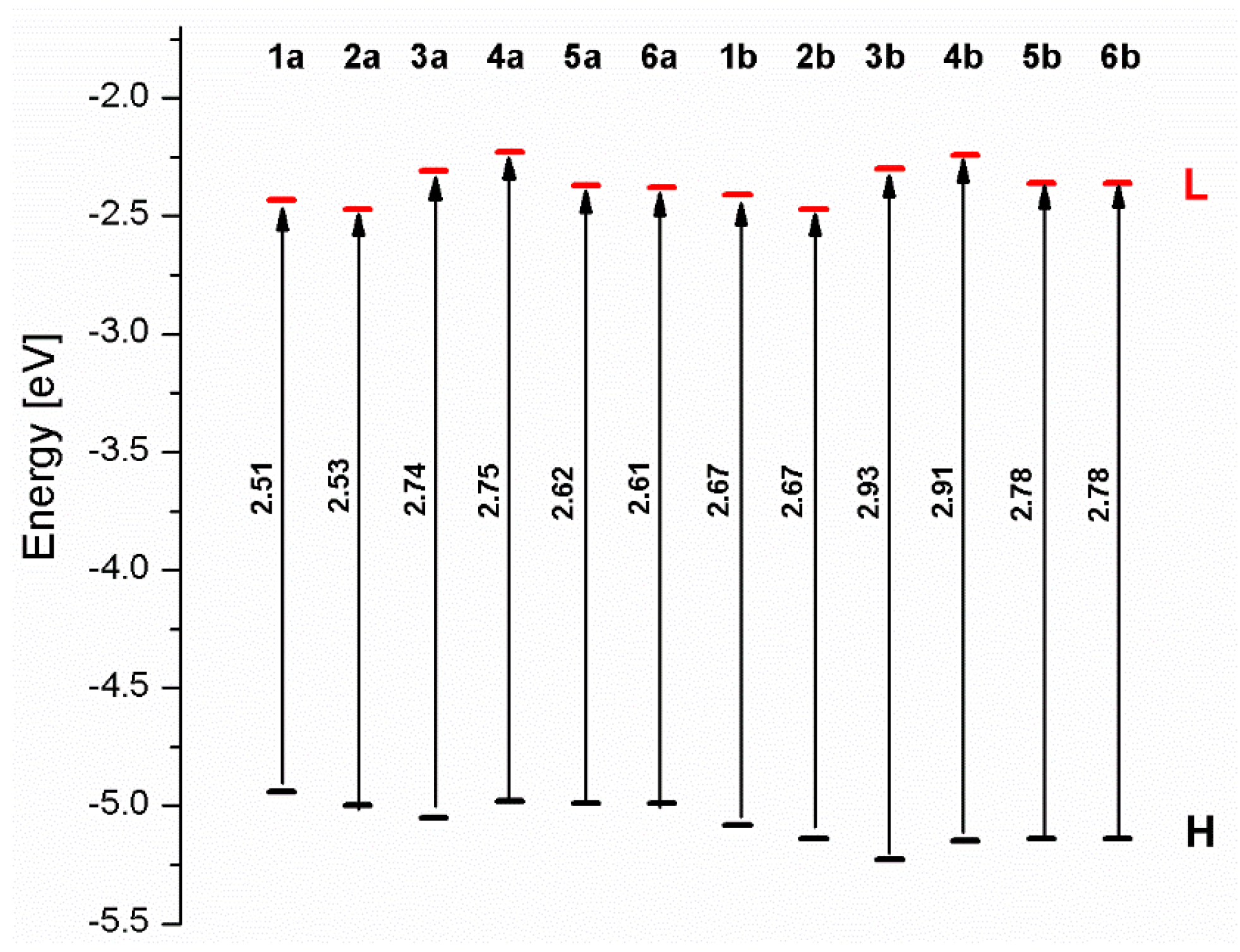
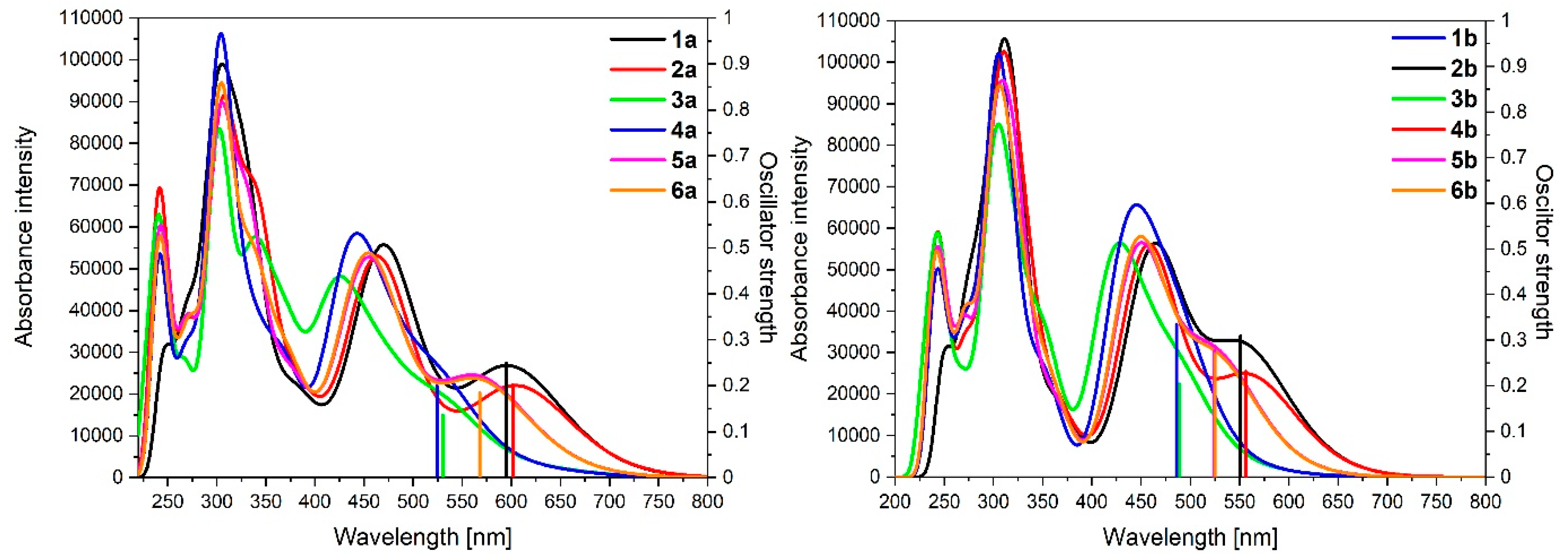
3.1. Mononuclear Osmium and Ruthenium Complexes
3.2. Dinuclear Osmium and Ruthenium Complexes
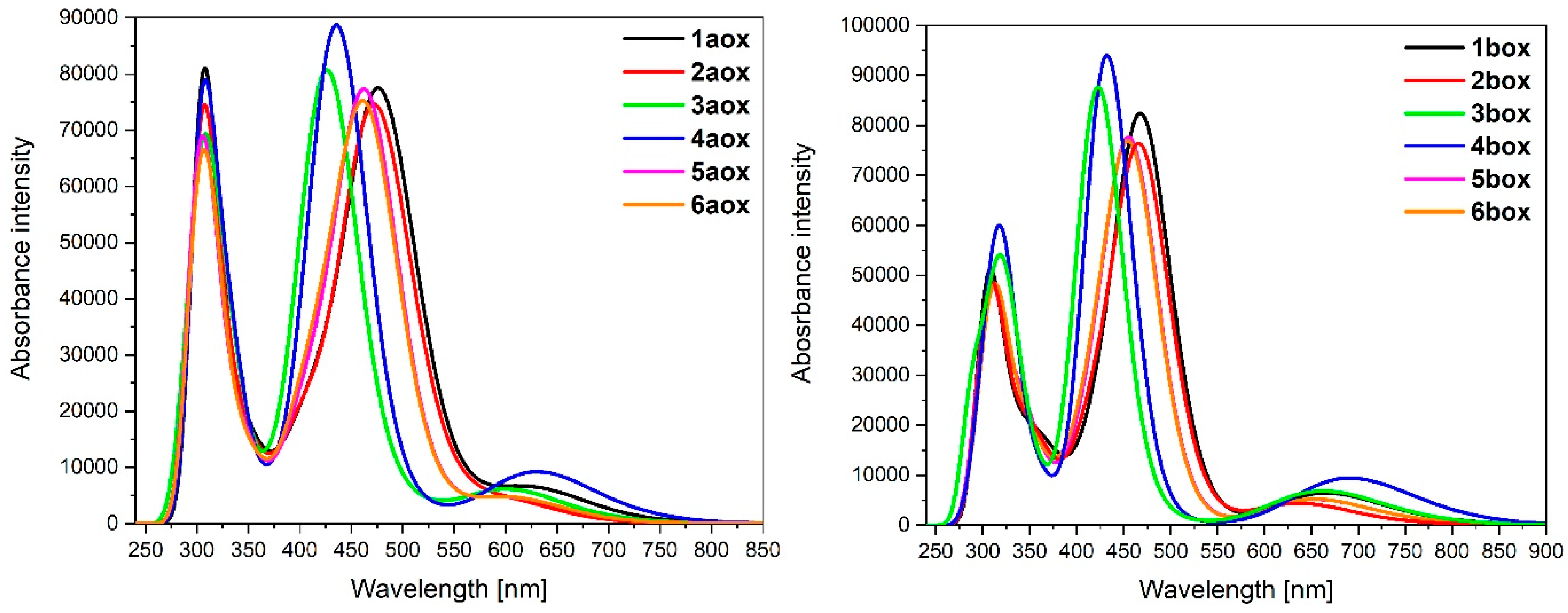
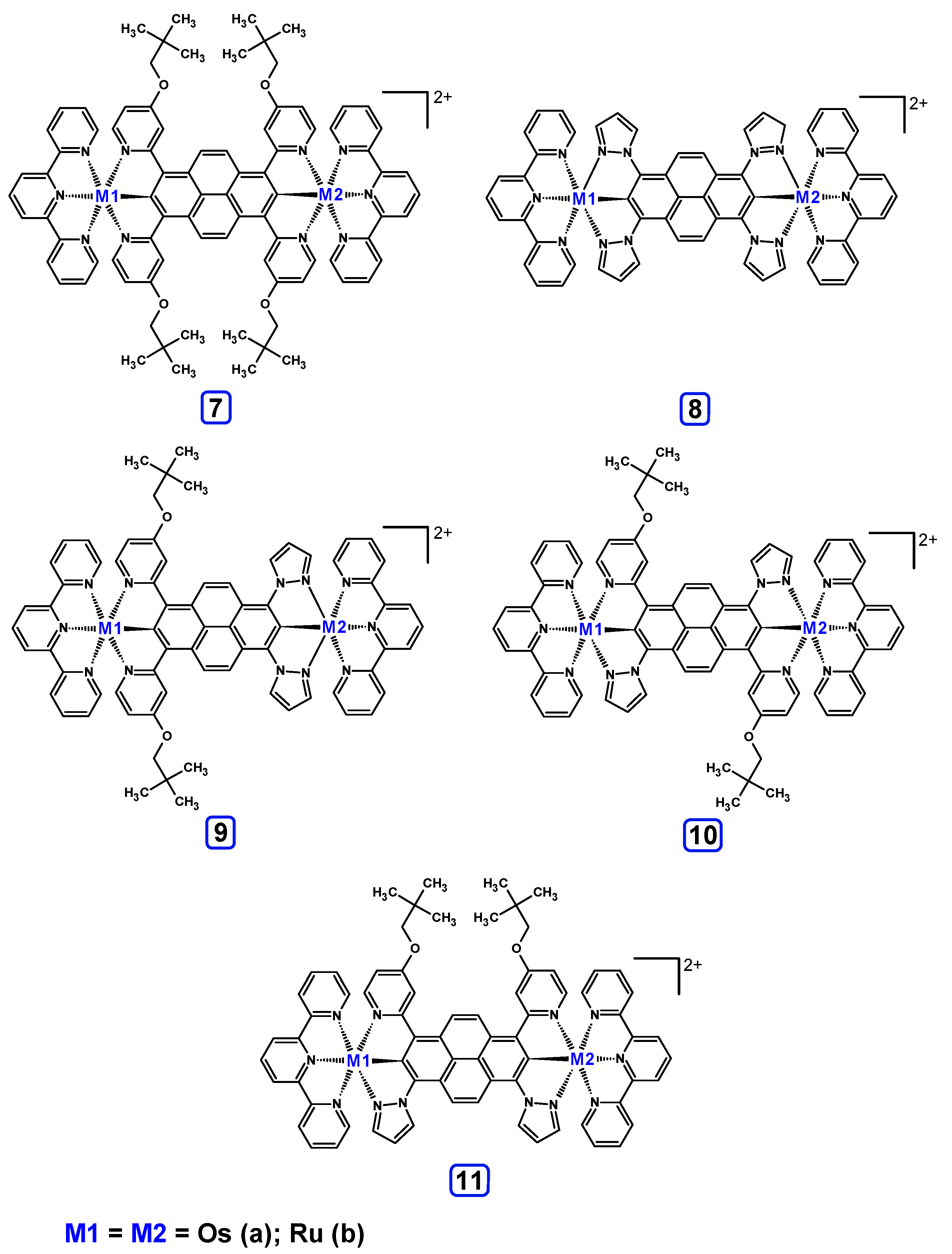
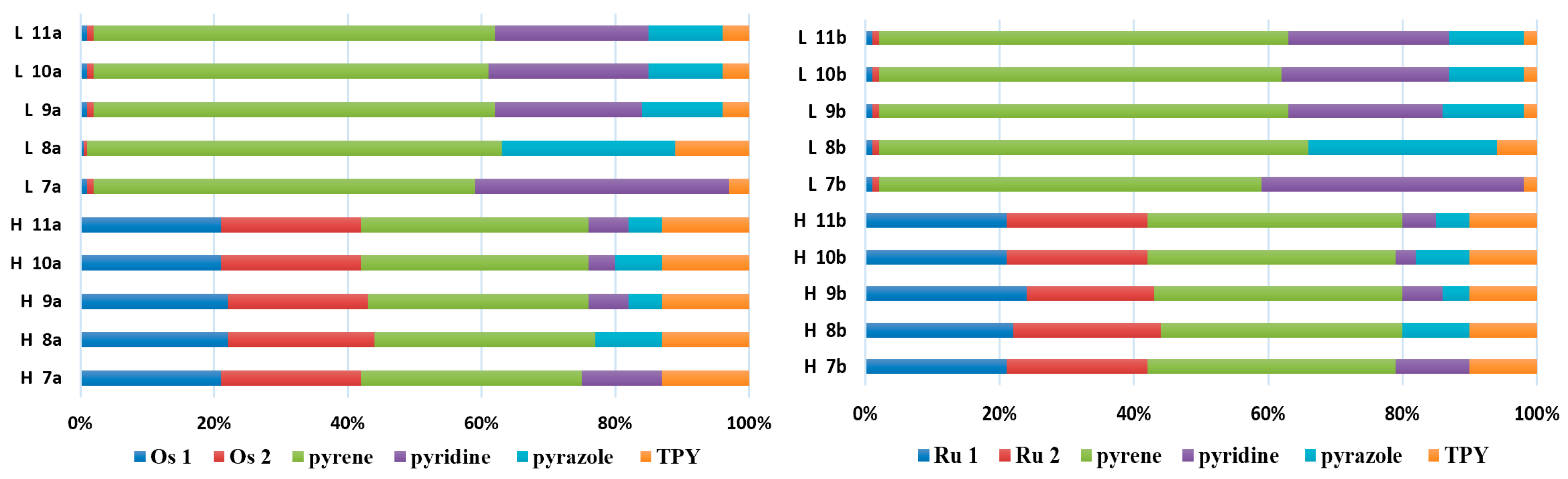
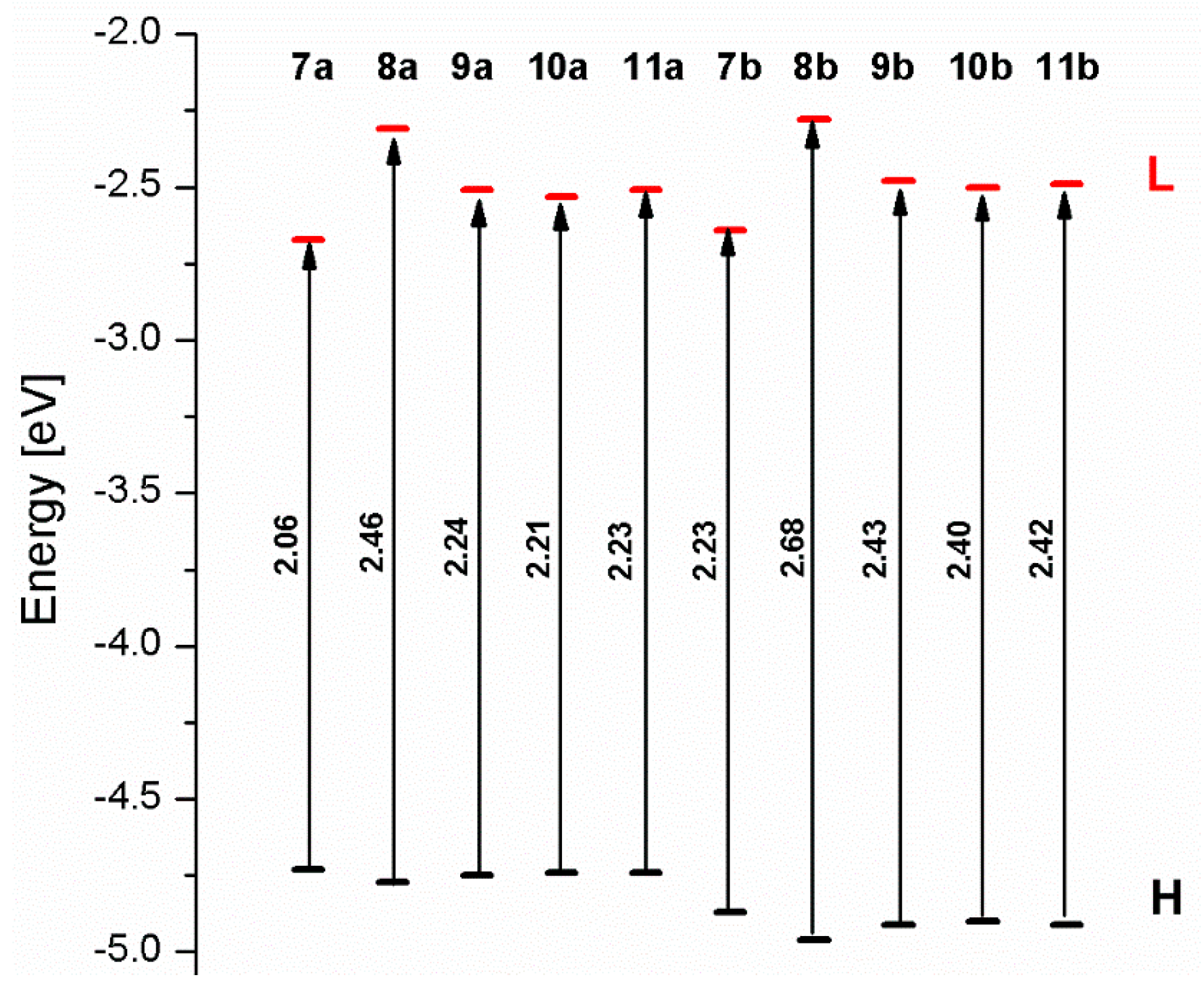
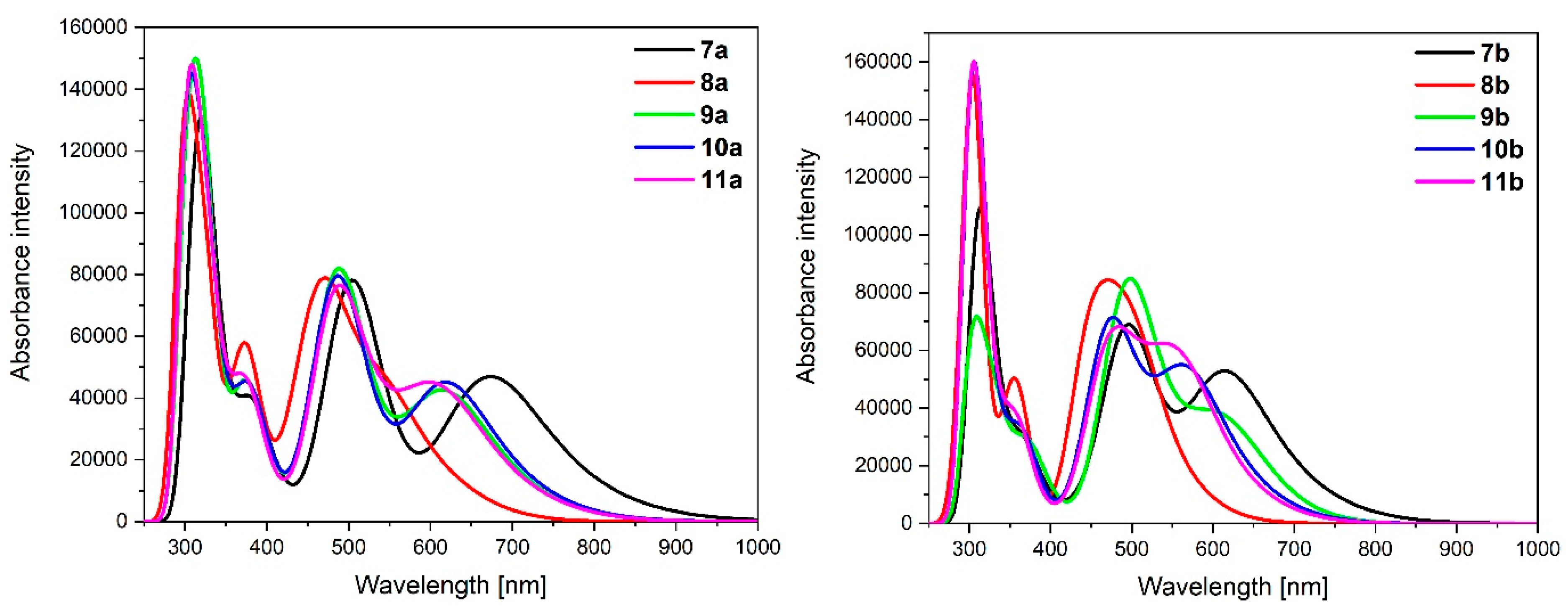
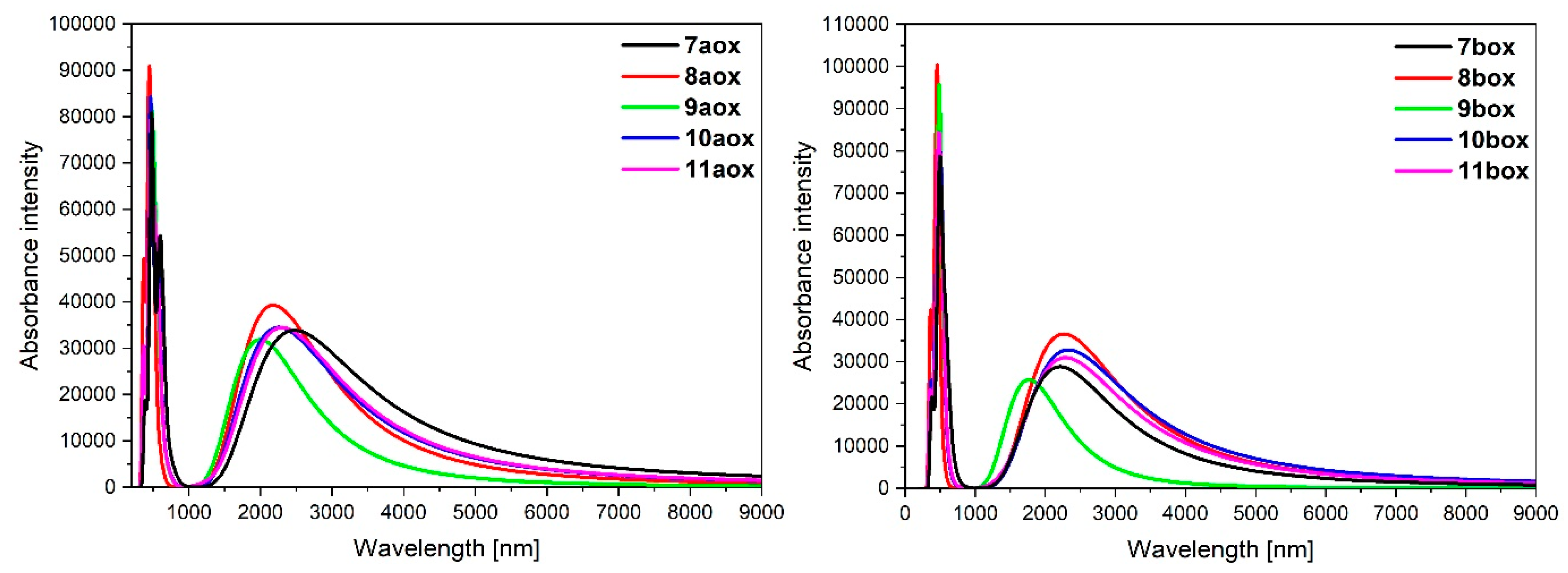
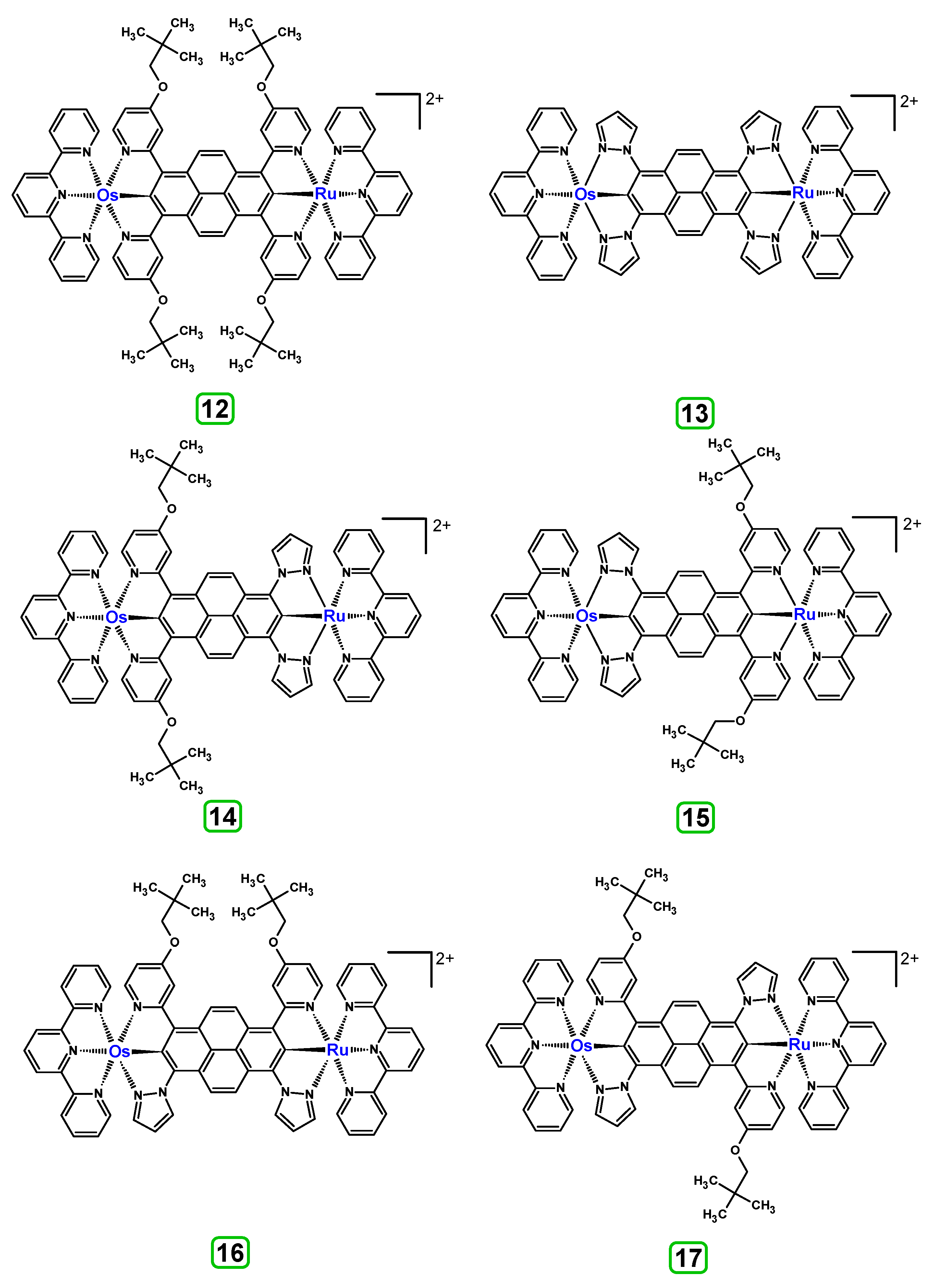
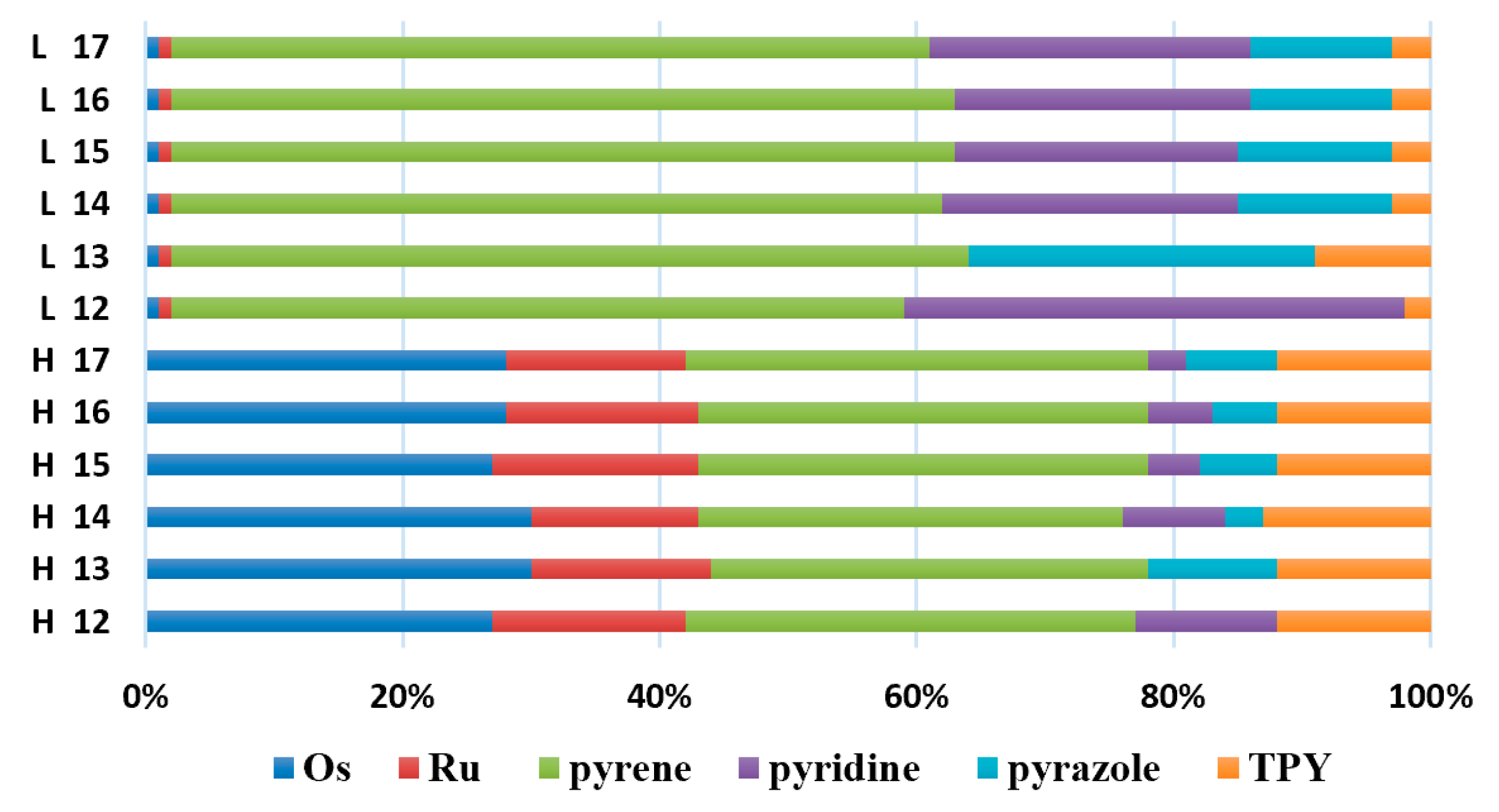
3.3. Dinuclear Mixed-Metal Osmium/Ruthenium Complexes
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Yao, C.J.; Sui, L.Z.; Xie, H.Y.; Xiao, W.J.; Zhong, Y.W.; Yao, J. Electronic Coupling between Two Cyclometalated Ruthenium Centers Bridged by 1,3,6,8-Tetra(2-Pyridyl)Pyrene (Tppyr). Inorg. Chem. 2010, 49, 8347–8350. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, W.-W.; Zheng, R.-H.; Shi, Q.; Zhong, Y.-W.; Yao, J. Electronic Coupling between Two Cyclometalated Ruthenium Centers Bridged by 1,3,6,8-Tetrakis(1-Butyl-1H-1,2,3-Triazol-4-yl)Pyrene. Inorg. Chem. 2011, 50, 7074–7079. [Google Scholar] [CrossRef]
- Zych, D.; Slodek, A.; Golba, S.; Krompiec, S. Cyclometalated Ruthenium, Osmium, and Iridium Complexes Bridged by an NCN-Pyrene-NCN Derivative—Synthesis and Comparison of Optical, Thermal, and Electrochemical Properties. Eur. J. Inorg. Chem. 2018, 2018, 1581–1588. [Google Scholar] [CrossRef]
- Zych, D. 1,3-Di(Hetero)Aryl-7-Substituted Pyrenes—An Undiscovered Area of Important Pyrene Derivatives. Multidiscip. Digit. Publ. Inst. Proc. 2019, 41, 28. [Google Scholar] [CrossRef] [Green Version]
- Cerón-Camacho, R.; Roque-Ramires, M.A.; Ryabov, A.D.; Le Lagadec, R. Cyclometalated Osmium Compounds and beyond: Synthesis, Properties, Applications. Molecules 2021, 26, 1563. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, T.; Ozawa, H.; Suzuki, T.; Nakabayashi, T.; Kanaizuka, K.; Haga, M. Photoresponsive Molecular Memory Films Composed of Sequentially Assembled Heterolayers Containing Ruthenium Complexes. Chem. Eur. J. 2016, 22, 1658–1667. [Google Scholar] [CrossRef]
- Haga, M.A.; Kobayashi, K.; Terada, K. Fabrication and Functions of Surface Nanomaterials Based on Multilayered or Nanoarrayed Assembly of Metal Complexes. Coord. Chem. Rev. 2007, 2007, 2688–2701. [Google Scholar] [CrossRef]
- Brunschwig, B.S.; Creutz, C.; Sutin, N. Optical Transitions of Symmetrical Mixed-Valence Systems in the Class II-III Transition Regime. Chem. Soc. Rev. 2002, 31, 168–184. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, D.M.; Keene, F.R. Current Trends and Future Challenges in the Experimental, Theoretical and Computational Analysis of Intervalence Charge Transfer (IVCT) Transitions. Chem. Soc. Rev. 2006, 35, 424–440. [Google Scholar] [CrossRef] [PubMed]
- Vogler, L.M.; Scott, B.; Brewer, K.J. Investigation of the Photochemical, Electrochemical, and Spectroelectrochemical Properties of an Iridium(III)/Ruthenium(II) Mixed-Metal Complex Bridged by 2,3,5,6-Tetrakis(2-Pyridyl)Pyrazine. Inorg. Chem. 1993, 32, 898–903. [Google Scholar] [CrossRef]
- Vogler, L.M.; Brewer, K.J. Building Block Approach to the Construction of Long-Lived Osmium(II) and Ruthenium(II) Multimetallic Complexes Incorporating the Tridentate Bridging Ligand 2,3,5,6-Tetrakis(2-Pyridyl)Pyrazine. Inorg. Chem. 1996, 35, 818–824. [Google Scholar] [CrossRef]
- Lee, J.D.; Vrana, L.M.; Bullock, E.R.; Brewer, K.J. A Tridentate-Bridged Ruthenium−Rhodium Complex as a Stereochemically Defined Light-Absorber−Electron-Acceptor Dyad. Inorg. Chem. 1998, 37, 3575–3580. [Google Scholar] [CrossRef]
- Zhao, S.; Arachchige, S.M.; Slebodnick, C.; Brewer, K.J. Synthesis and Study of the Spectroscopic and Redox Properties of RuII,PtII Mixed-Metal Complexes Bridged by 2,3,5,6-Tetrakis(2-Pyridyl)Pyrazine. Inorg. Chem. 2008, 47, 6144–6152. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.W.; Vrana, L.M.; Brewer, K.J. Using Spectroelectrochemistry to Probe the Light Absorbing Properties of Polymetallic Complexes Containing the Tridentate Bridging Ligand 2,3,5,6-Tetrakis(2-Pyridyl)Pyrazine. J. Organomet. Chem. 1998, 554, 29–40. [Google Scholar] [CrossRef]
- Nagashima, T.; Nakabayashi, T.; Suzuki, T.; Kanaizuka, K.; Ozawa, H.; Zhong, Y.-W.; Masaoka, S.; Sakai, K.; Haga, M. Tuning of Metal–Metal Interactions in Mixed-Valence States of Cyclometalated Dinuclear Ruthenium and Osmium Complexes Bearing Tetrapyridylpyrazine or -Benzene. Organometallics 2014, 33, 4893–4904. [Google Scholar] [CrossRef]
- Zych, D.; Slodek, A.; Pająk, M.; Krompiec, S.; Spólnik, G.; Danikiewicz, W. Mono- and Diruthenium, Symmetrical and Unsymmetrical Complexes Bridged by Pyrene Derivatives: Experimental and Theoretical Studies. Eur. J. Inorg. Chem. 2017, 2017, 3868–3877. [Google Scholar] [CrossRef]
- Yao, C.-J.; Nie, H.-J.; Yang, W.-W.; Yao, J.; Zhong, Y.-W. Combined Experimental and Computational Study of Pyren-2,7-Diyl-Bridged Diruthenium Complexes with Various Terminal Ligands. Inorg. Chem. 2015, 54, 4688–4698. [Google Scholar] [CrossRef] [PubMed]
- Zych, D.; Slodek, A.; Matuszczyk, D.; Golba, S. Comprehensive Study of Mononuclear Osmium Complexes with Various Pyrene Ligands. Eur. J. Inorg. Chem. 2018, 2018, 5117–5128. [Google Scholar] [CrossRef]
- Zych, D.; Slodek, A.; Frankowska, A. Is It Worthwhile to Deal with 1,3-Disubstituted Pyrene Derivatives?—Photophysical, Optical and Theoretical Study of Substitution Position Effect of Pyrenes Containing Tetrazole Groups. Comput. Mater. Sci. 2019, 165, 101–113. [Google Scholar] [CrossRef]
- Zych, D.; Slodek, A. Pyrene Derivatives with Two Types of Substituents at Positions 1-, 3-, 6-, and 8-—Fad or Necessity? RSC Adv. 2019, 9, 24015–24024. [Google Scholar] [CrossRef] [Green Version]
- Zych, D. Non-K Region Disubstituted Pyrenes (1,3-, 1,6- and 1,8-) by (Hetero)Aryl Groups—Review. Molecules 2019, 24, 2551. [Google Scholar] [CrossRef] [Green Version]
- Zych, D.; Slodek, A. Double NCN-Cyclometalating Pyrene Derivatives with Two Kinds of Substituents—Experimental and Theoretical Investigations. J. Mol. Struct. 2019, 1202, 127282. [Google Scholar] [CrossRef]
- Wolfgang, K.; Klein, A.; Glöckle, M. Exploration of Mixed-Valence Chemistry: Inventing New Analogues of the Creutz-Taube Ion. Acc. Chem. Res. 2000, 33, 755–763. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.E.A.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Scalmani, G.; Frisch, M.J. Continuous Surface Charge Polarizable Continuum Models of Solvation. I. General Formalism. J. Chem. Phys. 2010, 132, 114110. [Google Scholar] [CrossRef] [PubMed]
- O’boyle, N.M.; Tenderholt, A.L.; Langner, K.M. Cclib: A Library for Package-Independent Computational Chemistry Algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef]
- Yao, C.-J.; Zhong, Y.-W.; Yao, J. Five-Stage Near-Infrared Electrochromism in Electropolymerized Films Composed of Alternating Cyclometalated Bisruthenium and Bis-Triarylamine Segments. Inorg. Chem. 2013, 52, 10000–10008. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HOMO | LUMO | HOMO | LUMO | ||
|---|---|---|---|---|---|
| 1a |  |  | 1b |  |  |
| 2a | 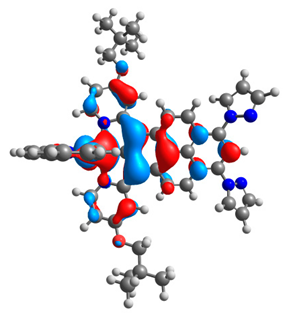 |  | 2b | 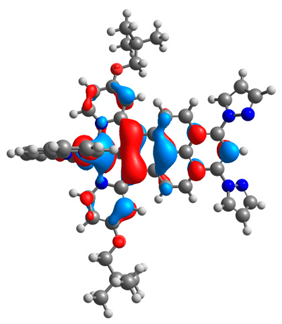 | 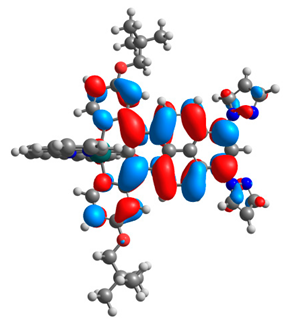 |
| 3a | 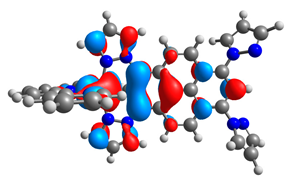 | 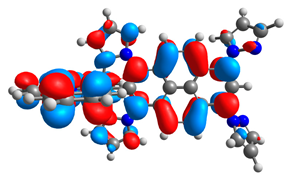 | 3b | 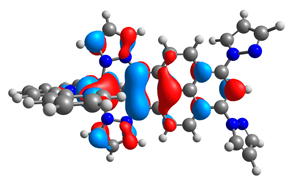 | 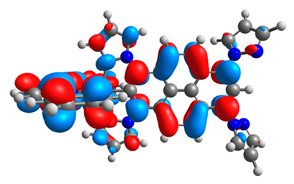 |
| 4a | 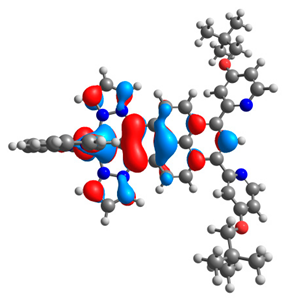 | 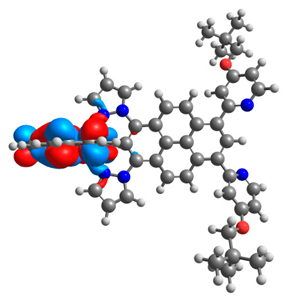 | 4b | 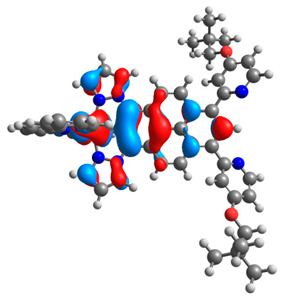 | 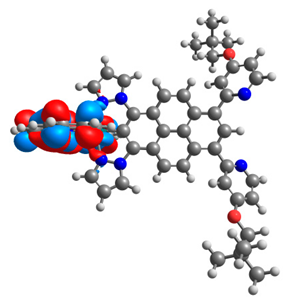 |
| 5a | 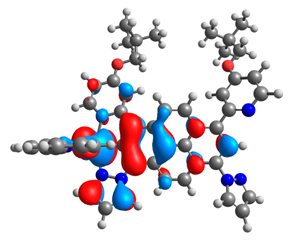 | 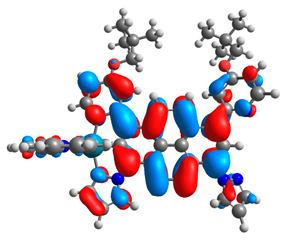 | 5b |  |  |
| 6a | 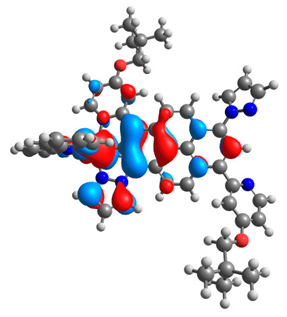 | 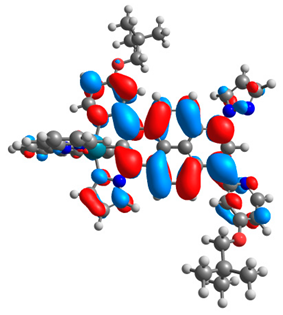 | 6b |  | 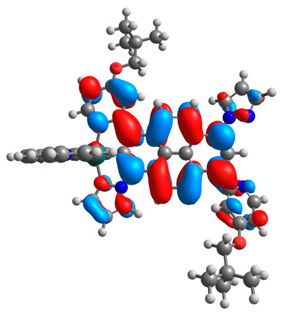 |
| 1a | 2a | 3a | 4a | 5a | 6a | 1b | 2b | 3b | 4b | 5b | 6b | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HOMO (eV) | −4.94 | −5.00 | −5.05 | −4.98 | −4.99 | −4.99 | −5.08 | −5.14 | −5.23 | −5.15 | −5.14 | −5.14 |
| LUMO (eV) | −2.43 | −2.47 | −2.31 | −2.23 | −2.37 | −2.38 | −2.41 | −2.47 | −2.30 | −2.24 | −2.36 | −2.36 |
| ΔE (eV) | 2.51 | 2.53 | 2.74 | 2.75 | 2.62 | 2.61 | 2.67 | 2.67 | 2.93 | 2.91 | 2.78 | 2.78 |
| M(II)-C (Å) | 1.994 | 1.992 | 1.998 | 2.000 | 1.995 | 1.995 | 1.974 | 1.972 | 1.980 | 1.981 | 1.976 | 1.976 |
| Calculated Wavelengths [nm] | Oscillator Strengths | Dominant Transitions (Contribution) | |
|---|---|---|---|
| 1a | 595.16 | 0.2499 | H-1→LUMO (93%) |
| 2a | 601.37 | 0.2030 | H-1→LUMO (95%) |
| 3a | 529.94 | 0.1365 | H-1→LUMO (37%), H-1→L + 1 (31%), H-1→L + 2 (19%) |
| 4a | 524.38 | 0.1993 | H-1→L + 1 (72%), H-1→L + 2 (16%) |
| 5a | 567.98 | 0.1843 | H-1→LUMO (80%), H-1→L + 2 (10%) |
| 6a | 568.11 | 0.1790 | H-1→LUMO (79%), H-1→L + 2 (10%) |
| 1b | 550.77 | 0.3083 | H-1→LUMO (84%), H-3→LUMO (10%) |
| 2b | 556.08 | 0.2317 | H-1→LUMO (88%) |
| 3b | 488.65 | 0.2036 | H-1→L + 1 (44%), H-1→LUMO (36%) |
| 4b | 485.74 | 0.3345 | H-1→L + 1 (66%), H-2→L + 1 (21%) |
| 5b | 524.27 | 0.2768 | H-1→LUMO (78%), H-3→LUMO (11%) |
| 6b | 524.56 | 0.2686 | H-1→LUMO (77%), H-3→LUMO (11%) |
| Hole (HOTO) | Electron (LUTO) | |
|---|---|---|
| 1a S4 2.083 eV (0.250) 97% | 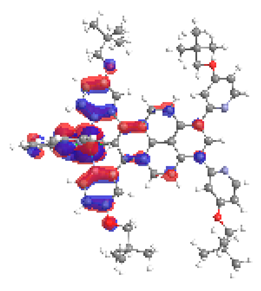 |  |
| 0.57/0.09/0.18/‒/0.00/‒/0.16 | 0.02/0.64/0.23/‒/0.08/‒/0.03 | |
| 2a S4 2.062 eV (0.203) 98% | 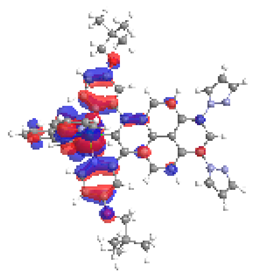 | 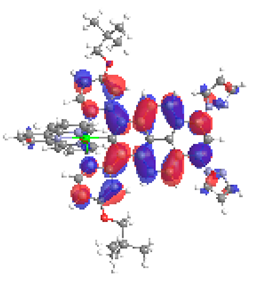 |
| 0.58/0.07/0.18/‒/‒/0.00/0.17 | 0.01/0.67/0.24/‒/‒/0.05/0.03 | |
| 3a S6 2.340 eV (0.137) 87% | 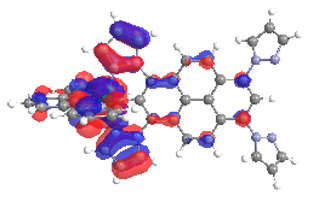 |  |
| 0.57/0.08/‒/0.19/‒/0.00/0.16 | 0.02/0.77/‒/0.15/‒0.05/0.01 | |
| 4a S6 2.364 eV (0.199) 88% | 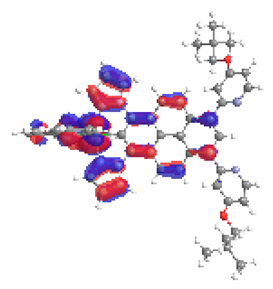 | 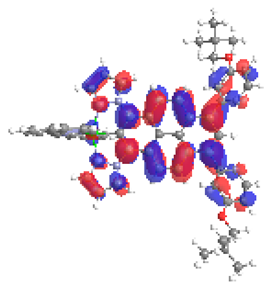 |
| 0.54/0.14/‒/0.17/0.00/‒/0.15 | 0.01/0.72/‒/0.15/0.12/‒/0.00 | |
| 5a S5 2.183 eV (0.184) 95% | 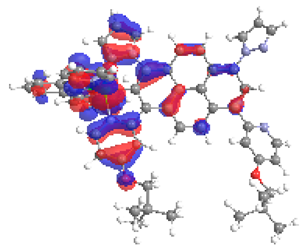 | 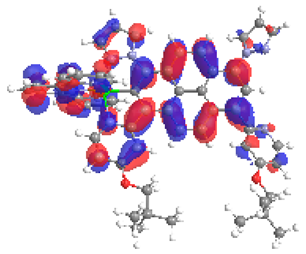 |
| 0.57/0.13/0.10/0.06/0.00/0.00/0.14 | 0.00/0.51/0.12/0.05/0.04/0.02/0.26 | |
| 6a S5 2.182 eV (0.179) 95% |  | 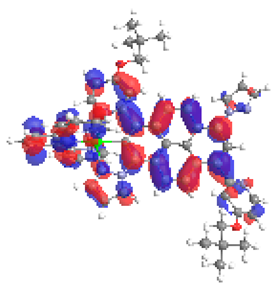 |
| 0.56/0.13/0.10/0.06/0.00/0.00/0.15 | 0.00/0.52/0.12/0.05/0.04/0.02/0.25 |
| Hole (HOTO) | Electron (LUTO) | |
|---|---|---|
| 1b S5 2.251 eV (0.308) 94% | 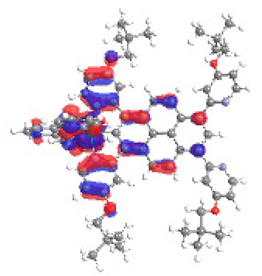 | 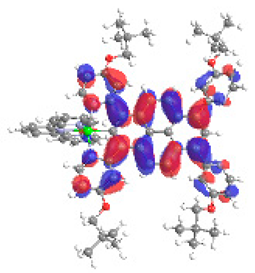 |
| 0.58/0.13/0.16/‒/0.00/‒/0.12 | 0.00/0.66/0.23/‒/0.09/‒/0.02 | |
| 2b S4 2.230 eV (0.232) 96% | 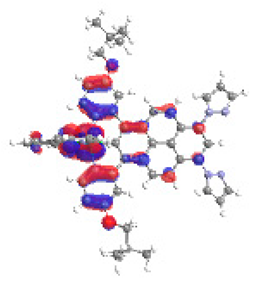 | 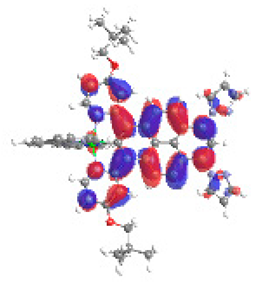 |
| 0.61/0.09/0.17/‒/‒/0.00/0.12 | 0.01/0.69/0.24/‒/‒/0.05/0.02 | |
| 3b S6 2.537 eV (0.204) 91% | 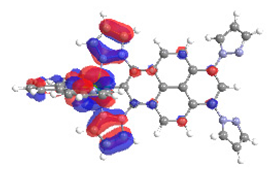 |  |
| 0.62/0.06/‒/0.18/‒/0.00/0.13 | 0.00/0.72/‒/0.14/‒0.05/0.08 | |
| 4b S6 2.553 eV (0.335) 88% |  | 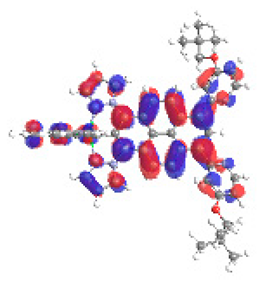 |
| 0.58/0.13/‒/0.17/0.01/‒/0.11 | 0.00/0.65/‒/0.14/0.10/‒/0.10 | |
| 5b S5 2.365 eV (0.277) 96% |  | 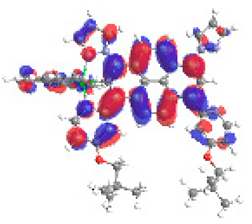 |
| 0.58/0.14/0.09/0.07/0.00/0.00/0.11 | 0.00/0.64/0.14/0.05/0.05/0.02/0.08 | |
| 6b S5 2.364 eV (0.269) 96% | 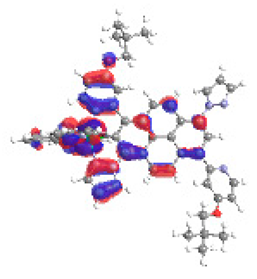 | 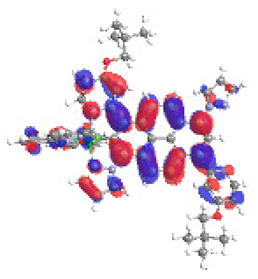 |
| 0.58/0.14/0.09/0.07/0.00/0.00/0.11 | 0.00/0.65/0.14/0.06/0.05/0.02/0.08 |
| 1a | 2a | 3a | 4a | 5a | 6a |
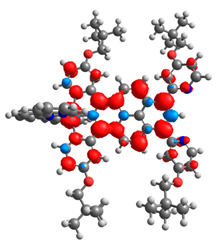 | 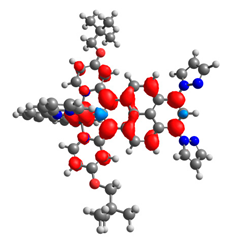 |  |  | 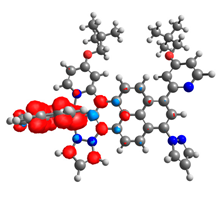 | 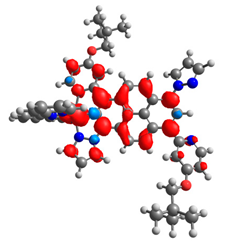 |
| 0.410 | 0.750 | 0.838 | 0.043 | 0.860 | 0.728 |
| 1b | 2b | 3b | 4b | 5b | 6b |
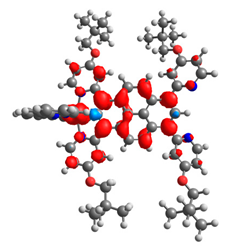 | 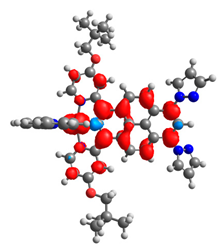 | 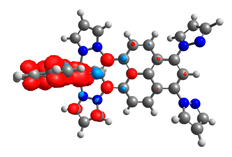 | 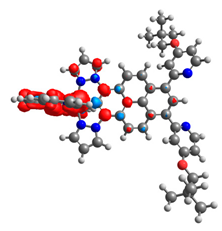 | 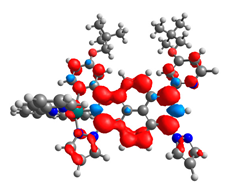 | 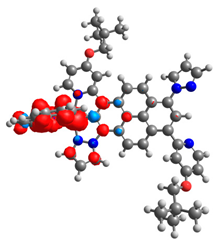 |
| 0.751 | 0.767 | 0.877 | 0.866 | 0.092 | 0.889 |
| 1aox | 2aox | 3aox | 4aox | 5aox | 6aox | 1box | 2box | 3box | 4box | 5box | 6box | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HOSO [eV] | α | −5.75 | −5.81 | −5.86 | −5.77 | −5.77 | −5.78 | −5.78 | −5.84 | −5.87 | −5.78 | −5.80 | −5.80 |
| β | −5.64 | −5.71 | −5.79 | −5.68 | −5.69 | −5.69 | −5.68 | −5.75 | −5.81 | −5.70 | −5.73 | −5.73 | |
| LUSO [eV] | α | −2.82 | −2.88 | −2.90 | −2.86 | −2.86 | −2.86 | −2.83 | −2.89 | −2.88 | −2.84 | −2.85 | −2.85 |
| β | −4.12 | −4.15 | −4.28 | −4.23 | −4.16 | −4.17 | −4.24 | −4.27 | −4.46 | −4.40 | −4.31 | −4.31 | |
| ΔE [eV] | α | 2.93 | 2.93 | 2.96 | 2.91 | 2.91 | 2.92 | 2.95 | 2.95 | 2.99 | 2.94 | 2.95 | 2.95 |
| β | 1.52 | 1.56 | 1.51 | 1.45 | 1.53 | 1.52 | 1.44 | 1.48 | 1.35 | 1.30 | 1.42 | 1.42 | |
| M-C [Å] | 1.975 | 1.981 | 1.980 | 1.970 | 1.982 | 1.982 | 1.951 | 1.957 | 1.953 | 1.946 | 1.958 | 1.957 | |
| Calculated Wavelengths (nm) | Oscillator Strengths | Dominant Transitions (Contribution) | |
|---|---|---|---|
| 1aox | 626.88 | 0.0811 | H-3(β)→LUSO(β) (64%), H-7(β)→LUSO(β) (11%) |
| 2aox | 601.78 | 0.0488 | H-3(β)→LUSO(β) (55%), H-5(β)→LUSO(β) (12%), H-7(β)→LUSO(β) (12%) |
| 3aox | 601.75 | 0.0785 | H-3(β)→LUSO(β) (47%), H-7(β)→LUSO(β) (27%) |
| 4aox | 628.12 | 0.1183 | H-5(β)→LUSO(β) (16%), H-3(β)→LUSO(β) (53%) |
| 5aox | 607.38 | 0.0594 | H-3(β)→LUSO(β) (58%), H-7(β)→LUSO(β) (15%) |
| 6aox | 607.08 | 0.0600 | H-3(β)→LUSO(β) (44%), H-4(β)→LUSO(β) (13%), H-6(β)→LUSO(β) (10%), H-7(β)→LUSO(β) (10%) |
| 1box | 662.66 | 0.0862 | H-3(β)→LUSO(β) (57%), HOSO(α)→LUSO(α) (11%) |
| 2box | 636.34 | 0.0592 | H-3(β)→LUSO(β) (49%), H-5(β)→LUSO(β) (13%), H-7(β)→LUSO(β) (10%) |
| 3box | 645.95 | 0.0616 | H-3(β)→LUSO(β) (32%), HOSO(α)→L + 1(α) (23%), H-7(β)→LUSO(β) (17%), HOSO(β)→L + 3(β) (11%) |
| 4box | 667.77 | 0.0540 | HOSO(α)→L + 1(α) (36%), HOSO(β)→L + 3(β) (24%), H-2(β)→LUSO(β) (24%) |
| 5box | 650.87 | 0.0683 | H-3(β)→LUSO(β) (48%), H-7(β)→LUSO(β) (12%) |
| 6box | 650.09 | 0.0687 | H-3(β)→LUSO(β) (33%), H-6(β)→LUSO(β) (14%), H-4(β)→LUSO(β) (13%) |
| HOMO | LUMO | HOMO | LUMO | ||
|---|---|---|---|---|---|
| 7a | 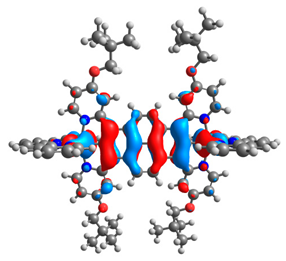 | 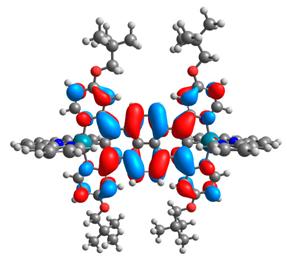 | 7b | 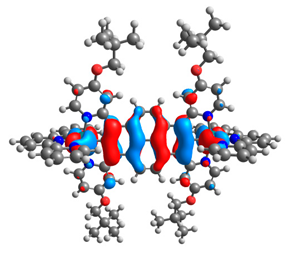 | 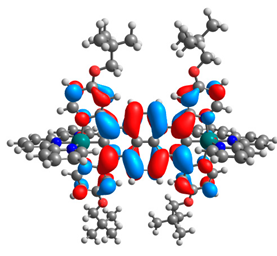 |
| 8a | 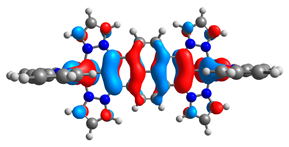 |  | 8b |  | 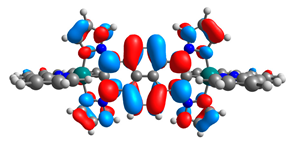 |
| 9a | 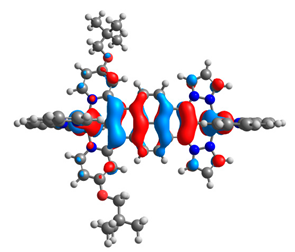 | 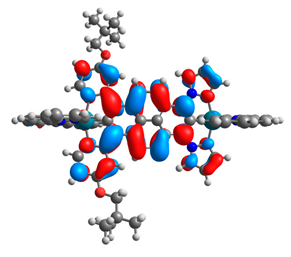 | 9b | 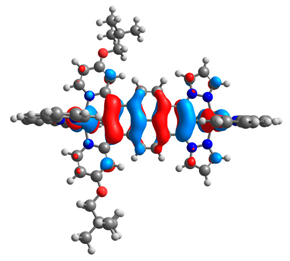 |  |
| 10a |  | 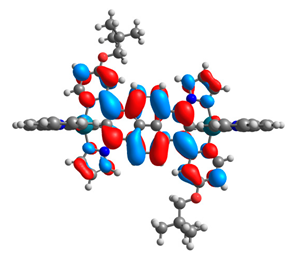 | 10b | 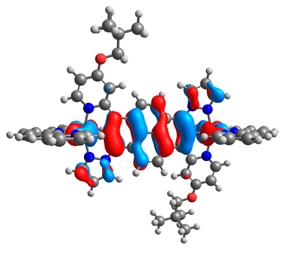 |  |
| 11a | 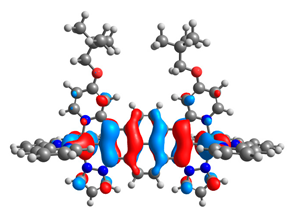 | 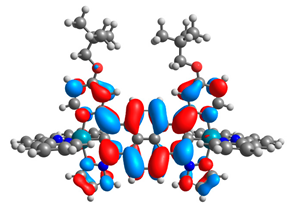 | 11b | 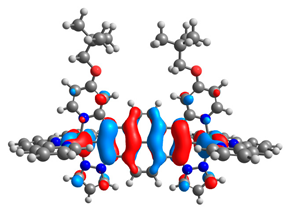 | 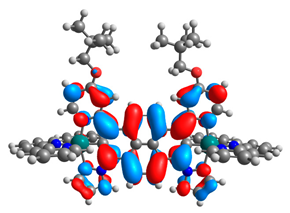 |
| 7a | 8a | 9a | 10a | 11a | 7b | 8b | 9b | 10b | 11b | |
|---|---|---|---|---|---|---|---|---|---|---|
| HOMO [eV] | −4.73 | −4.77 | −4.75 | −4.74 | −4.74 | −4.87 | −4.96 | −4.91 | −4.90 | −4.91 |
| LUMO [eV] | −2.67 | −2.31 | −2.51 | −2.53 | −2.51 | −2.64 | −2.28 | −2.48 | −2.50 | −2.49 |
| ΔE [eV] | 2.06 | 2.46 | 2.24 | 2.21 | 2.23 | 2.23 | 2.68 | 2.43 | 2.40 | 2.42 |
| M1-C/M2-C [Å] | 1.995/ 1.995 | 2.001/ 2.001 | 1.995/ 2.002 | 1.997/ 1.997 | 1.997/ 1.997 | 1.975/ 1.975 | 1.982/ 1.982 | 1.974/ 1.983 | 1.978/ 1.978 | 1.978/ 1.978 |
| Calculated Wavelengths (nm) | Oscillator Strengths | Dominant Transitions (Contribution) | |
|---|---|---|---|
| 7a | 771.43 | 0.1088 | HOMO→LUMO (99%) |
| 8a | 620.45 | 0.0652 | HOMO→LUMO (89%) |
| 9a | 696.97 | 0.0975 | HOMO→LUMO (97%) |
| 10a | 705.26 | 0.0882 | HOMO→LUMO (89%) |
| 11a | 699.02 | 0.0736 | HOMO→LUMO (77%), HOMO→L + 1 (11%) |
| 7b | 702.22 | 0.1203 | HOMO→LUMO (98%) |
| 8b | 562.93 | 0.0695 | HOMO→LUMO (85%) |
| 9b | 653.75 | 0.0574 | HOMO→LUMO (96%) |
| 10b | 641.54 | 0.0903 | HOMO→LUMO (78%), HOMO→L + 1 (14%) |
| 11b | 634.74 | 0.0849 | HOMO→LUMO (76%), HOMO→L + 2 (16%) |
| 7a | 8a | 9a | 10a | 11a |
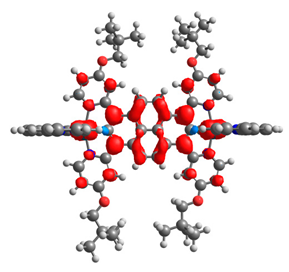 |  |  | 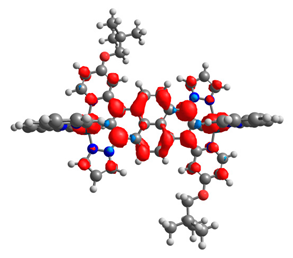 |  |
| 0.345/0.345 | 0.331/0.331 | 0.540/0.155 | 0.320/0.320 | 0.306/0.376 |
| 7b | 8b | 9b | 10b | 11b |
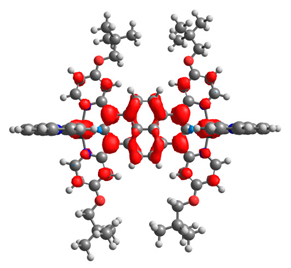 | 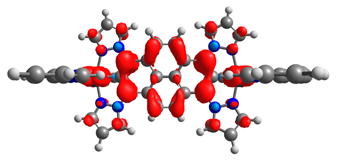 | 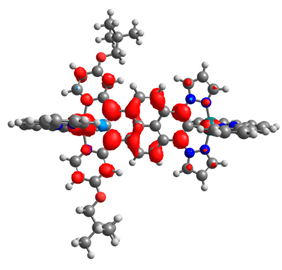 | 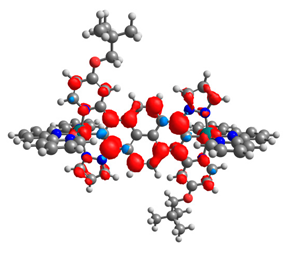 |  |
| 0.333/0.333 | 0.316/0.316 | 0.610/0.090 | 0.167/0.167 | 0.320/0.331 |
| β-HOSO | β-LUSO | β-HOSO | β-LUSO | ||
|---|---|---|---|---|---|
| 7aox |  | 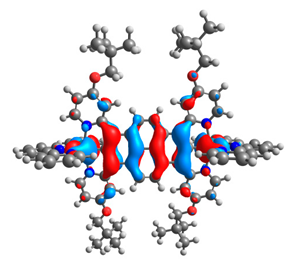 | 7box | 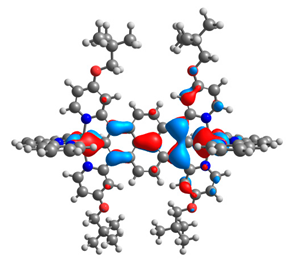 | 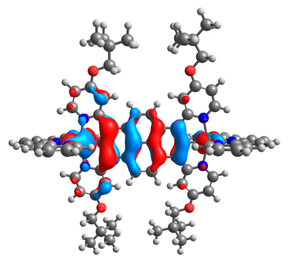 |
| 8aox |  |  | 8box | 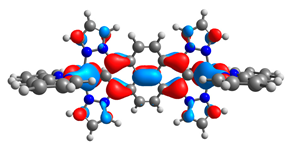 |  |
| 9aox |  |  | 9box | 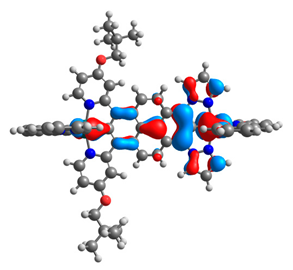 | 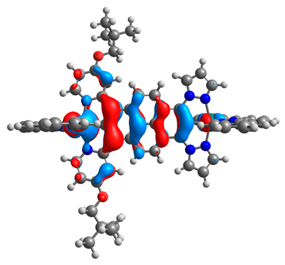 |
| 10aox | 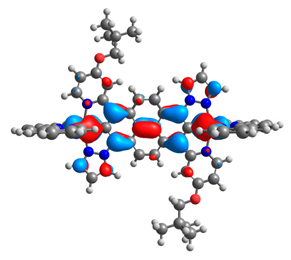 |  | 10box | 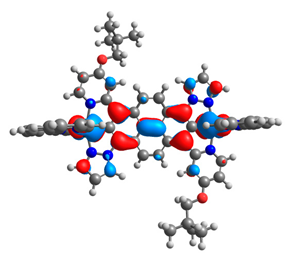 |  |
| 11aox | 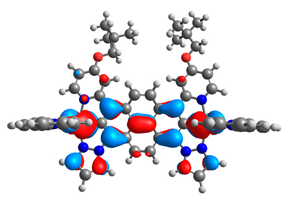 | 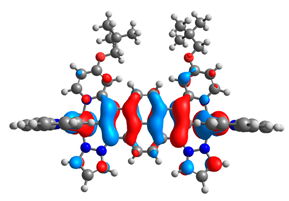 | 11box | 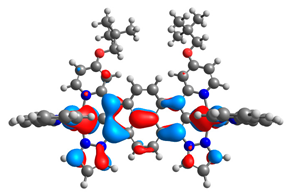 |  |
| 7aox | 8aox | 9aox | 10aox | 11aox | 7box | 8box | 9box | 10box | 11box | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HOSO [eV] | α | −5.35 | −5.45 | −5.31 | −5.39 | −5.38 | −5.47 | −5.70 | −5.46 | −5.61 | −5.57 |
| β | −5.13 | −5.22 | −5.20 | −5.19 | −5.17 | −5.28 | −5.39 | −5.39 | −5.34 | −5.32 | |
| LUSO [eV] | α | −3.02 | −2.70 | −2.88 | −2.90 | −2.88 | −3.02 | −2.71 | −2.88 | −2.90 | −2.88 |
| β | −4.13 | −4.20 | −4.13 | −4.16 | −4.16 | −4.25 | −4.40 | −4.25 | −4.33 | −4.32 | |
| ΔE [eV] | α | 2.33 | 2.75 | 2.43 | 2.49 | 2.50 | 2.45 | 2.99 | 2.58 | 2.71 | 2.69 |
| β | 1.00 | 1.02 | 1.07 | 1.03 | 1.01 | 1.03 | 0.99 | 1.14 | 1.01 | 1.00 | |
| M-C [Å] | 1.965/ 1.965 | 1.966/ 1.966 | 1.963/ 1.977 | 1.965/ 1.965 | 1.967/ 1.965 | 1.938/ 1.938 | 1.945/ 1.945 | 1.940/ 1.962 | 1.942/ 1.942 | 1.948/ 1.940 | |
| Calculated Wavelengths (nm) | Oscillator Strengths | Dominant Transitions (Contribution) | |
|---|---|---|---|
| 7aox | 2479.19 | 0.4520 | HOSO(β)→LUSO(β) (93%) |
| 8aox | 2172.11 | 0.5407 | HOSO(β)→LUSO(β) (96%) |
| 9aox | 1988.84 | 0.4308 | HOSO(β)→LUSO(β) (94%) |
| 10aox | 2444.48 | 0.2490 | HOSO(β)→LUSO(β) (61%), H-2(β)→LUSO(β) (36%) |
| 2082.02 | 0.2460 | H-2(β)→LUSO(β) (63%), HOSO(β)→LUSO(β) (34%) | |
| 11aox | 2289.64 | 0.4195 | HOSO(β)→LUSO(β) (83%), H-2(β)→LUSO(β) (13%) |
| 7box | 2265.79 | 0.1105 | H-5(β)→LUSO(β) (70%), HOSO(β)→LUSO(β) (27%) |
| 2189.37 | 0.2830 | HOSO(β)→LUSO(β) (69%), H-5(β)→LUSO(β) (27%) | |
| 8box | 2260.42 | 0.5043 | HOSO(β)→LUSO(β) (96%) |
| 9box | 1765.15 | 0.3460 | HOSO(β)→LUSO(β) (95%) |
| 10box | 2431.54 | 0.3392 | HOSO(β)→LUSO(β) (78%), H-2(β)→LUSO(β) (19%) |
| 2069.85 | 0.1275 | H-2(β)→LUSO(β) (79%), HOSO(β)→LUSO(β) (18%) | |
| 11box | 2295.15 | 0.4071 | HOSO(β)→LUSO(β) (92%) |
| HOMO-1 | HOMO | LUMO | LUMO+1 | |
|---|---|---|---|---|
| 12 | 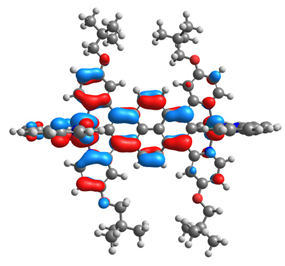 |  | 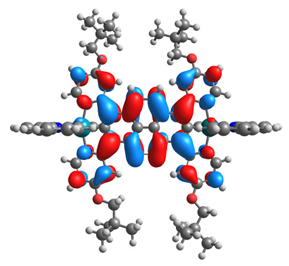 | 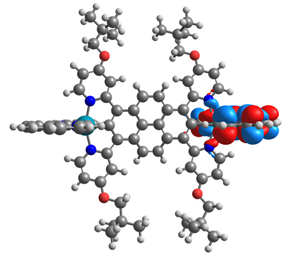 |
| 13 | 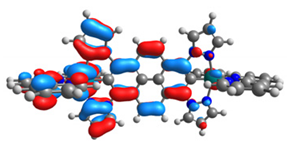 |  |  |  |
| 14 | 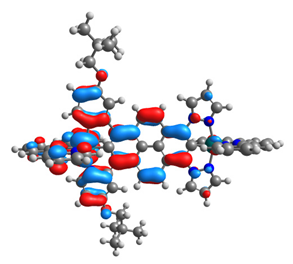 | 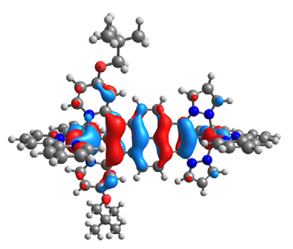 | 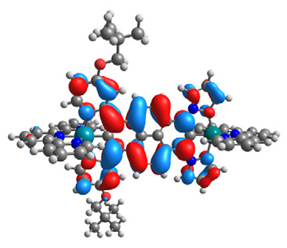 |  |
| 15 | 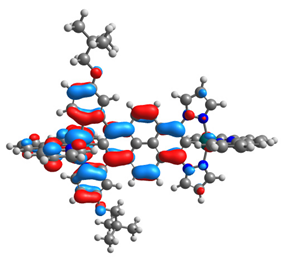 | 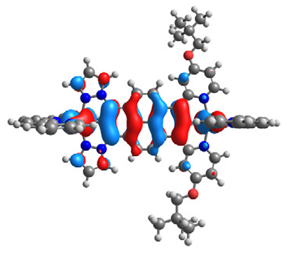 | 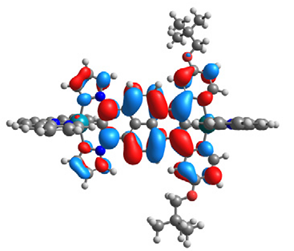 | 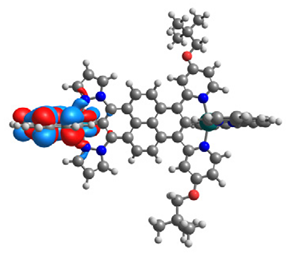 |
| 16 | 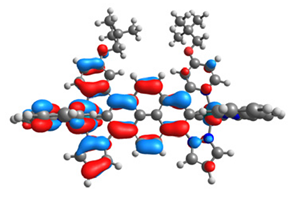 |  | 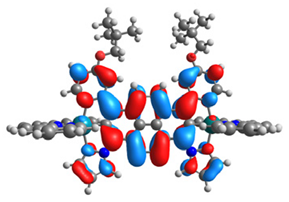 | 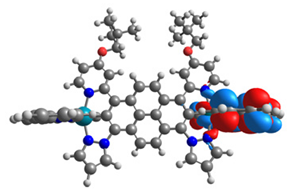 |
| 17 | 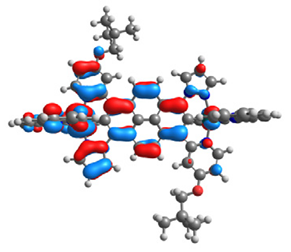 |  | 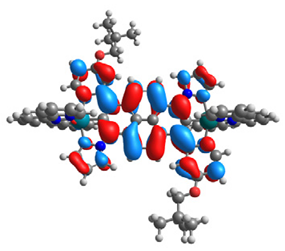 | 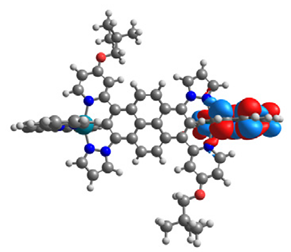 |
| 12 | 13 | 14 | 15 | 16 | 17 | |
|---|---|---|---|---|---|---|
| HOMO [eV] | −4.79 | −4.85 | −4.82 | −4.81 | −4.82 | −4.81 |
| LUMO [eV] | −2.66 | −2.30 | −2.50 | −2.49 | −2.50 | −2.52 |
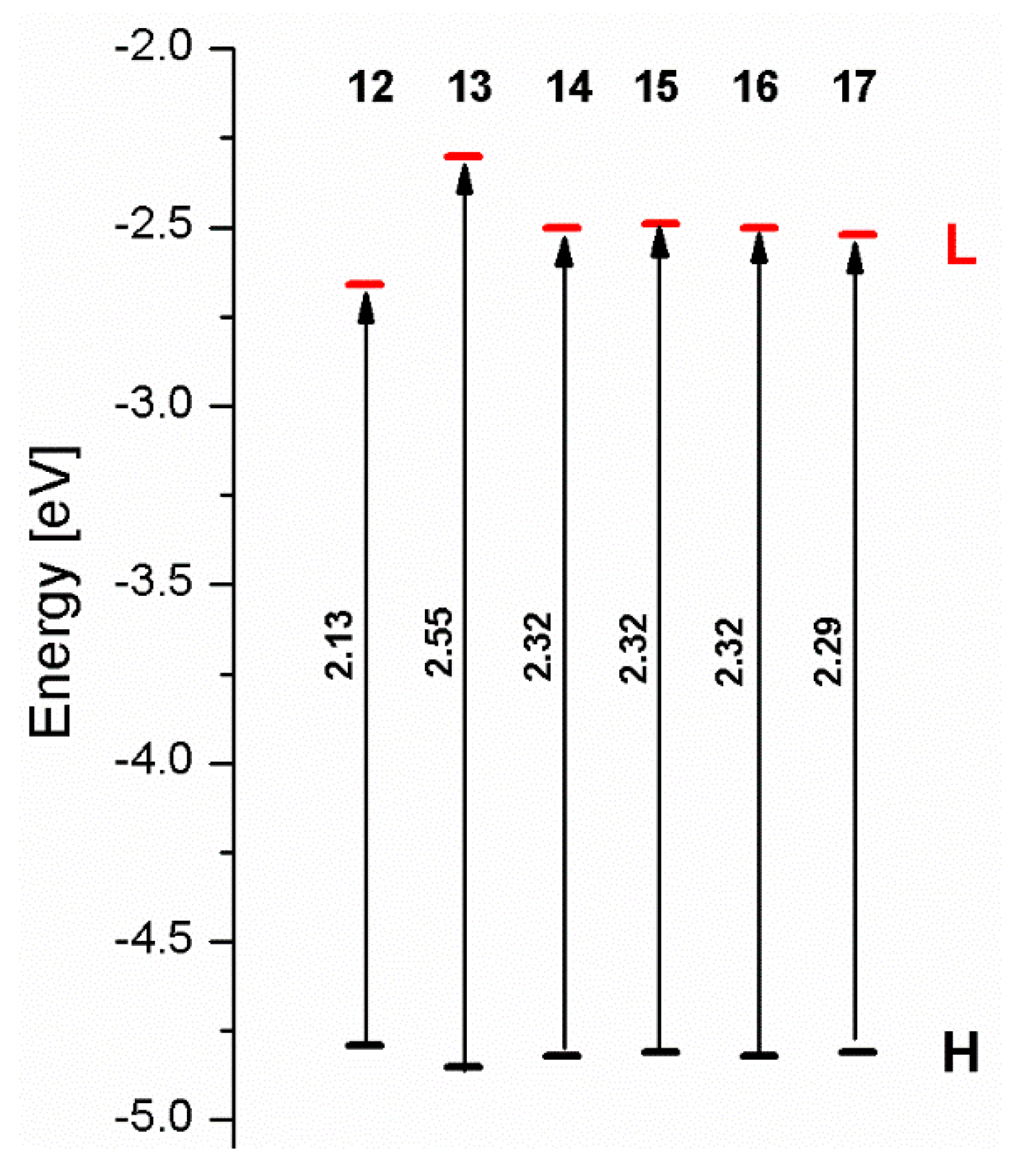
| ΔE [eV] | 2.13 | 2.55 | 2.32 | 2.32 | 2.32 | 2.29 |
| Os(II)-C [Å] | 1.995 | 2.001 | 1.994 | 2.002 | 1.997 | 1.997 |
| Ru(II)-C [Å] | 1.975 | 1.982 | 1.983 | 1.974 | 1.978 | 1.978 |
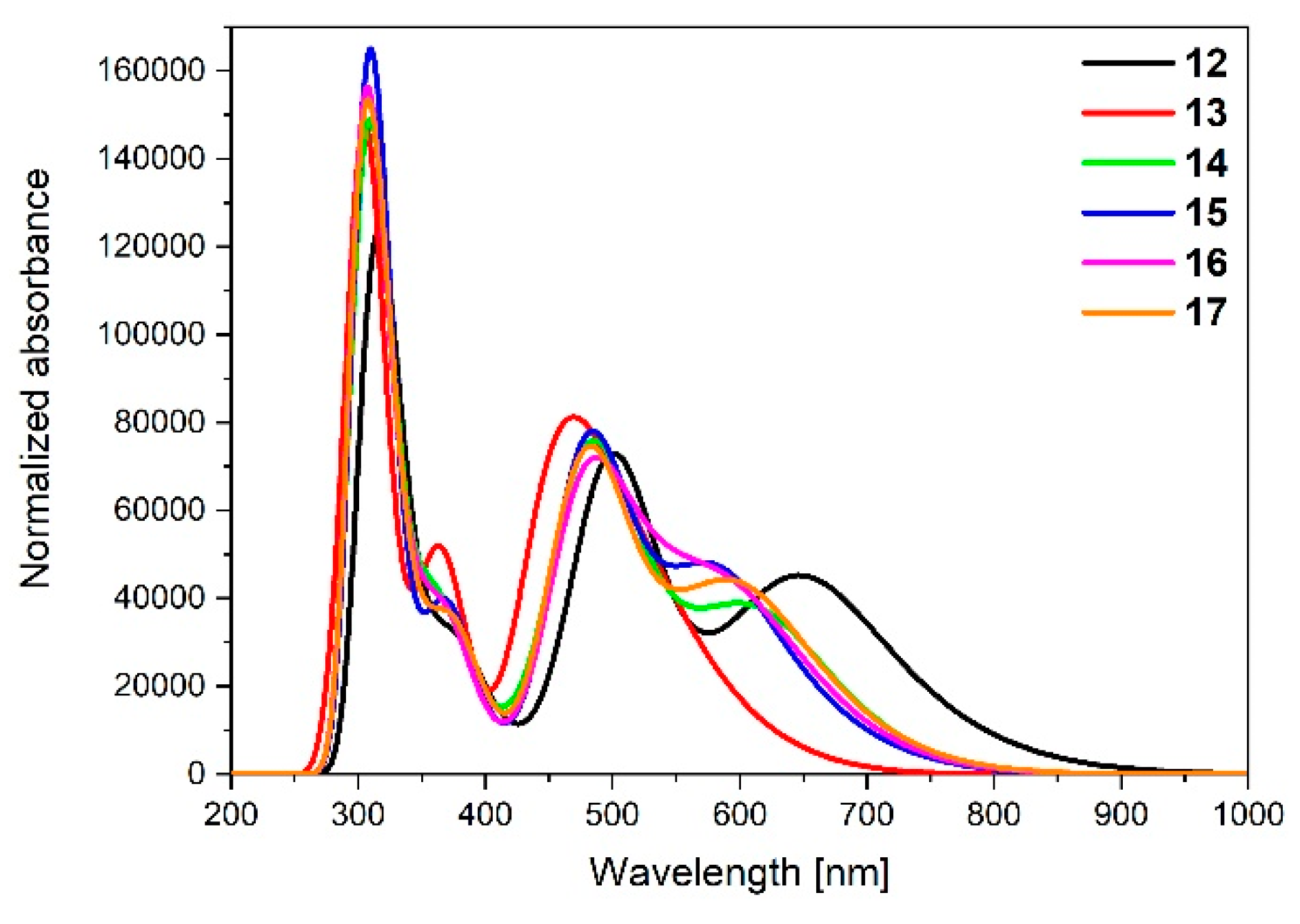
| Calculated Wavelengths (nm) | Oscillator Strengths | Dominant Transitions (Contribution) | |
|---|---|---|---|
| 12 | 739.63 | 0.1111 | HOMO→LUMO (98%) |
| 13 | 588.50 | 0.0647 | HOMO→LUMO (53%), HOMO→L + 4 (41%) |
| 14 | 673.24 | 0.0982 | HOMO→LUMO (97%) |
| 15 | 664.69 | 0.1011 | HOMO→LUMO (98%) |
| 16 | 670.55 | 0.0950 | HOMO→LUMO (97%) |
| 17 | 674.93 | 0.1012 | HOMO→LUMO (91%) |
| 12 | 13 | 14 | 15 | 16 | 17 |
|---|---|---|---|---|---|
 | 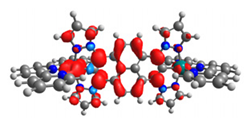 | 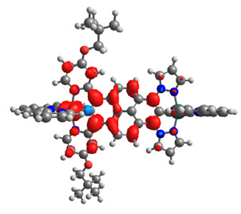 | 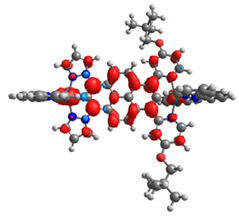 | 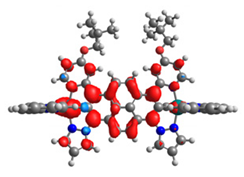 | 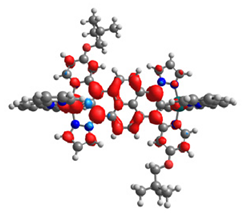 |
| 0.593/0.106 | 0.579/0.089 | 0.643/0.064 | 0.500/0.164 | 0.594/0.096 | 0.539/0.110 |
| 12ox | 13ox | ||
|---|---|---|---|
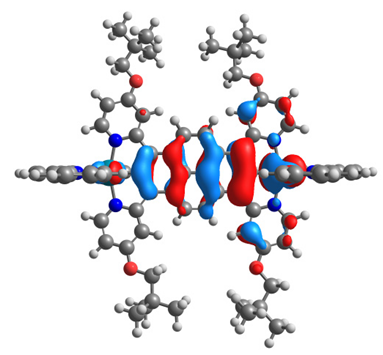 |  | 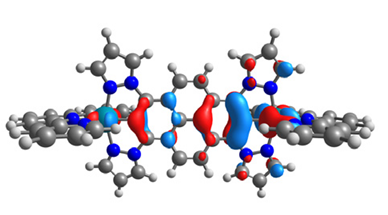 | 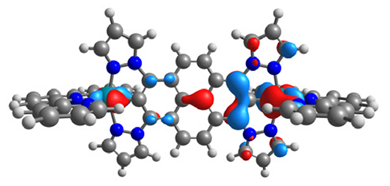 |
| α-HOSO | β-HOSO | α-HOSO | β-HOSO |
 |  |  | 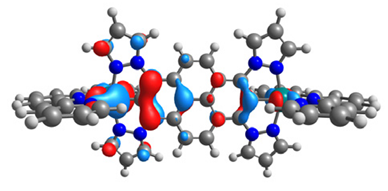 |
| α-LUSO | β-LUSO | α-LUSO | β-LUSO |
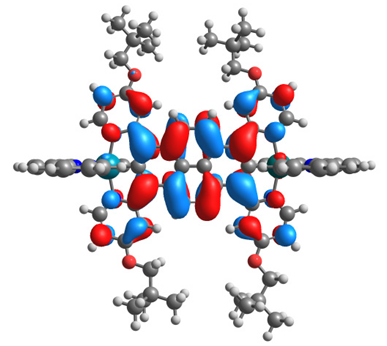
| 14ox | 15ox | ||
 |  | 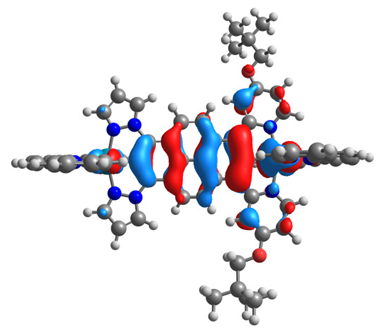 | 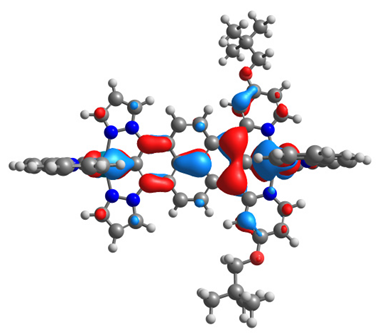 |
| α-HOSO | β-HOSO | α-HOSO | β-HOSO |
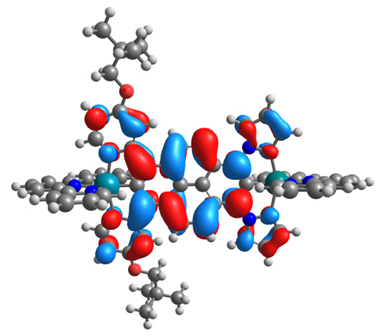 |  |  | 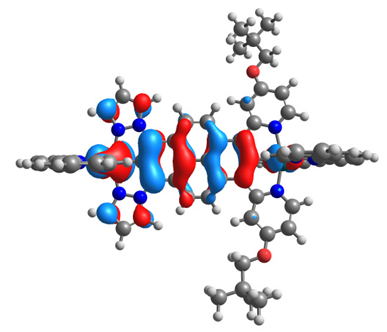 |
| α-LUSO | β-LUSO | α-LUSO | β-LUSO |
| 16ox | 17ox | ||
 |  | 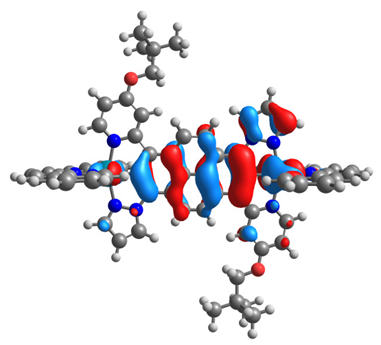 |  |
| α-HOSO | β-HOSO | α-HOSO | β-HOSO |
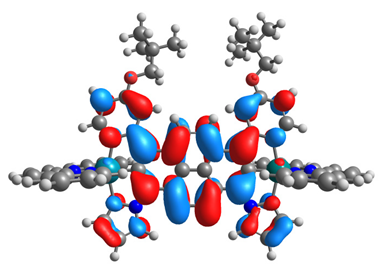 | 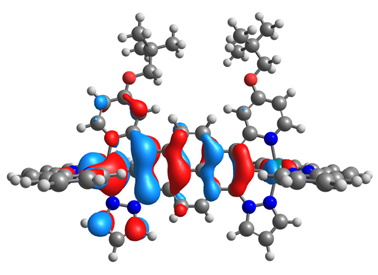 | 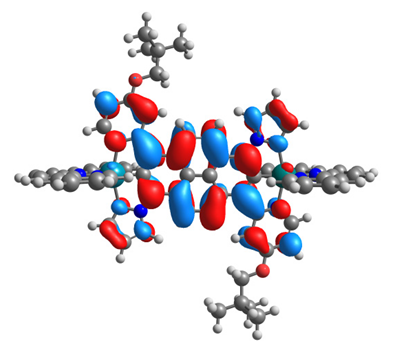 |  |
| α-LUSO | β-LUSO | α-LUSO | β-LUSO |
| 12ox | 13ox | 14ox | 15ox | 16ox | 17ox | ||
|---|---|---|---|---|---|---|---|
| HOSO [eV] | α | −5.32 | −5.45 | −5.38 | −5.40 | −5.37 | −5.38 |
| β | −5.31 | −5.41 | −5.39 | −5.31 | −5.34 | −5.36 | |
| LUSO [eV] | α | −3.01 | −2.80 | −2.88 | −2.87 | −2.88 | −2.90 |
| β | −4.11 | −4.22 | −4.12 | −4.20 | −4.16 | −4.16 | |
| ΔE [eV] | α | 2.31 | 2.65 | 2.50 | 2.53 | 2.49 | 2.48 |
| β | 1.20 | 1.19 | 1.27 | 1.11 | 1.18 | 1.20 | |
| Os-C [Å] | 1.965 | 1.965 | 1.967 | 1.965 | 1.966 | 1.964 | |
| Ru-C [Å] | 1.957 | 1.961 | 1.966 | 1.951 | 1.960 | 1.958 | |
| Calculated Wavelengths (nm) | Oscillator Strengths | Dominant Transitions (Contribution) | |
|---|---|---|---|
| 12ox | 1654.67 | 0.3155 | HOSO(β)→LUSO(β) (95%) |
| 13ox | 1618.17 | 0.3813 | HOSO(β)→LUSO(β) (97%) |
| 14ox | 1476.53 | 0.3011 | HOSO(β)→LUSO(β) (93%) |
| 15ox | 1876.27 | 0.1030 | H-1(β)→LUSO(β) (63%), HOSO(β)→LUSO(β) (26%) |
| 1873.72 | 0.2977 | HOSO(β)→LUSO(β) (70%), H-1(β)→LUSO(β) (23%) | |
| 16ox | 1668.25 | 0.2636 | HOSO(β)→LUSO(β) (75%), H-1(β)→LUSO(β) (19%) |
| 17ox | 1630.30 | 0.3009 | HOSO(β)→LUSO(β) (84%), H-1(β)→LUSO(β) (11%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zych, D. DFT/TD-DFT Framework of Mixed-Metal Complexes with Symmetrical and Unsymmetrical Bridging Ligands—Step-By-Step Investigations: Mononuclear, Dinuclear Homometallic, and Heterometallic for Optoelectronic Applications. Materials 2021, 14, 7783. https://doi.org/10.3390/ma14247783
Zych D. DFT/TD-DFT Framework of Mixed-Metal Complexes with Symmetrical and Unsymmetrical Bridging Ligands—Step-By-Step Investigations: Mononuclear, Dinuclear Homometallic, and Heterometallic for Optoelectronic Applications. Materials. 2021; 14(24):7783. https://doi.org/10.3390/ma14247783
Chicago/Turabian StyleZych, Dawid. 2021. "DFT/TD-DFT Framework of Mixed-Metal Complexes with Symmetrical and Unsymmetrical Bridging Ligands—Step-By-Step Investigations: Mononuclear, Dinuclear Homometallic, and Heterometallic for Optoelectronic Applications" Materials 14, no. 24: 7783. https://doi.org/10.3390/ma14247783
APA StyleZych, D. (2021). DFT/TD-DFT Framework of Mixed-Metal Complexes with Symmetrical and Unsymmetrical Bridging Ligands—Step-By-Step Investigations: Mononuclear, Dinuclear Homometallic, and Heterometallic for Optoelectronic Applications. Materials, 14(24), 7783. https://doi.org/10.3390/ma14247783