
Photoresponsive Amide-Based Derivatives of Azobenzene-4,4′-Dicarboxylic Acid—Experimental and Theoretical Studies

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. General
2.2. Synthesis and Characterization of the Compounds
2.2.1. Compound L1
2.2.2. Compound L2
2.3. Theoretical Calculations
2.4. Photoisomerization Studies
2.5. Ligand–Ion Interactions Studies
2.6. Complexes Preparation for Spectroscopic Experiments
3. Results and Discussion
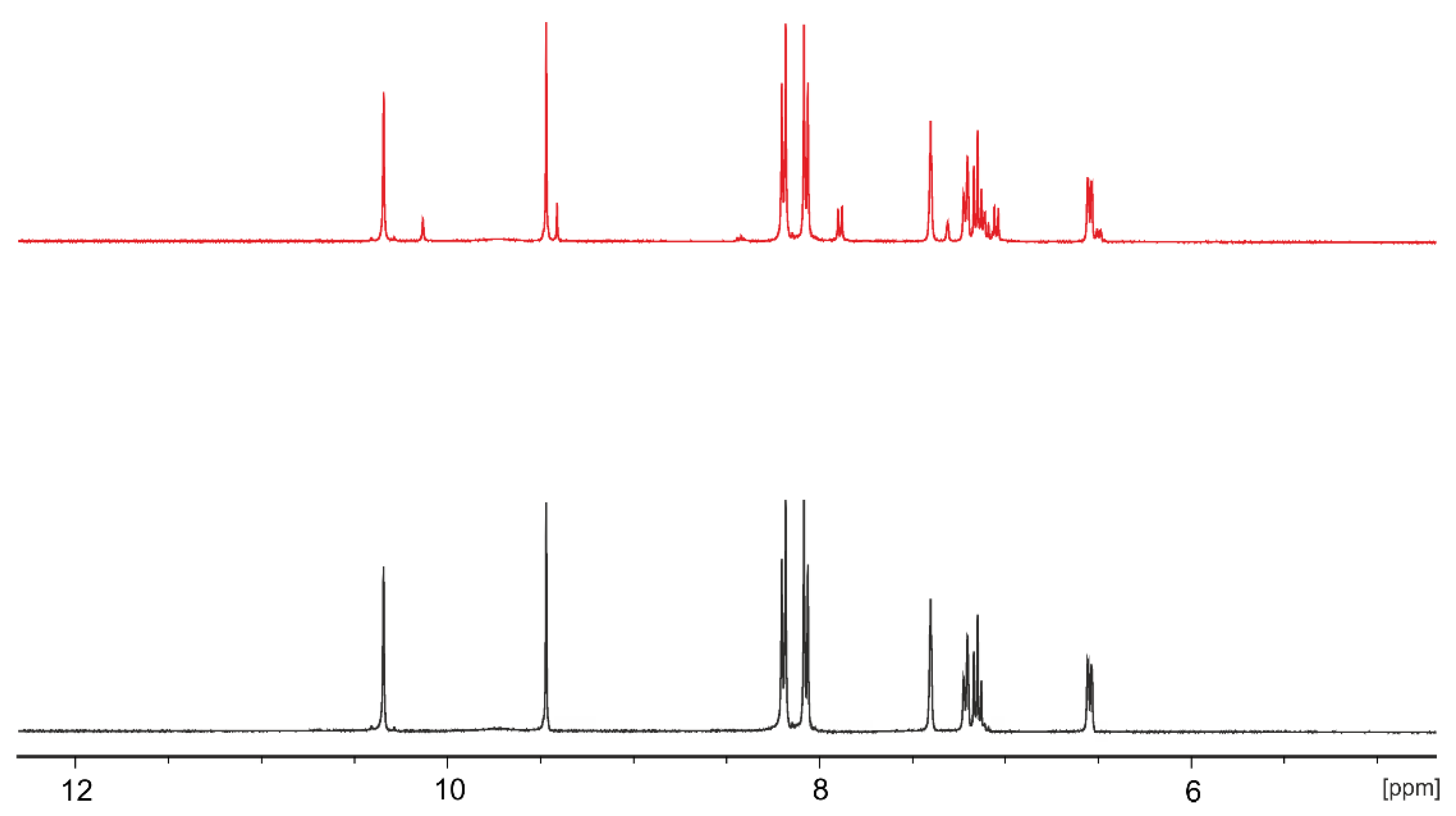
3.1. Synthesis and Characterization
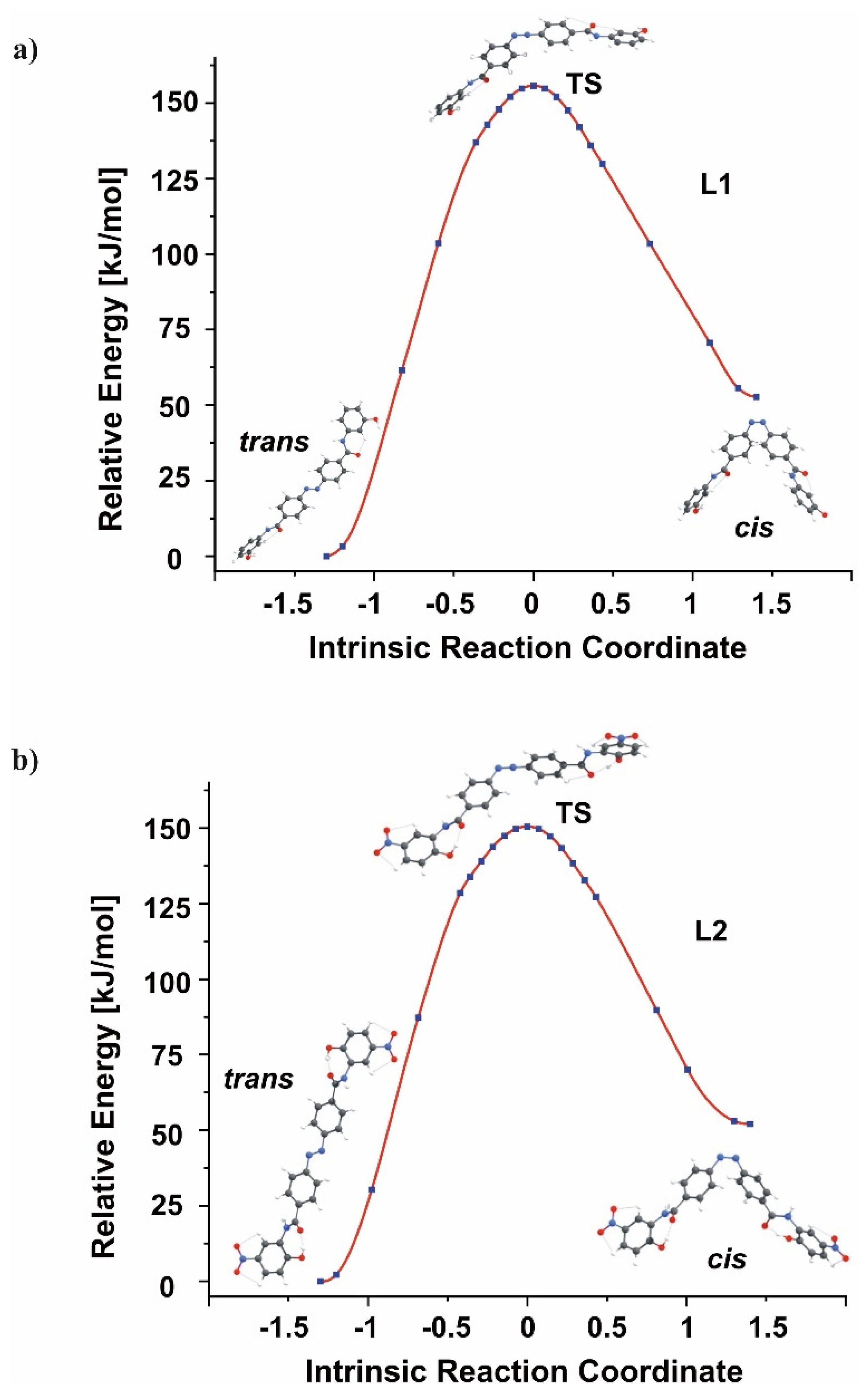
3.2. Isomerization Studies
3.3. Interactions with Ions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mahimwalla, Z.; Yager, K.G.; Mamiya, J.I.; Shishido, A.; Priimagi, A.; Barrett, C.J. Azobenzene Photomechanics: Prospects and Potential Applications. Polym. Bull. 2012, 69, 967–1006. [Google Scholar] [CrossRef]
- Wei, M.; Gao, Y.; Li, X.; Serpe, M.J. Stimuli-responsive polymers and their applications. Polym. Chem. 2017, 8, 127–143. [Google Scholar] [CrossRef] [Green Version]
- Manrique-Juárez, M.D.; Rat, S.; Salmon, L.; Molnár, G.; Quintero, C.M.; Nicu, L.; Shepherd, H.J.; Bousseksou, A. Switchable molecule-based materials for micro- and nanoscale actuating applications: Achievements and prospects. Coord. Chem. Rev. 2016, 308, 395–408. [Google Scholar] [CrossRef]
- Dumartin, M.; Lipke, M.C.; Stoddart, J.F. A Redox-Switchable Molecular Zipper. J. Am. Chem. Soc. 2019, 141, 18308–18317. [Google Scholar] [CrossRef] [PubMed]
- Tylkowski, B.; Trojanowska, A.; Marturano, V.; Nowak, M.; Marciniak, L.; Giamberini, M.; Ambrogi, V.; Cerruti, P. Power of light—Functional complexes based on azobenzene molecules. Coord. Chem. Rev. 2017, 351, 205–217. [Google Scholar] [CrossRef]
- Yamada, H.; Yagai, S. (Eds.) Light-Active Functional Organic Materials; Jenny Stanford Publishing: Singapore, 2019; ISBN 9789814800150. [Google Scholar]
- Zhang, Z.; Wang, W.; Jin, P.; Xue, J.; Sun, L.; Huang, J.; Zhang, J.; Tian, H. A building-block design for enhanced visible-light switching of diarylethenes. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakharov, A.V.; Yadykov, A.V.; Lvov, A.G.; Mitina, E.A.; Shirinian, V.Z. Photochemical rearrangement of diarylethenes: Synthesis of functionalized phenanthrenes. Org. Biomol. Chem. 2020, 18, 3098–3103. [Google Scholar] [CrossRef]
- Kortekaas, L.; Browne, W.R. The evolution of spiropyran: Fundamentals and progress of an extraordinarily versatile photochrome. Chem. Soc. Rev. 2019, 48, 3406–3424. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.A.; Kharbash, R.; Kim, Y. Chemo- and biosensing applications of spiropyran and its derivatives—A review. Anal. Chim. Acta 2020, 1110, 199–223. [Google Scholar] [CrossRef]
- Benkhaya, S.; M’rabet, S.; El Harfi, A. Classifications, properties, recent synthesis and applications of azo dyes. Heliyon 2020, 6. [Google Scholar] [CrossRef] [Green Version]
- Cardona, M.A.; Makuc, D.; Szacilowski, K.; Plavec, J.; Magri, D.C. Water-Soluble Colorimetric Amino[bis(ethanesulfonate)] Azobenzene pH Indicators: A UV-Vis Absorption, DFT, and 1H-15N NMR Spectroscopy Study. ACS Omega 2017, 2, 6159–6166. [Google Scholar] [CrossRef]
- García-Amorós, J.; Velasco, D. Recent advances towards azobenzene-based light-driven real-time information-transmitting materials. Beilstein J. Org. Chem. 2012, 8, 1003–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eom, T.; Yoo, W.; Kim, S.; Khan, A. Biologically activatable azobenzene polymers targeted at drug delivery and imaging applications. Biomaterials 2018, 185, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Begum, N.; Kaur, S.; Xiang, Y.; Yin, H.; Mohiuddin, G.; Rao, N.V.S.; Pal, S.K. Photoswitchable Bent-Core Nematic Liquid Crystals with Methylated Azobenzene Wing Exhibiting Optic-Field-Enhanced Fréedericksz Transition Effect. J. Phys. Chem. C 2019. [Google Scholar] [CrossRef]
- Jaunet-Lahary, T.; Chantzis, A.; Chen, K.J.; Laurent, A.D.; Jacquemin, D. Designing efficient azobenzene and azothiophene nonlinear optical photochromes. J. Phys. Chem. C 2014, 118, 28831–28841. [Google Scholar] [CrossRef]
- Mabhai, S.; Dolai, M.; Dey, S.; Dhara, A.; Das, B.; Jana, A. A novel chemosensor based on rhodamine and azobenzene moieties for selective detection of Al3+ ions. New J. Chem. 2018, 42, 10191–10201. [Google Scholar] [CrossRef]
- Liu, J.L.; Wada, S.; Wang, J.Y. Two azobenzene derivatives CAB/ACB as reusable sunscreen: UV absorptive capacity and biosafety evaluation. RSC Adv. 2018, 8, 13274–13283. [Google Scholar] [CrossRef] [Green Version]
- Vapaavuori, J.; Bazuin, C.G.; Priimagi, A. Supramolecular design principles for efficient photoresponsive polymer-azobenzene complexes. J. Mater. Chem. C 2018, 6, 2168–2188. [Google Scholar] [CrossRef] [Green Version]
- Merino, E. Synthesis of azobenzenes: The coloured pieces of molecular materials. Chem. Soc. Rev. 2011, 40, 3835–3853. [Google Scholar] [CrossRef]
- Katsonis, N.; Lubomska, M.; Pollard, M.M.; Feringa, B.L.; Rudolf, P. Synthetic light-activated molecular switches and motors on surfaces. Prog. Surf. Sci. 2007, 82, 407–434. [Google Scholar] [CrossRef]
- Beharry, A.A.; Woolley, G.A. Azobenzene photoswitches for biomolecules. Chem. Soc. Rev. 2011, 40, 4422–4437. [Google Scholar] [CrossRef]
- Barrett, C.; Natansohn, A.; Rochon, P. Cis-Trans Thermal Isomerization Rates of Bound and Doped Azobenzenes in a Series of Polymers. Chem. Mater. 1995, 7, 899–903. [Google Scholar] [CrossRef]
- Shibaev, V.; Bobrovsky, A.; Boiko, N. Photoactive liquid crystalline polymer systems with light-controllable structure and optical properties. Prog. Polym. Sci. 2003, 28, 729–836. [Google Scholar] [CrossRef]
- Bandara, H.M.D.; Burdette, S.C. Photoisomerization in different classes of azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. [Google Scholar] [CrossRef] [PubMed]
- Lubbe, A.S.; Szymanski, W.; Feringa, B.L. Recent developments in reversible photoregulation of oligonucleotide structure and function. Chem. Soc. Rev. 2017, 46, 1052–1079. [Google Scholar] [CrossRef]
- Ar, Y.; Kumar, S. Writing with Light: Recent Advances in Optical Storage Property of Azobenzene Derivatives. Gen. Chem. 2018, 4–9. [Google Scholar] [CrossRef]
- Nishioka, H.; Liang, X.; Kashida, H.; Asanuma, H. 2′,6′-Dimethylazobenzene as an efficient and thermo-stable photo-regulator for the photoregulation of DNA hybridization. Chem. Commun. 2007, 4354–4356. [Google Scholar] [CrossRef]
- Korbus, M.; Balasubramanian, G.; Müller-Plathe, F.; Kolmar, H.; Meyer-Almes, F.J. Azobenzene switch with a long-lived cis-state to photocontrol the enzyme activity of a histone deacetylase-like amidohydrolase. Biol. Chem. 2014, 395, 401–412. [Google Scholar] [CrossRef]
- Mourot, A.; Herold, C.; Kienzler, M.A.; Kramer, R.H. Understanding and improving photo-control of ion channels in nociceptors with azobenzene photo-switches. Br. J. Pharmacol. 2018, 175, 2296–2311. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Liang, S.; Xu, W.C.; Xu, G.; Wu, S. Photoresponsive polymers with multi-Azobenzene groups. Polym. Chem. 2019, 10, 4389–4401. [Google Scholar] [CrossRef] [Green Version]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of Ab initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Miertuš, S.; Tomasi, J. Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem. Phys. 1982, 65, 239–245. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Casida, M.E.; Jamorski, C.; Casida, K.C.; Salahub, D.R. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J. Chem. Phys. 1998, 108, 4439–4449. [Google Scholar] [CrossRef]
- Scalmani, G.; Frisch, M.J.; Mennucci, B.; Tomasi, J.; Cammi, R.; Barone, V. Geometries and properties of excited states in the gas phase and in solution: Theory and application of a time-dependent density functional theory polarizable continuum model. J. Chem. Phys. 2006, 124. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- BindFit v0.5. Available online: http://app.supramolecular.org/bindfit/ (accessed on 4 February 2021).
- Thordarson, P. Determining association constants from titration experiments in supramolecular chemistry. Chem. Soc. Rev. 2011, 40, 1305–1323. [Google Scholar] [CrossRef]
- Wiberg, K.B. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Wagner-Wysiecka, E.; Łukasik, N.; Biernat, J.F.; Luboch, E. Azo Group(s) in Selected Macrocyclic Compounds. J. Incl. Phenom. Macrocycl. Chem. 2018, 90, 189–257. [Google Scholar] [CrossRef] [Green Version]
- Heath, H. A new sensitive chemical actinometer—II. Potassium ferrioxalate as a standard chemical actinometer. Proc. R. Soc. Lond. Ser. A. Math. Phys. Sci. 1956, 235, 518–536. [Google Scholar] [CrossRef]
- Wezenberg, S.J.; Feringa, B.L. Photocontrol of Anion Binding Affinity to a Bis-urea Receptor Derived from Stiff-Stilbene. Org. Lett. 2017, 19, 324–327. [Google Scholar] [CrossRef] [Green Version]
- Dąbrowa, K.; Jurczak, J. Tetra-(meta-butylcarbamoyl)azobenzene: A Rationally Designed Photoswitch with Binding Affinity for Oxoanions in a Long-Lived Z-State. Org. Lett. 2017, 19, 1378–1381. [Google Scholar] [CrossRef]
- Dabrowa, K.; Niedbala, P.; Jurczak, J. Anion-tunable control of thermal Z → E isomerisation in basic azobenzene receptors. Chem. Commun. 2014, 50, 15748–15751. [Google Scholar] [CrossRef] [Green Version]
- Weston, C.E.; Richardson, R.D.; Haycock, P.R.; White, A.J.P.; Fuchter, M.J. Arylazopyrazoles: Azoheteroarene photoswitches offering quantitative isomerization and long thermal half-lives. J. Am. Chem. Soc. 2014, 136, 11878–11881. [Google Scholar] [CrossRef] [PubMed]
- Stricker, L.; Böckmann, M.; Kirse, T.M.; Doltsinis, N.L.; Ravoo, B.J. Arylazopyrazole Photoswitches in Aqueous Solution: Substituent Effects, Photophysical Properties, and Host–Guest Chemistry. Chem. Eur. J. 2018, 24, 8639–8647. [Google Scholar] [CrossRef]
- Łukasik, N.; Wagner-Wysiecka, E. Anion binding by p-aminoazobenzene-derived aromatic amides: Spectroscopic and electrochemical studies. Photochem. Photobiol. Sci. 2017, 16, 1570–1579. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Casas, C.; Yatsimirsky, A.K. Detailing hydrogen bonding and deprotonation equilibria between anions and urea/thiourea derivatives. J. Org. Chem. 2008, 73, 2275–2284. [Google Scholar] [CrossRef]
- Łukasik, N.; Chojnacki, J.; Luboch, E.; Okuniewski, A.; Wagner-Wysiecka, E. Photoresponsive, amide-based derivative of embonic acid for anion recognition. J. Photochem. Photobiol. A Chem. 2020, 390, 112307. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | λmax E (nm) | εE(M−1 cm−1) | ΔA (%) | ΦE-Z (%) | |
|---|---|---|---|---|---|
| π→π* | n→π* | ||||
| L1 | 347 | ~460 | 1.3 × 104 | 25.0 | 6.19 ± 0.13 |
| L2 | 335 | 448 | 3.1 × 104 | 10.1 | 2.79 ± 0.22 |
| Compound | Ea | ΔH ‡ | ΔS ‡ | ΔG ‡ |
|---|---|---|---|---|
| L1 | 98.6 ± 1.8 | 95.8 ± 1.4 | −31.2 ± 2.8 | 105.1 ± 1.4 |
| L2 | 70.9 ± 0.7 | 68.2 ± 1.1 | −101.2 ± 3.4 | 98.4 ± 4.4 |
| Stoichiometry (S:A) | L1 | L2 | ||||
|---|---|---|---|---|---|---|
| F− | F− | AcO− | BzO− | H2PO4− | Cu2+ | |
| 1:1 | 5.65 ± 0.03 | 5.63 ± 0.12 | 5.66 ± 0.07 | |||
| 1:2 | 4.51 ± 0.01 | 4.38 ± 0.09 | ||||
| 2:1 | 5.22 ± 0.57 | |||||
| Stoichiometry (S:A) | F− | AcO− | BzO− | H2PO4− | Cu2+ |
|---|---|---|---|---|---|
| 1:1 | 5.76 ± 0.07 | ||||
| 1:2 | 9.15 ± 0.05 | 6.31 ± 0.04 | 6.21 ± 0.11 | 9.87 ± 0.16 |
| Sample Code | t1/2 (Free) | t1/2 (F−) | t1/2 (AcO−) | t1/2 (BzO−) | t1/2 (H2PO4−) | t1/2 (Cu2+) |
|---|---|---|---|---|---|---|
| L1 | 94 | 41 | - | 51 | - | - |
| L2 | 15 | 49 | 47 | 62 | 42 | 52 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Łukasik, N.; Hemine, K.; Anusiewicz, I.; Skurski, P.; Paluszkiewicz, E. Photoresponsive Amide-Based Derivatives of Azobenzene-4,4′-Dicarboxylic Acid—Experimental and Theoretical Studies. Materials 2021, 14, 3995. https://doi.org/10.3390/ma14143995
Łukasik N, Hemine K, Anusiewicz I, Skurski P, Paluszkiewicz E. Photoresponsive Amide-Based Derivatives of Azobenzene-4,4′-Dicarboxylic Acid—Experimental and Theoretical Studies. Materials. 2021; 14(14):3995. https://doi.org/10.3390/ma14143995
Chicago/Turabian StyleŁukasik, Natalia, Koleta Hemine, Iwona Anusiewicz, Piotr Skurski, and Ewa Paluszkiewicz. 2021. "Photoresponsive Amide-Based Derivatives of Azobenzene-4,4′-Dicarboxylic Acid—Experimental and Theoretical Studies" Materials 14, no. 14: 3995. https://doi.org/10.3390/ma14143995
APA StyleŁukasik, N., Hemine, K., Anusiewicz, I., Skurski, P., & Paluszkiewicz, E. (2021). Photoresponsive Amide-Based Derivatives of Azobenzene-4,4′-Dicarboxylic Acid—Experimental and Theoretical Studies. Materials, 14(14), 3995. https://doi.org/10.3390/ma14143995