
Syntheses, Structures and Properties of Alkali and Alkaline Earth Metal Diamond-Like Compounds Li2MgMSe4 (M = Ge, Sn)

 ,
, 
Abstract
:1. Introduction
2. Experimental Sections
2.1. Chemical Syntheses
2.2. Single-Crystal X-ray Diffractions
2.3. Powder X-ray Diffraction (PXRD)
2.4. UV–Vis–NIR Diffuse Reflectance Spectroscopy
2.5. Raman Spectroscopy
2.6. Theoretical Calculations
3. Results and Discussion
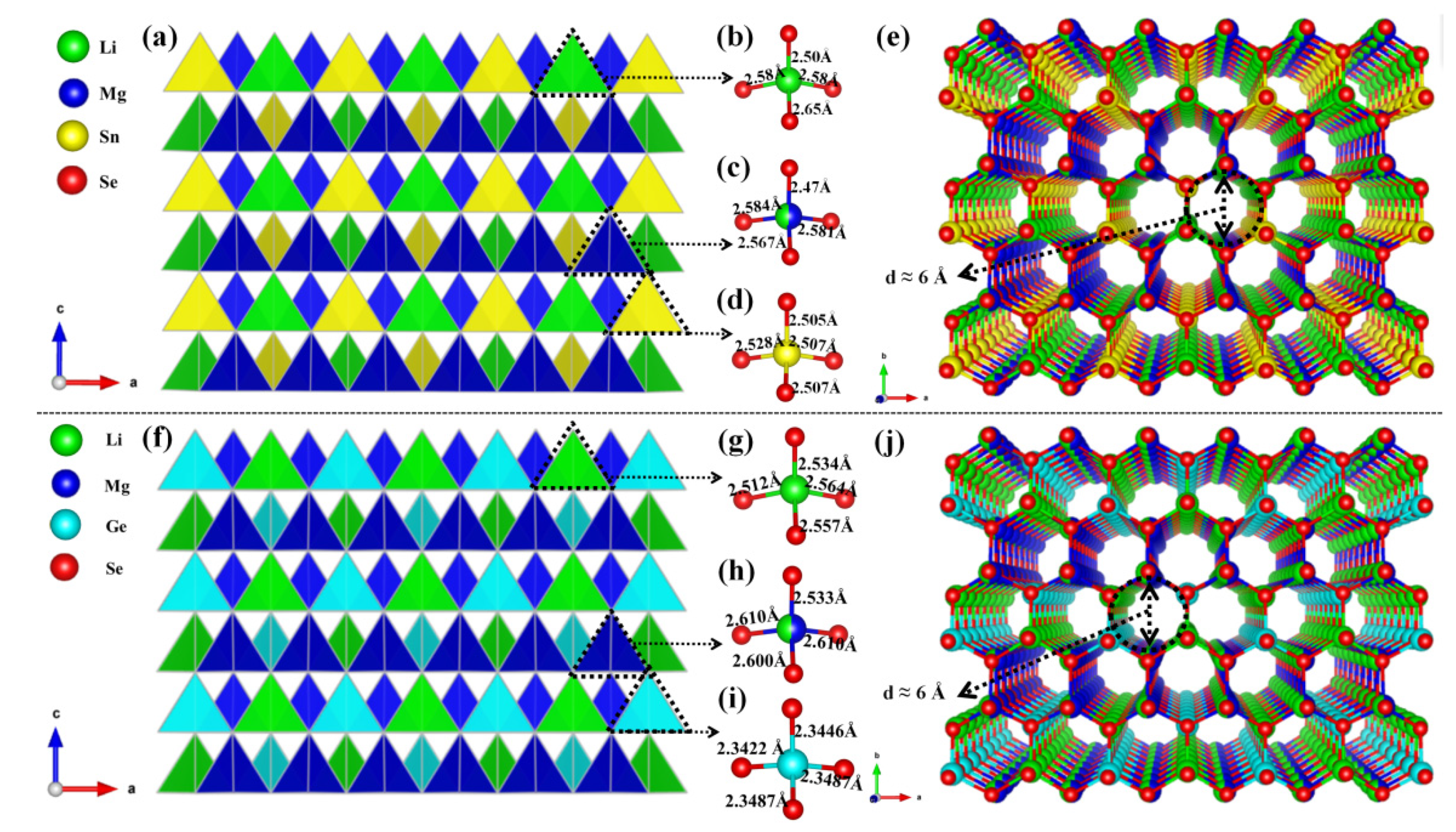
3.1. Crystal Structure
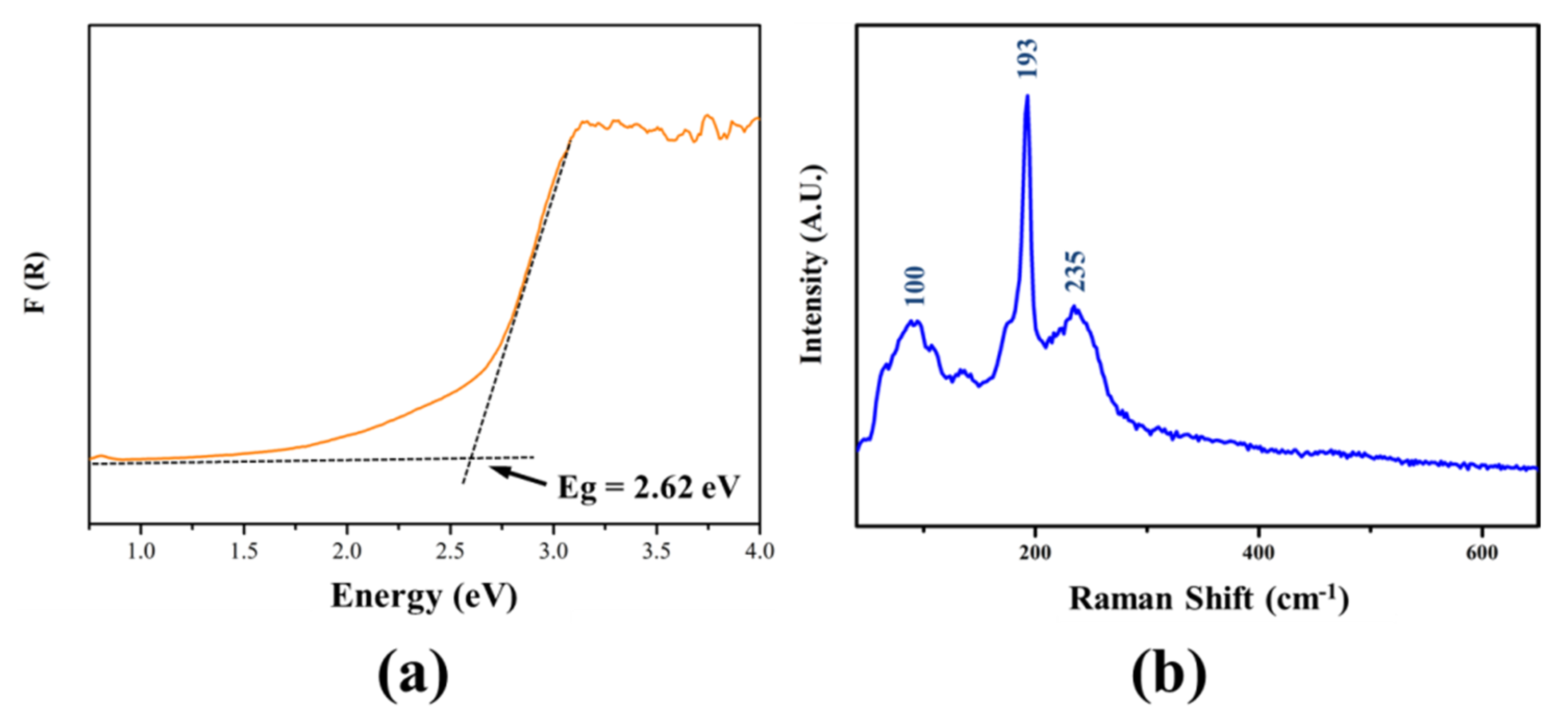
3.2. Optical Properties
3.3. Theoretical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Geng, L.; Meng, C.; Lu, H.; Luo, Z.; Lin, C.; Cheng, W. Bi2Te(IO3)O5Cl: A novel polar iodate oxychloride exhibiting a second-order nonlinear optical response. Dalton Trans. 2015, 44, 2469–2475. [Google Scholar] [CrossRef]
- Lu, X.; Chen, Z.; Shi, X.; Jing, Q.; Lee, M. Two Pyrophosphates with Large Birefringences and Second-Harmonic Responses as Ultraviolet Nonlinear Optical Materials. Angew. Chem. Int. Ed. 2020, 59, 17648–17656. [Google Scholar] [CrossRef]
- Liang, F.; Kang, L.; Lin, Z.; Wu, Y. Mid-Infrared Nonlinear Optical Materials Based on Metal Chalcogenides: Structure–Property Relationship. Cryst. Growth Des. 2017, 17, 2254–2289. [Google Scholar] [CrossRef]
- Kotb, H.; Khater, H.; Saber, O.; Ahmad, M. Sintering Temperature, Frequency, and Temperature Dependent Dielectric Properties of Na0.5Sm0.5Cu3Ti4O12 Ceramics. Materials 2021, 14, 4805. [Google Scholar] [CrossRef] [PubMed]
- Rong, L.; Xu, Z.; Sun, J.; Guo, G. New methyl formate synthesis method: Coal to methyl formate. J. Energy Chem. 2018, 27, 238–242. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Yang, G.; Humphrey, M.G.; Zhang, C. Recent Advances in Ultraviolet and Deep-Ultraviolet Second-Order Non-linear Optical Crystals. Coord. Chem. Rev. 2018, 375, 459–488. [Google Scholar] [CrossRef]
- Mutailipu, M.; Zhang, M.; Zhang, B.B.; Wang, L.Y.; Yang, Z.H.; Zhou, X.; Pan, S.L. SrB5O7F3 Functionalized with [B5O9F3]6− Chromophores: Accelerating the Rational Design of Deep-Ultraviolet Nonlinear Optical Materials. Angew. Chem. Int. Ed. 2018, 57, 6095–6099. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, B.; Yang, Z.; Pan, S. Cation-Tuned Synthesis of Fluorooxoborates: Towards Optimal Deep-Ultraviolet Nonlinear Optical Materials. Angew. Chem. Int. Ed. 2018, 57, 2150–2154. [Google Scholar] [CrossRef]
- Huang, Y.; Gao, L.; Yu, H.; Yang, Z.; Li, J.; Pan, S. Na6MQ4 (M = Zn, Cd; Q = S, Se): Promising New Ternary Infrared Nonlinear Optical Materials. Chem. Eur. J. 2021, 27, 6538–6544. [Google Scholar] [CrossRef]
- Xu, F.; Peng, G.; Lin, C.; Zhao, D.; Li, B.-X.; Zhang, G.; Yang, S.; Ye, N. Na3Sc2(PO4)2F3: Rational design and synthesis of an alkali rare-earth phosphate fluoride as an ultraviolet nonlinear optical crystal with an enlarged birefringence. J. Mater. Chem. C 2020, 8, 4965–4972. [Google Scholar] [CrossRef]
- Lee, H.; Ok, K.M. Na2Mg1-xZnxSiO4 (0 ≤ x ≤ 1): Noncentrosymmetric Sodium Metal Silicate Solid Solutions with Ultraviolet Nonlinear Optical Properties. Bull. Korean Chem. Soc. 2020, 41, 139–142. [Google Scholar] [CrossRef]
- Kee, J.; Ok, K.M. Hydrogen-Bond-Driven Synergistically Enhanced Hyperpolarizability: Chiral Coordination Polymers with Nonpolar Structure Exhibiting Unusually Strong Second-Harmonic Generation. Angew. Chem. Int. Ed. 2021, 60, 20656–20660. [Google Scholar] [CrossRef]
- Wu, K.; Yang, Z.; Pan, S. The first quaternary diamond-like semiconductor with 10-membered LiS4 rings exhibiting excellent nonlinear optical performances. Chem. Commun. 2017, 53, 3010–3013. [Google Scholar] [CrossRef]
- Abudurusuli, A.; Huang, J.; Wang, P.; Yang, Z.; Pan, S.; Li, J. Li4MgGe2S7: The First Alkali and Alkaline-Earth Diamond-Like Infrared Nonlinear Optical Material with Exceptional Large Band Gap. Angew. Chem. Int. Ed. 2021. [Google Scholar] [CrossRef]
- Lekse, J.W.; Moreau, M.A.; McNerny, K.L.; Yeon, J.; Halasyamani, S.; Aitken, J. Second-Harmonic Generation and Crystal Structure of the Diamond-like Semiconductors Li2CdGeS4 and Li2CdSnS4. Inorg. Chem. 2009, 48, 7516–7518. [Google Scholar] [CrossRef]
- Zhang, J.H.; Clark, D.J.; Weiland, A.; Stoyko, S.S.; Kim, Y.S.; Jang, J.I.; Aitken, J.A. Li2CdGeSe4 and Li2CdSnSe4: Biaxial Nonlinear Optical Materials with Strong Infrared Second-Order Responses and Laser-Induced Damage Thresholds Influenced by Photoluminescence. Inorg. Chem. Front. 2017, 4, 1472–1484. [Google Scholar] [CrossRef]
- Zhang, J.-H.; Clark, D.J.; Brant, J.A.; Sinagra, C.W.; Kim, Y.S.; Jang, J.I.; Aitken, J.A. Infrared nonlinear optical properties of lithium-containing diamond-like semiconductors Li2ZnGeSe4 and Li2ZnSnSe4. Dalton Trans. 2015, 44, 11212–11222. [Google Scholar] [CrossRef]
- Ritscher, A.; Hoelzel, M.; Lerch, M. The order-disorder transition in Cu2ZnSnS4-A neutron scattering investigation. J. Solid State Chem. 2016, 238, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Liang, F.; Kang, L.; Lin, Z.; Wu, Y.; Chen, C. Analysis and prediction of mid-IR nonlinear optical metal sulfides with diamond-like structures. Coord. Chem. Rev. 2017, 333, 57–70. [Google Scholar] [CrossRef]
- Kang, L.; Liang, F.; Jiang, X.; Lin, Z.; Chen, C. First-Principles Design and Simulations Promote the Development of Nonlinear Optical Crystals. Accounts Chem. Res. 2019, 53, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-M.; Xue, H.-G.; Guo, S.-P. Multinary metal chalcogenides with tetrahedral structures for second-order nonlinear optical, photocatalytic, and photovoltaic applications. Coord. Chem. Rev. 2018, 368, 115–133. [Google Scholar] [CrossRef]
- Pauling, L. The principles determining the structure of complex ionic crystals. J. Am. Chem. Soc. 1929, 51, 1010–1026. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, B.; Yu, H.; Hu, Z.; Wang, J.; Wu, Y.; Halasyamani, P.S. Designing Silicates as Deep-UV Nonlinear Optical (NLO) Materials using Edge-Sharing Tetrahedra. Angew. Chem. Int. Ed. 2020, 59, 8922–8926. [Google Scholar] [CrossRef]
- Zhang, J.-H.; Clark, D.J.; Brant, J.A.; Rosmus, K.A.; Grima, P.; Lekse, J.W.; Jang, J.I.; Aitken, J.A. α-Li2ZnGeS4: A Wide-Bandgap Diamond-like Semiconductor with Excellent Balance between Laser-Induced Damage Threshold and Second Harmonic Generation Response. Chem. Mater. 2020, 32, 8947–8955. [Google Scholar] [CrossRef]
- Brant, J.A.; Clark, D.J.; Kim, Y.S.; Jang, J.I.; Zhang, J.-H.; Aitken, J.A. Li2CdGeS4, A Diamond-Like Semiconductor with Strong Second-Order Optical Nonlinearity in the Infrared and Exceptional Laser Damage Threshold. Chem. Mater. 2014, 26, 3045–3048. [Google Scholar] [CrossRef]
- Li, G.; Chu, Y.; Zhou, Z. From AgGaS2 to Li2ZnSiS4: Realizing Impressive High Laser Damage Threshold Together with Large Second-Harmonic Generation Response. Chem. Mater. 2018, 30, 602–606. [Google Scholar] [CrossRef]
- Rosmus, K.A.; Brant, J.A.; Wisneski, S.D.; Clark, D.J.; Kim, Y.S.; Jang, J.I.; Brunetta, C.D.; Zhang, J.-H.; Srnec, M.N.; Aitken, J.A. Optical Nonlinearity in Cu2CdSnS4 and α/β-Cu2ZnSiS4: Diamond-like Semiconductors with High Laser-Damage Thresholds. Inorg. Chem. 2014, 53, 7809–7811. [Google Scholar] [CrossRef]
- Chen, J.; Chen, H.; Xu, F.; Cao, L.; Jiang, X.; Yang, S.; Sun, Y.; Zhao, X.; Lin, C.; Ye, N. Mg2In3Si2P7: A Quaternary Diamond-like Phosphide Infrared Nonlinear Optical Material Derived from ZnGeP2. J. Am. Chem. Soc. 2021, 143, 10309–10316. [Google Scholar] [CrossRef]
- SAINT, version 7.60A; Bruker Analytical X-ray Instruments Incorporated: Madison, WI, USA, 2008.
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Mei, D.J.; Cao, W.Z.; Wang, N.Z.; Jiang, X.X.; Zhao, J.; Wang, W.K.; Dang, J.H.; Zhang, S.Y.; Wu, Y.D.; Rao, P.H.; et al. Breaking Through the “3.0 eV wall” of Energy Band Gap in Mid-Infrared Nonlinear Optical Rare Earth Chalcogenides by Charge-Transfer Engineering. Mater. Horiz. 2021, 8, 2330–2334. [Google Scholar] [CrossRef]
- Jiang, J.Q.; Mei, D.J.; Gong, P.F.; Lin, Z.S.; Zhong, J.B.; Wu, Y.D. Wide Band Gap Design of New Chalcogenide Compounds: KSrPS4 and CsBaAsS4. RSC Adv. 2017, 7, 38044–38051. [Google Scholar] [CrossRef] [Green Version]
- Larson, A.C.; Dreele, R.B.V. General Structure Analysis System (GSAS); Los Alamos National Laboratory, University of California: Oakland, CA, USA, 2004; pp. 86–748. [Google Scholar]
- Kortüm, G. Reflectance Spectroscopy; Springer: Berlin, Germany, 1969. [Google Scholar]
- Tauc, J. Absorption edge and internal electric fields in amorphous semiconductors. Mater. Res. Bull. 1970, 5, 721–729. [Google Scholar] [CrossRef]
- Clark, S.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Ceperley, D.; Alder, B.J. Ground State of the Electron Gas by a Stochastic Method. Phys. Rev. Lett. 1980, 45, 566–569. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048–5079. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Bellaiche, L.; Vanderbilt, D. Virtual crystal approximation revisited: Application to dielectric and piezoelectric properties of perovskites. Phys. Rev. B 2000, 61, 7877–7882. [Google Scholar] [CrossRef] [Green Version]
- Winkler, B.; Pickard, C.; Milman, V. Applicability of a quantum mechanical virtual crystal approximation’ to study Al/Si-disorder. Chem. Phys. Lett. 2002, 362, 266–270. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.W.; Zhang, M.J.; Zhao, Z.Y.; Zeng, H.Y.; Zheng, F.K.; Guo, G.C.; Huang, J.S. Synthesis, Structure, and Optical Properties of the Quaternary Diamond-Like Compounds I2-II-IV-VI4 (I = Cu; II = Mg; IV = Si, Ge; VI = S, Se). J. Solid State Chem. 2013, 204, 251–256. [Google Scholar] [CrossRef]
- Chen, J.; Hu, C.L.; Mao, F.F.; Zhang, X.H.; Yang, B.P.; Mao, J.G. LiMg(IO3)3: An Excellent SHG Material Designed by Single-Site Aliovalent Substitution. Chem. Sci. 2019, 10, 10870–10875. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhang, M.; Pan, S.L.; Wang, Y.; Zhang, H.; Chen, Z.H. Li0.8Mg2.1B2O5F: The First Borate Fluoride with Magnesium-Oxygen-Fluorine Octahedral Chains. Dalton Trans. 2014, 43, 2828–2834. [Google Scholar] [CrossRef] [PubMed]
- Abudurusuli, A.; Li, J.J.; Tong, T.H.; Yang, Z.H.; Pan, S.L. LiBa4Ga5Q12 (Q = S, Se): Noncentrosymmetric Metal Chalcogenides with a Cesium Chloride Topological Structure Displaying a Remarkable Laser Damage Threshold. Inorg. Chem. 2020, 59, 5674–5682. [Google Scholar] [CrossRef]
- Abudurusuli, A.; Wu, K.; Pan, S.L. Four New Quaternary Chalcogenides A2Ba7Sn4Q16 (A = Li, Na; Q = S, Se): Syntheses, Crystal Structures Determination, Nonlinear Optical Performances Investigation. New J. Chem. 2018, 42, 3350–3355. [Google Scholar] [CrossRef]
- Wu, K.; Su, X.; Yang, Z.; Pan, S. An investigation of new infrared nonlinear optical material: BaCdSnSe4, and three new related centrosymmetric compounds: Ba2SnSe4, Mg2GeSe4, and Ba2Ge2S6. Dalton Trans. 2015, 44, 19856–19864. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Pan, S.L.; Yang, Z.H. Ba2GeS4 and Mg2SnS4: Synthesis, Structures, Optical Properties and Electronic Structures. RSC Adv. 2015, 5, 33646–33652. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical Formula | Li2MgSnSe4 | Li2MgGeSe4 |
|---|---|---|
| Formula weight | 472.72 g/mol | 426.62 g/mol |
| Temperature | 296.15 K | 153 (2) K |
| Crystal system | Orthorhombic | Orthorhombic |
| Space group | Pmn21 (No. 31) | Pmn21 (No. 31) |
| Unit cell dimensions | a = 8.402 (14) Å b = 7.181 (12) Å c = 6.728 (11) Å | a = 8.2961 (7) Å b = 7.0069 (5) Å c = 6.6116 (6) Å |
| Volume | 405.9 (12) Å3 | 384.33 (5) Å3 |
| Z | 2 | 2 |
| Calculated density | 3.867 g/cm3 | 3.686 g/cm3 |
| Absorption coefficient | 21.047 mm−1 | 22.891 mm−1 |
| Goodness-of-fit on F2 | 0.993 | 1.110 |
| Final R indices [Fo2 > 2σ(Fo2)] [a] | R1 = 0.0349; wR2 = 0.0750 | R1 = 0.0350; wR2 = 0.0783 |
| R indices | R1 = 0.0401; wR2 = 0.0784 | R1 = 0.0413; wR2 = 0.0831 |
| Largest diff. peak and hole | 2.08 e·A−3 and −0.93 e·A−3 | 1.55 e·A−3 and −2.46 e·A−3 |
| Compound | Eg (cal./eV) | d15 (pm/v) | d24 (pm/v) | d33 (pm/v) | Δn@1064 nm |
|---|---|---|---|---|---|
| Li2MgSnSe4 | 2.42 | −4.68 | −5.81 | 12.19 | 0.011 |
| Li2MgGeSe4 | 2.4 | 5.53 | 7.14 | −14.77 | 0.012 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, H.; Zhang, K.; Abudurusuli, A.; Bai, C.; Yang, Z.; Lai, K.; Li, J.; Pan, S. Syntheses, Structures and Properties of Alkali and Alkaline Earth Metal Diamond-Like Compounds Li2MgMSe4 (M = Ge, Sn). Materials 2021, 14, 6166. https://doi.org/10.3390/ma14206166
Gao H, Zhang K, Abudurusuli A, Bai C, Yang Z, Lai K, Li J, Pan S. Syntheses, Structures and Properties of Alkali and Alkaline Earth Metal Diamond-Like Compounds Li2MgMSe4 (M = Ge, Sn). Materials. 2021; 14(20):6166. https://doi.org/10.3390/ma14206166
Chicago/Turabian StyleGao, Hongbo, Kewang Zhang, Ailijiang Abudurusuli, Chen Bai, Zhihua Yang, Kangrong Lai, Junjie Li, and Shilie Pan. 2021. "Syntheses, Structures and Properties of Alkali and Alkaline Earth Metal Diamond-Like Compounds Li2MgMSe4 (M = Ge, Sn)" Materials 14, no. 20: 6166. https://doi.org/10.3390/ma14206166
APA StyleGao, H., Zhang, K., Abudurusuli, A., Bai, C., Yang, Z., Lai, K., Li, J., & Pan, S. (2021). Syntheses, Structures and Properties of Alkali and Alkaline Earth Metal Diamond-Like Compounds Li2MgMSe4 (M = Ge, Sn). Materials, 14(20), 6166. https://doi.org/10.3390/ma14206166