3. Results
FTIR qualitatively evaluated the chemical composition of the samples, and
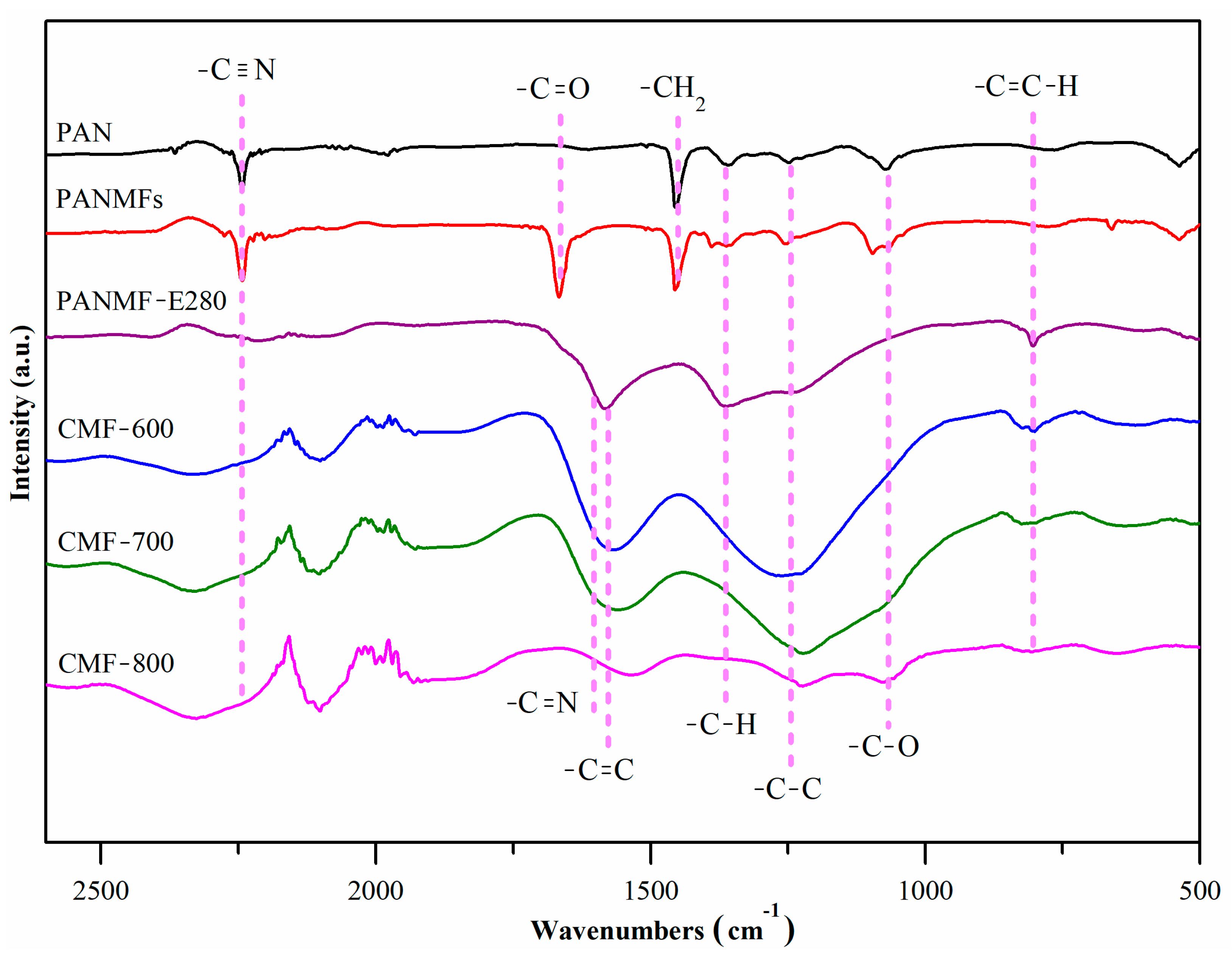
Figure 2 shows the infrared spectra in the interval between 2500 cm
−1 and 500 cm
−1. The precursor polymer (polyacrylonitrile, PAN) had (i) two well defined signals: ~2245 cm
−1 (nitrile groups, −C≡N) and ~1454 cm
−1 (aliphatic groups,
in CH
2); and (ii) three weak signals: at 1360 cm
−1 (
in CH), at 1240 cm
−1 (-C-C), and at 1070 cm
−1 (-C-O) [
25]. In addition to the five PAN signals, the PANMFs samples also presented one more signal in the region between 1600 cm
−1 and 1700 cm
−1, centered at ~1670 cm
−1, ascribed to the carbonyl groups (-C=O). This could be due to residues of Dimethylformamide (DMF), which was used as a solvent to prepare the PAN microfibers, because the signal disappears when the sample was heated.
When PANMFs were stabilized at 280 °C, the intensity of the signal in ~2245 cm
−1 (nitrile groups, −C≡N) decreased significantly and two broad and well defined signals were formed. The first one lay between 1480 cm
−1 and 1630 cm
−1, with a well defined center in 1574 cm
−1 (-C=C); however, there were also the -C=N bonds within this broad signal, at 1590 cm
−1. The onset of both bonds (-C=C and -C=N) suggests that cyclization and dehydrogenation had started; these processes co-occur during the stabilization of the materials at 280 °C (a temperature used in this work). The second signal was between 1180 cm
−1 and 1420 cm
−1, with two centers in 1240 cm
−1 (-C-C) and 1360 cm
−1 (-C-H). One weak signal could be observed at ~805 cm
−1, which is characteristic of aromatic rings (C=C-H) and carbon-hydrogen bonds (-C-H, ~1360 cm
−1) [
26,
27,
28]. The decrease in the number of nitrile groups, loss of hydrogen and formation of aromatic structures are believed to result from the cyclization (-C=N-), dehydrogenation (-C=C-) and oxidation (-C=O) of PANMFs occurring during the stabilization stage.
The carbonization at 600 °C (CMF-600) and 700 °C (CMF-700) of the PANMFs led to: (i) increments in the signals between 1480–1630 cm
−1 (-C=C, ~1574 cm
−1 and -C=N, 1590 cm
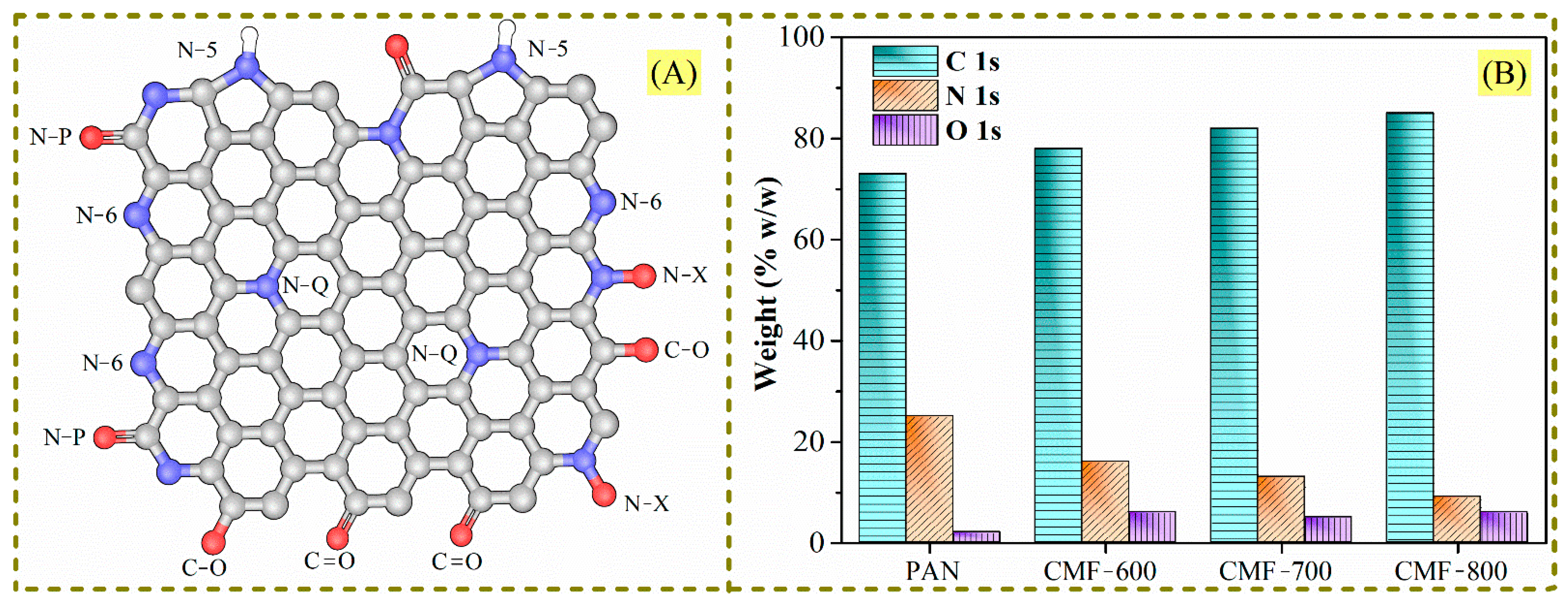
−1), although as the calcination temperature increased there was a trending shift towards the formation of -C=C bonds and a decrease in -C=N bonds, which is consistent with the XPS results reported in Ojeda et al. [
13], showing that the amount of carbon increases as the amount of nitrogen decreases (
Figure 1B); (ii) an increment in the signal between 1180–1420 cm
−1, although the signal for -C-H (1360 cm
−1) disappeared and the signal for -C-C bonds is shifted to a single center located at ~1240 cm
−1. This corroborates that dehydrogenation had occurred, and adjacent polymer chains were joined by C-C bonds, resulting in longer carbon sheets, which is the objective of the 90 min carbonization process [
17]. Nevertheless, when PANMFs were carbonized at 800 °C (CMF-800), both signals decreased because, at this temperature, the material had reached a higher carbon content and had significantly decreased in nitrogen compared to the CMF-600 and CMF-700 samples, which was confirmed by XRD and Raman analysis, as described in the following sections.
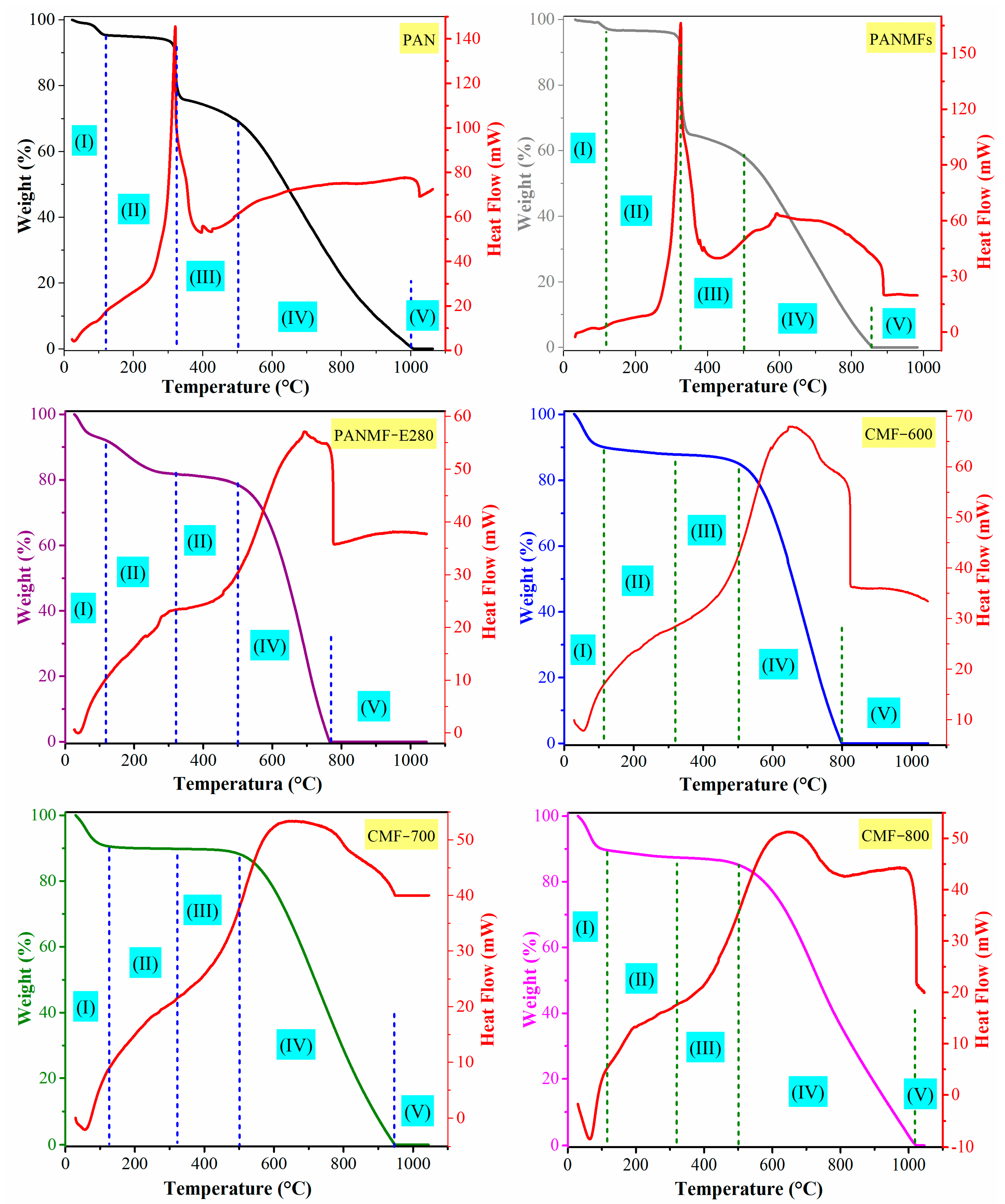
The TG/DTA curves for the studied samples are shown in
Figure 3, and they were performed under nitrogen flow. All materials showed at least one exothermic and sharp peak in the DTA curve. In the case of PAN and PANMFs, this peak occured at 320 °C, above 320 °C, PANMFs showed a significant chemical transformation; therefore, it is recommended that the stabilization process for PANMFs be carried out at temperatures below 320 °C. The peak for the rest of the materials that had already been calcined appeared at temperatures above 500 °C (
Figure 3), which accounts for the process of dehydrogenation and denitrogenation [
29,
30]. The increase in carbonization temperature generated broader peaks (CMF-800 > CMF-700 > CMF-600), which may be a result of a lower concentration of functional groups (as evidenced by XPS), so that the energy release became slower. A narrower peak means a large amount of heat evolved at a shorter time.
The TG curve in
Figure 3 can be divided into five regions based on mass loss [
25,
31]. The first part is up to 120 °C, where the weight loss is mainly due to the desorption of physisorbed water. The second part is up to 320 °C, where PAN and PANMF materials experienced a more pronounced weight loss due to the occurring cyclization and dehydrogenation [
29,
30], PANMF-E280 experienced a smaller mass loss than PAN and PANMFs. On this very same range, the calcined materials (CMF-600, CMF-700, and CMF-800) presented minimum losses (less than 2%
w/w), which means that the stabilization process had been successful on these materials. In the third part, up to 500 °C, in addition to dehydrogenation, denitrogenation took place. Materials that had not been carbonized (PAN, PANMFs and PANMF-E280) showed a larger mass loss in this temperature range (320 °C to 500 °C), while materials carbonized at 600, 700 and 800 °C showed a smaller percent loss. The fourth part accounts for temperatures above 500 °C and up to 800 or 1000 °C; the mass loss was due to eliminating of some volatile components such as H
2, CH
4, CO, CO
2, H
2O, N
2, NH
3 and HCN [
29,
30]. At this stage, all materials showed a mass loss of more than 50%
w/w. Finally, beyond 800 °C (PANMFs, PANMF-E280, and CMF-800) or 1000 °C (PAN, CMF-700, and CMF-800) the thermal degradation of PAN had been completed. Considering the losses in the region between 120 °C and 320 °C, PANMFs lost 30% in mass, the characteristic region where the cyclization and dehydrogenation of the stabilization stage occurs; when the material was stabilized, the loss in PANMF-E280 decreased down to 10% and, for the case of the materials that were carbonized at 600, 700 and 800 °C, the loss was 2% in all cases, which is a sign of a good stabilization process. Likewise, the mass loss in the region between 320 °C and 500 °C was 3%, 2%, and 2%, respectively, for carbonized materials.
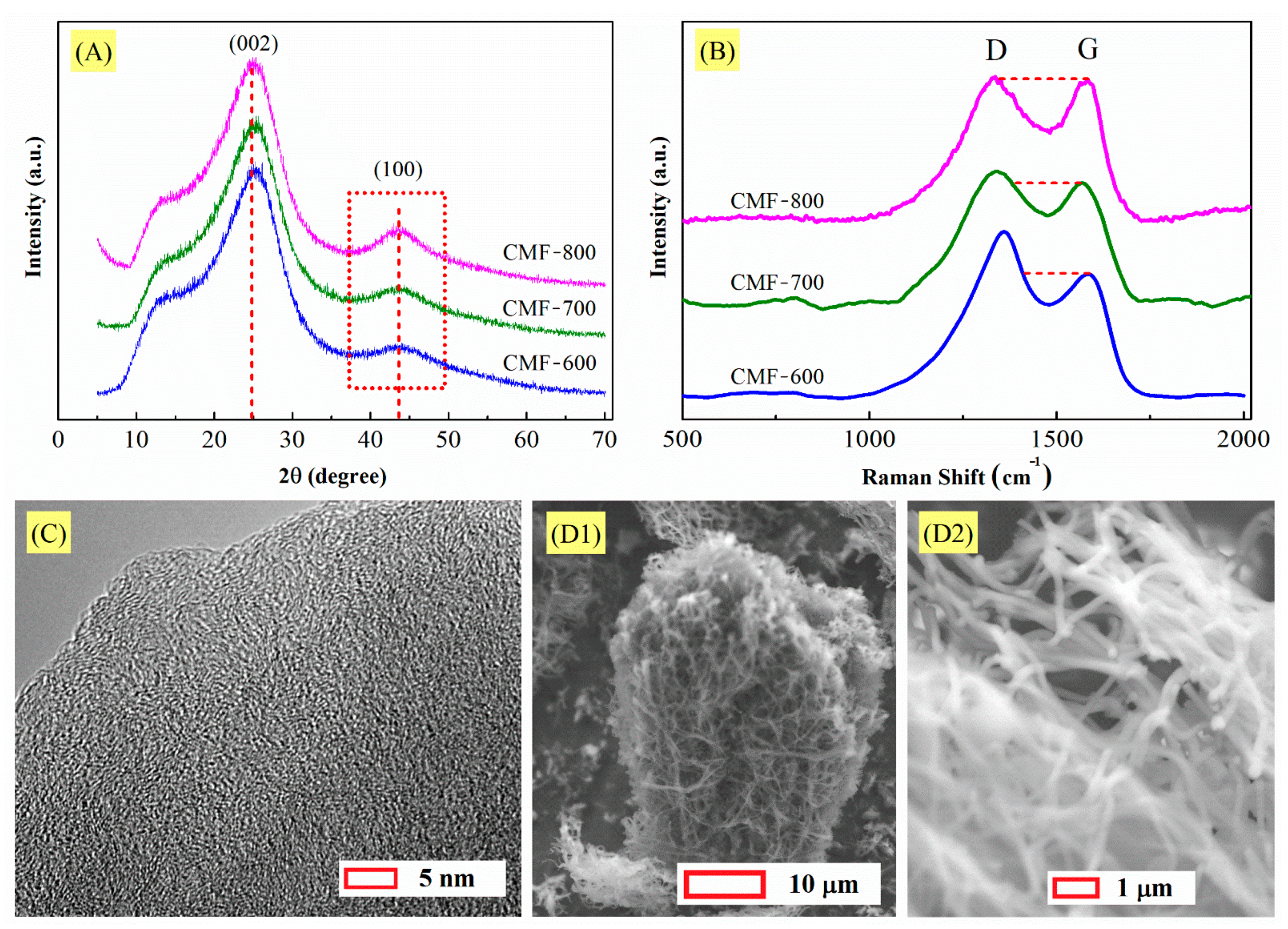
XRD, Raman spectra, SEM images and TEM imagen for carbonized materials are shown in
Figure 4. In the XRD patterns of
Figure 4A, all materials exhibited the signal at 2θ = 25°, which corresponds to the graphitic (002) planes [
13,
32,
33]. The high amplitude of the peak between 5° and 35° indicates that the structure of the carbon microfibers is composed mainly of amorphous carbon. This will be observed in the signal at 2θ = 44 ° in the plane (100), attributed to the lateral extent of graphitic domains. The spacing of the crystal layers (
) was calculated using Bragg’s Law [
34]:
where
λ is the X-ray wavelength (Cu Kα: 0.154 nm),
θ is the incident angle (the angle between the incident ray and the scattering plane), and
n is an integer, in this case,
n = 1.
From the XRD results, it is also possible to calculate the microcrystallite sizes (L) in the a-axes (stack width, La) and c-axes (stack highest, Lc) directions, as determined from the full width at half maximum (FWHM) of the diffraction peak according to the Scherer equation [
35]:
where
L is microcrystallite size,
λ is the wavelength of the incident X-rays (Cu Kα: 0.154 nm),
θ is the incident angle (Bragg angle),
is the FWHM in radians, and finally,
K is the Scherer constant, which depends on the lattice dimension,
K = 1.84 for La ((100) plane) and
K = 0.91 for Lc ((002) plane); the crystallite size along the a-axis,
, corresponds to the extent of a hexagonal net plane, and the one along the c-axis,
, to a width of stacking layer of hexagonal net layers [
36,
37]. Both parameters, the spacing of the crystal layers (
) and microcrystallite size (L), are present in
Table 1.
The peaks of all materials vary only in the height of the signals, the centers being located almost at the same angle. Therefore, the value for d
002 and d
100 should be very similar in the three materials (
Figure 4A). For example, d
002 was 0.360 nm, 0.359 nm, and 0.358 nm for CMF-600, CMF-700, and CMF-800, respectively. Although there is minimal variation, d
002 tended to decrease, which means a closer graphite interplanar spacing (0.3354 nm). Carbon materials with a turbostratic structure usually present a much larger spacing between adjacent layers than 0.3354 nm because of weak van der Waals interaction due to no regularity in stacking [
37]. In general, heat treatment at high temperatures gradually decreased the value for d
002, approaching 0.3354 nm, because the graphitic three dimensional stacking of layers occurs randomly in the crystallite; this parameter (d
002) has commonly been used as a measure of the development of graphitic structure in carbon materials [
37]. In the case of d
100, the value was the same, 0.208 nm, for all materials. The decrease in FWHM when the carbonization temperature rose suggests an increase in their crystallinity; furthermore,
and
presented the same behavior and this increase in crystal size can be interpreted as a decrease in the disorder between the carbon sheets in the carbon microfibers.
Based on the XRD results, it can be observed that due to the temperature used for the synthesis of the materials, the degree of amorphicity is high. Raman spectroscopy results confirm this point. Generally, this technique allows to characterize the structure development of carbon material. The spectra in
Figure 4B indicate a structural change in the CMFs with increasing carbonization temperature. Two peaks can be identified, G (Graphite) and D (Disorder). The G band centered at 1580 cm
−1 is associated with the sp
2-hybridized carbon, and the D band at 1360 cm
−1 is attributed to the disorder related to the sp
3-hybridized carbon. The intensity ratio of the D band to G band (I
D/I
G = R) expresses the sp
3/sp
2 carbon ratio, indicating the degree of disorder. It depends on the degree of graphitization and the alignment of the graphitic planes. The R-value is sensitive to the ratio of the concentration of graphite edge planes and/or crystal boundaries relative to standard graphite planes, i.e., the lower the R-value, the higher is the amount of sp
2 (graphite) clusters that exist on the sample; there is an empirical formula for the relationship between the R-value and the crystallite domain size,
, of graphite as
[
32,
38]. Both R and La were calculated, and they are shown in
Table 1. The R values are relatively high as compared to those usually presented by carbon materials with a higher degree of graphitization (lower than 1.0) [
32,
38], which means that the I
D/I
G ratio is not conclusive at these low carbonization temperatures. Nevertheless, R-values corroborate the findings previously described by several studies about carbon: they decrease with an increase in carbonization temperature. By simple visual inspection, it can be observed that the G-band signal increases while the D-band signal decreases. In the case of carbon microfibers synthesized from PAN and by the electrospinning method, Andrei et al. have shown that temperatures above 1200 °C allow obtaining materials with a significant degree of graphitization and values close to 1.0 for R [
39].
Thus, XRD and Raman patterns suggest that the carbon microfibers are a disordered arrangement of carbon layers and the TEM micrograph in
Figure 4C confirms this conclusion by revealing randomly oriented carbon layers. Some of these layers seem to be larger than the others. As the carbonization temperature increases, these layers tend to become oriented in one direction only. It is also important to mention that the stabilization process to obtain carbon microfibers is crucial because it triggers the oxidation and dehydrogenation of the polymeric nanofibers; oxygen is an initiator for the formation of active centers for cyclization that increases the activation energy and stimulates the dehydrogenation process [
29,
30]. In this context, SEM micrographs in
Figure 4D1,D2) confirm that an adequate temperature (280 °C) had been used for the stabilization process, which avoided the fusion of the microfibers during carbonization.
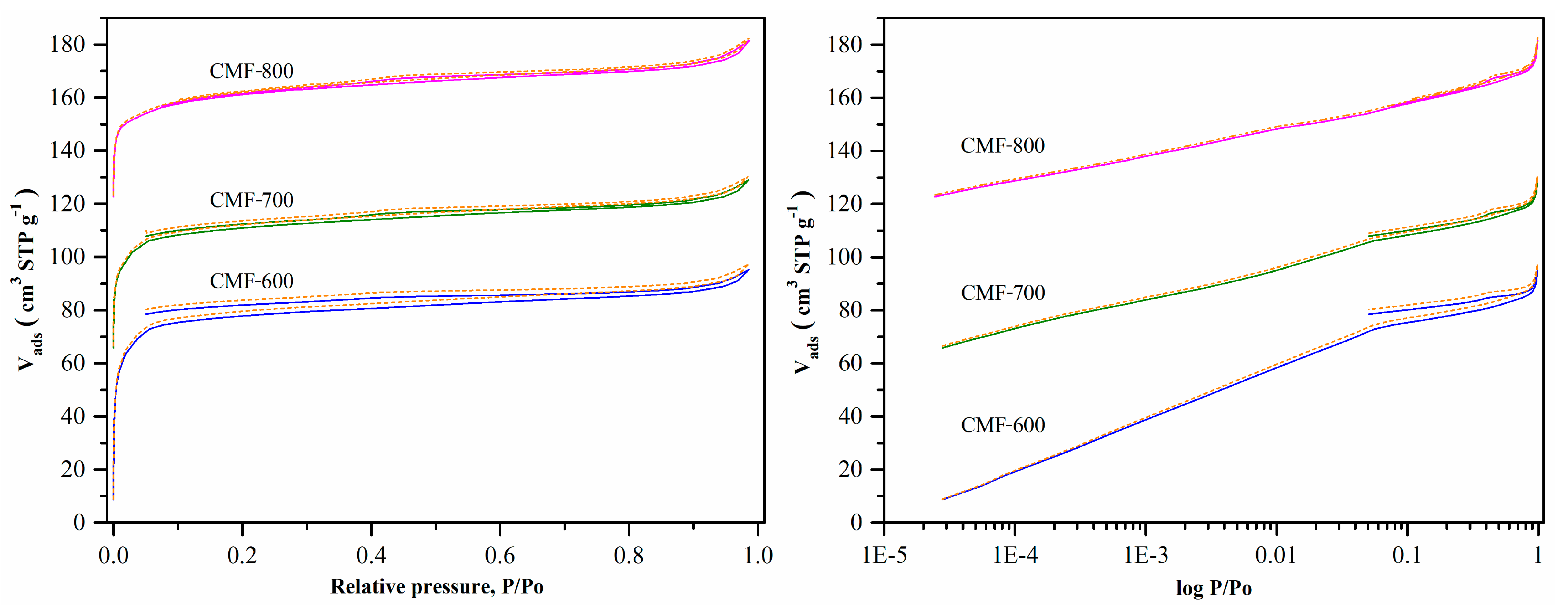
Nitrogen adsorption/desorption isotherms at −196.15 are shown in
Figure 5. The isotherms showed features of both Type IA (microporous) and Type IV (mesopores), with a narrow hysteresis loop H1, which is typical of slit like larger pores in activated carbons [
19]. The hysteresis cycle was subtle, and the high microporosity of the CMFs easily overshadowed it. The high adsorbed volume at a relative pressure of less than 0.1 indicates that the material is mainly microporous. However, the isotherms for CMF-600 and CMF-700 samples presented a particular behavior: the desorption curve does not match the adsorption curve at relative pressures lower than 0.4 (open loop for hysteresis), as did the isotherm of the CMF-800 sample.
To calculate textural parameters such as specific surface area and pore size distribution in the case of microporous materials such as carbons, it is very important to have data at low pressures, starting from 1 × 10
−4 (relative pressure), because the range considered for BET calculations in mesoporous materials (0.05 to 0.35) does not strictly apply.
Figure 5 shows the nitrogen isotherms at an equilibration time of 10 min and tolerance of 0 (±0.001 bar) and 9 (±0.01 bar). Note that the tolerance had no significant effect on the results shown for 0 (solid line) and 9 (dotted line). The one aspect not readily visible in the isotherm, but the acquisition time for these isotherms is shown in
Table S1. For the CMF-600 material, regardless of the programmed conditions (equilibrium time: 10 min and tolerance: 0 or 9), it took more than one hour to collect the first point and approximately 71 h to reach a relative pressure of 0.3 (81 h for a complete analysis). Beyond this relative pressure, the time it took to measure the rest of the isotherm was roughly equivalent to the 10 min program, so this isotherm took around three days to be completed. Further increasing the equilibration time, which means that the analysis takes more than three days, was impractical because the liquid nitrogen in the Dewar Flask (P/N 01879 9371) only lasts for four days. Hence, to evaluate the effect of the equilibration time on the hysteresis cycle, isotherms were obtained starting with a relative pressure of 1 × 10
−2. These isotherms probably cannot be considered to calculate textural parameters, but it will be possible to observe the impact that the equilibration time would have on the hysteresis cycle.
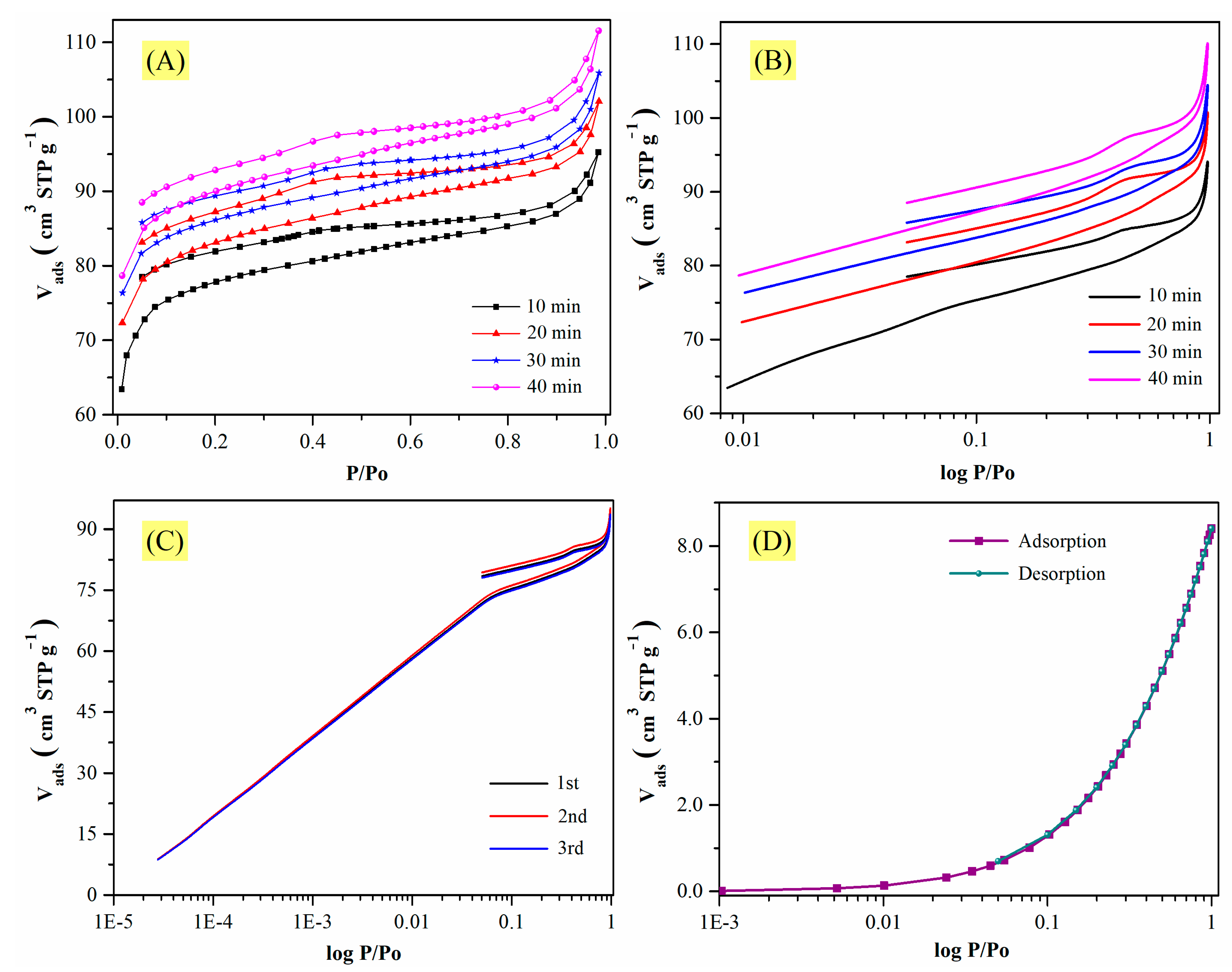
In
Figure 6A,B, the isotherms for CMF-600 are shown considering a tolerance of 0 (±0.001 bar) and equilibration times of 10, 20, 30, and 40 min, starting at a relative pressure of 1 × 10
−2. Note that there was a remarkable variation in the amounts adsorbed. As the equilibration time increased, so did the adsorbed volume. However, the trend of the hysteresis cycle remained the same, i.e., there was no closure between the adsorption curve and the desorption curve, although the gap decreased. For example, at a lower equilibrium time, at a relative pressure of 0.4, there was a difference of ~4.00 cm
3 (STP) g
−1, which decreased to ~3.00 cm
3 (STP) g
−1 when the equilibrium time increased, but not enough to close the hysteresis loop. Considering the equilibration time of 10, 20, 30, and 40 min and 89, 95, 98, and 104 cm
3 STP g
−1 as the amount adsorbed at a relative pressure of 0.95, respectively, the Pearson’s correlation coefficient was calculated. The value obtained was 0.9922, which means that both variables are strongly related, i.e., the larger the equilibrium time, the greater the adsorbed volume. Therefore, even though the equilibration time does have an impact on the N
2 uptake, it does not seem to be the only cause of the unclosed hysteresis loop.
The observed variations could also be due to poor material stability, and to verify this hypothesis the same sample underwent a triplicate analysis. Before each isotherm, the material was degassed at 200 °C for 6 h. The obtained isotherms are illustrated in
Figure 6C, and they show excellent reproducibility; therefore, the material is not only stable but also regenerable. Additionally,
Figure 6D shows a nitrogen adsorption isotherm for the same material (CMF-600) at 0 °C, where the closing of the hysteresis loop can be observed. So, in fact, the analysis temperature seems to be one of the most important factors influencing hysteresis closure. However, the purpose of this study is to explain why, at −196 °C, materials calcined at temperatures below 800 °C exhibit an open hysteresis loop.
The CMF-700 material had a similar behavior to CMF-600; although it seems that the desorption curve is closer to the adsorption curve, the gap between them remains (
Figure 5). The analysis conditions were the same (equilibration time: 10 min and tolerance: 0 and 9). Compared to CMF-600, the acquisition time decreased by 15 h since it took 56 h to reach the relative pressure of 0.3 and 70 h for the complete analysis (
Table S1). On the contrary, CMF-800 shows a closure between both curves (adsorption and desorption). The acquisition time is the time programmed for the analysis, with 25 h for the whole analysis (
Table S1). Since the detailed study of the CMF-600 material showed that the gap between adsorption and desorption branches persisted, regardless of the equilibration time, and the gap decreased for increasing carbonization temperatures, it is plausible to assume that the chemical composition of the samples may have a determining role in this phenomenon. Adsorption at low pressures for these materials takes place in the micropores, where diffusion is usually hindered. Yet, the material with the most significant micropore volume is CMF-800, which had no problems during the analysis.
In addition to the equilibration time during the analysis, the chemical composition (density of functional groups) could also be considered. Chemical interactions between adsorbate and adsorbent are ruled out, because nitrogen is a nonpolar molecule, and the analysis was carried out at −196.15 °C. On the other hand, it is possible that a higher content of nitrogen and oxygen functional groups within the structure of the carbon layers may lead to pore blocking, especially in small pores. As a matter of fact, the concentration of such functional groups is higher for samples carbonized at lower temperatures and carbon layers are more disordered (as confirmed by TEM). This may be the cause of the results shown in
Table S1, where a longer equilibration time is required for isotherm points at low pressures to achieve stability between equilibration time and tolerance. Having said that, it is plausible to propose that the chemical composition of the samples (directly related to the carbonization temperature) has an impact on the atypical behavior of the hysteresis loop for the CMF-600 and CMF-700 materials, which may be further confirmed with enthalpy estimations and ab initio calculations.
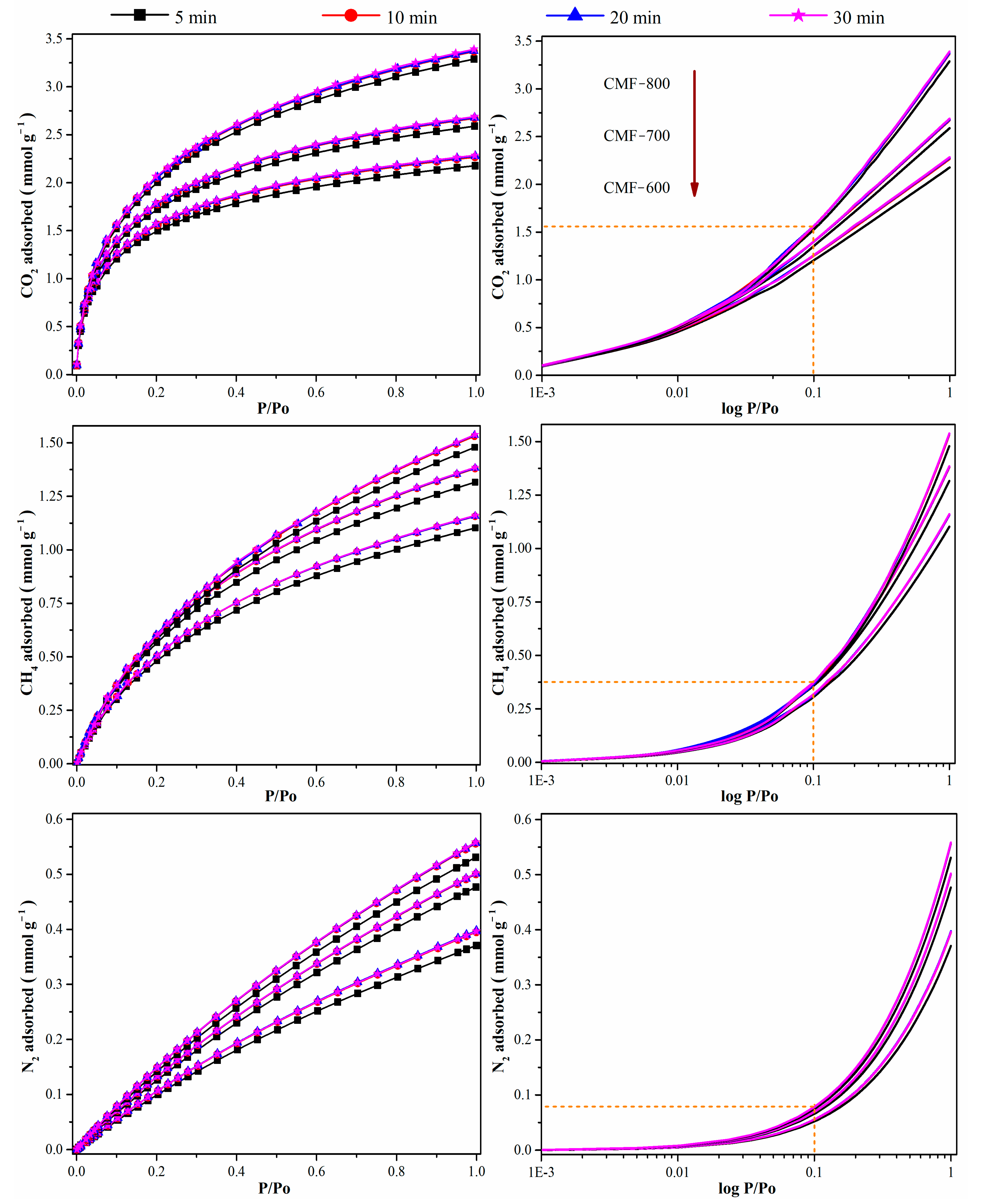
The impact of the equilibration time (5, 10, 20, and 30 min) of other molecules, such as CO
2, CH
4, and N
2 isotherms, at 0 °C on the CMFs is illustrated in
Figure 7. Unlike the nitrogen isotherms at −196 °C, the adsorption isotherms for CO
2, CH
4, and N
2 at 0 °C reached stability after an equilibration time of 10 min, regardless of the carbonization temperature of the carbon microfibers. The correlation coefficient between the maximum amount adsorbed at 1 bar and the equilibration time was ~0.8, which is far from 1.0. Therefore, if the measurements are performed at higher temperatures, these two variables (equilibration time at uptake a 1 bar) are independent form one another. The plots on the right side in
Figure 7 also show the preference of the CMFs towards CO
2, which would make it a promising material for CO
2 selectivity over CH
4 or N
2, where microporosity is one of the important parameters that improve selectivity [
40].
From the N
2 isotherms at −196.15 °C and CO
2 isotherms at 0 °C, some textural properties have been calculated that allow a better evaluation of the CMFs. The values of these properties are detailed in
Table 2 and the average pore size (pore size distribution, PSD) in
Figure 8. To calculate the PSD, it is commonly recommended to use the desorption curve; however, due to the behavior they were showing, some authors recommended using the adsorption curve for average pore size calculations [
41]. Additionally, in the case of microporous carbon materials, it had also been recommended to calculate the PSD using the CO
2 isotherms at 0 °C, because the diffusion is much faster and pores as small as 0.4 nm can be accessed [
19].
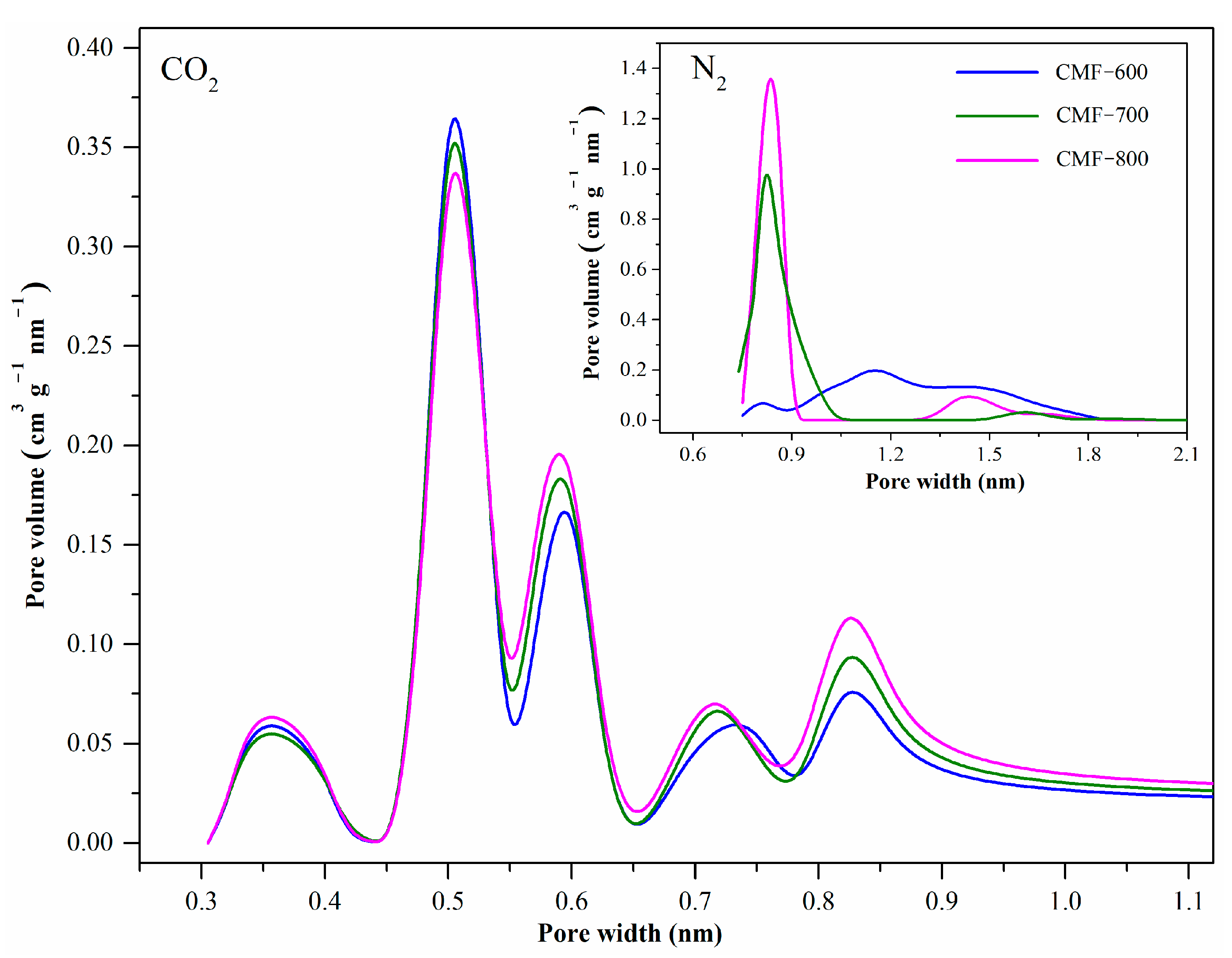
Figure 8 shows the pore size distributions (PSD) of CMFs by both N
2 isotherms at −196.15 °C and CO
2 isotherms at 0 °C. The results were obtained directly from the Quantachrome software, considering the NLFDT equilibrium model and slit pores for carbon materials. The nitrogen isotherms are only able to “sense” micropores down to approximately 0.8 nm. The CO
2 isotherms provide information of micropores down to 0.35 nm, even if there are a small fraction of pores with this diameter, comparable to the results from XRD (0.360 nm). The PSDs obtained from CO
2 isotherms showed five peaks. Considering the more pronounced peak, centered at 0.50 nm, C-600 shows the greatest pore volume. For the other average sizes (0.60, 0.75 and 0.85 nm), the order is as follows: CMF-800 > CMF-700 > CMF-600, which is consistent with the adsorbed volume of each of the materials (
Figure 7). Based on PSDs calculated by CO
2 isotherms, the CMF samples have supermicropores (2.0 nm > width > 0.7 nm) and ultramicropores (width < 0.7 nm) [
19,
42].
From the results in
Table 2, considering N
2 adsorption at −196.15 °C, the following observations can be made: (i) the specific surface area (by BET equation) increases from 296 m
2 g
−1 (CMF-600) to 635 m
2 g
−1 (CMF-800), and with it, the microporosity from 79% to 89%; (ii) the total volume is duplicated between the CMF-600 and CMF-800 material; (iii) the micropore volume increases with increasing carbonization temperature and this behavior is corroborated by three methods (t-plot, α-plot, and DR); (iv) the average pore diameter decreases with increasing calcination temperature and NLDFT and DR models demonstrate that these materials are highly microporous. Considering CO
2 isotherms, the micropore specific areas (by DR) have been calculated for the three samples and compared to A
BET(N
2) (see
Table 2). Note that A
BET(N
2)/A
DR(CO
2) increased for higher carbonization temperatures. Actually, A
DR(CO
2) increased moderately with higher carbonization temperatures, whereas A
BET(N
2) increased considerably. The average pore diameter remained almost constant, ~1.4 nm by DR model and ~0.5 nm by NLDFT model, which is consistent with the pore size distribution (
Figure 8) as calculated from CO
2 isotherms. Therefore, as carbonization temperature increases in this range (600–800 °C), the specific volume of ultramicropores (as sensed by CO
2) remains approximately the same, whereas the surface area and volume of micropores larger than 0.7 nm increases significantly. These values suggest that the microporosity cannot be directly related to hysteresis cycle behavior. Three different models (t-plot, alpha-plot, and DR) have been used to determine the microporosity in these materials and all of them confirm that CMF-800 has the largest micropore volume, the largest micropore area and the narrowest pores (as sensed by N
2 isotherms). Hence, in the specific case of carbon microfibers synthesized from PAN electrospinning, a higher microporosity, per se, does not lead to greater diffusion problems for the nitrogen molecule and should not be the cause of the open hysteresis loop.
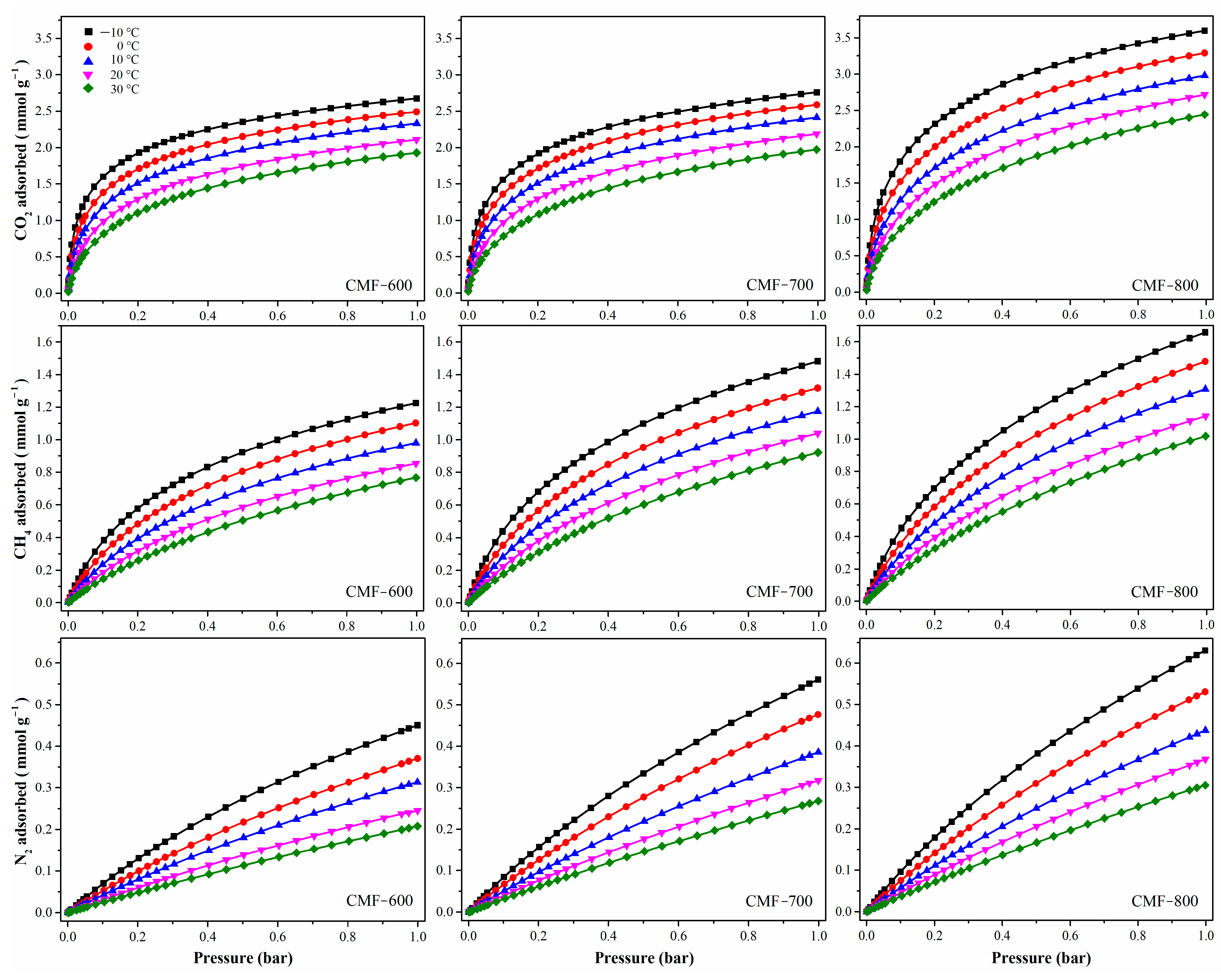
To evaluate the influence of the chemical composition, the interaction energy between the different gases (N2, CO2, and CH4) and the carbon microfibers (CMF-600, CMF-700, and CMF-800), two methods will be employed: (i) the isosteric enthalpy of adsorption and (ii) a theoretical analysis by molecular dynamics.
In this work, the isosteric enthalpy of adsorption has been calculated indirectly, that is, by using adsorption isotherms at different temperatures. It has been reported that when the process is dominated by physisorption, the temperatures selected for the analysis do not have much influence if the difference between them is not larger than 20 °C [
43]. Five temperatures have been considered (−10, 0, 10, 20, and 30 °C). The CO
2, CH
4, and N
2 isotherms for CMF-600, CMF-700, and CMF-800 are shown in
Figure 9; increments in the carbonization temperature also increase the adsorbed concentrations in all cases, CO
2, CH
4, and N
2 (
Table 3). Additionally, all materials adsorbed more CO
2 than CH
4 and N
2, particularly in the lower pressure range, which means that there is a higher affinity of the CMFs towards the CO
2 molecule. Actually, CO
2 selectivity on all samples (defined as the ratio CO
2/CH
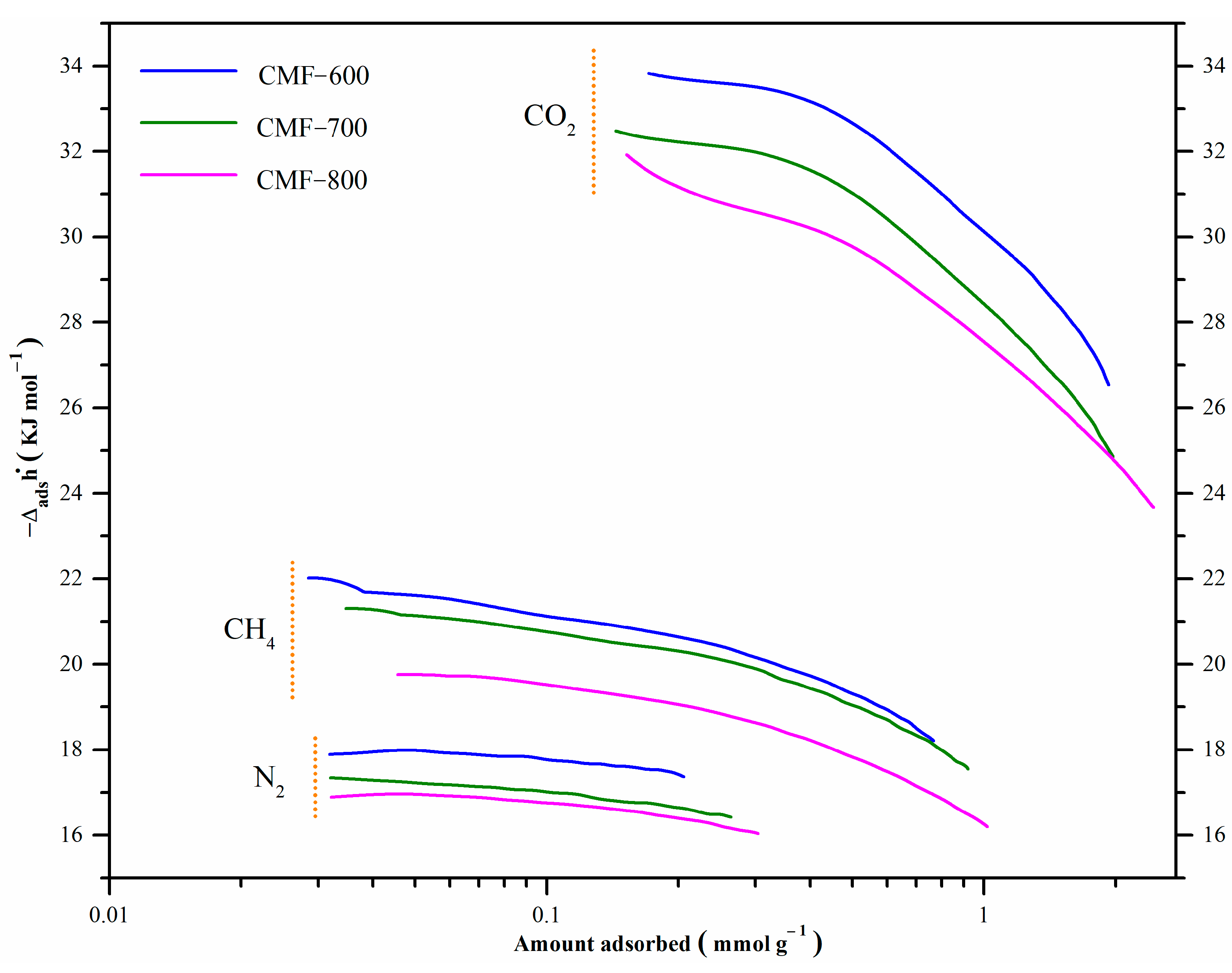
4) slightly increased with increasing temperature. Although selectivity tended to decrease with increasing carbonization temperature (and hence less functional groups), it remained above 2 for sample MCF800 regardless of the temperature. This behavior is consistent with the isosteric enthalpies of adsorption of the adsorbate under study, shown in
Figure 10. Additionally, Pearson’s correlation coefficient (
Table 3) has been calculated in an attempt to correlate the experiment temperature with (i) the amount adsorbed of each gas and (ii) CO
2 selectivity. In the first case, all values are close to −0.99; the negative value means that the variables are inversely related, i.e., with increasing temperature, the amount adsorbed will decrease. In the case of CO
2 selectivity, it shows a positive correlation, around 0.95, which means that both variables are directly related, so an increase in temperature also increases selectivity.
The isosteric enthalpy of CO
2 adsorption at low surface coverage was around −33.8, −32.3, and −31.4 kJmol
−1 for materials CMF-600, CMF-700, and CMF-800, respectively. These values decrease with an increment in surface coverage, which can be associated with the energy heterogeneity of the carbon microfibers surface adsorption sites. The isosteric enthalpy of adsorption of these materials followed the order: CMF-600 > CMF-700 > CMF-800, so the interaction of CO
2 with CMF-600 was stronger than with CMF-800, because it released a greater amount of energy. The behavior is similar in the isosteric enthalpy of CH
4: the material with higher affinity was CMF-600 with −21.6 KJmol
−1; CMF-700 and CMF-800 with −21.1 KJmol
−1 and −19.8 KJmol
−1, respectively. CMF-800 is the material with the highest surface area, highest microporosity, and highest uptakes for both CO
2 and CH
4. Sample CMF-800 was the one with the lowest adsorption enthalpies, and therefore the interaction energy between the three probe gas molecules and the sample is lower as compared to the interaction in CMF-600 and CMF-700 materials. Based on these results, it is evident that the isosteric enthalpy of adsorption is not strictly related to the microporosity or specific surface area. Regarding the chemical composition, CMF-800 is the material with the highest carbon content and the lowest content of both nitrogen and oxygen (
Table 1 and
Figure 1B). Therefore, the presence of functional groups derived from these heteroatoms seems to lead to stronger interactions of these probe molecules in CMF-600 and CMF-700 samples and thus higher adsorption enthalpies, although not the highest uptakes. Furthermore, it is interesting to observe the behavior of the isosteric enthalpies as a function of coverage. For example, in
Figure 9, the enthalpy curves for CO
2 show a pronounced decrease with increasing coverage, which means that the interaction between the CO
2 molecules with the surface groups of the CMFs is quite strong when the first CO
2 molecules are adsorbed. On the other hand, the slope of the curves for CH
4 is less pronounced as a result of a weaker interaction of this gas with the surface of the CMFs samples. In the case of N
2, there is practically no chemical interaction since the lines show a constant behavior. Therefore, these materials could be potentially used as CO
2 adsorbents in CO
2:CH
4 mixtures or in CO
2:N
2 mixtures.
In addition to the isosteric enthalpy of adsorption, the binding energies of CO
2 and CH
4 with graphitic materials were estimated by ab-initio calculations (periodic DFT, more details in
Section 2.4: computational study) in order to understand the possible intermolecular interactions. The CO
2 and CH
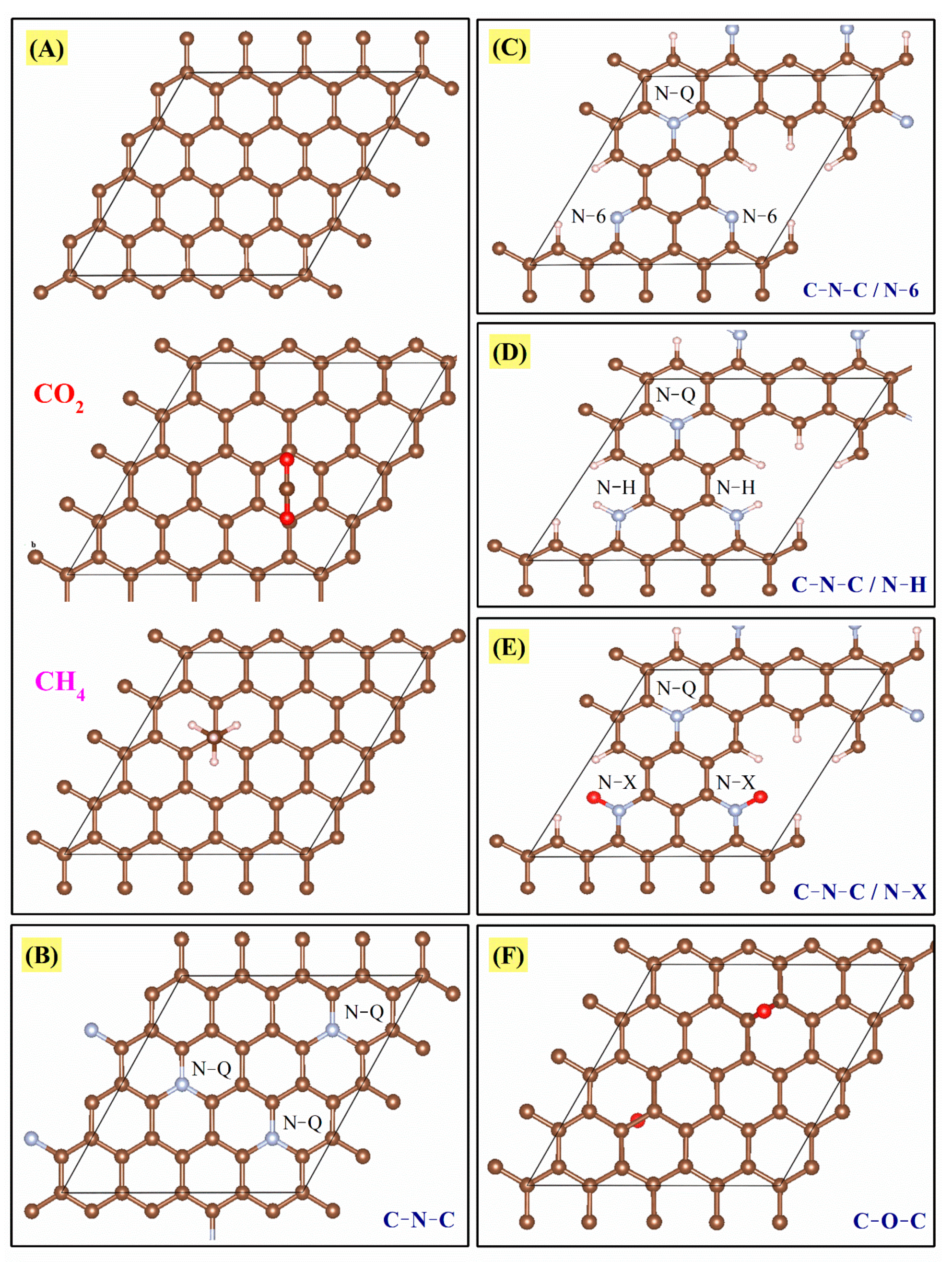
4 interaction energies were analyzed considering a graphene sheet containing the following groups: (i) pure carbon (C=C), (ii) C-N-C (quaternary-N), (iii) C-N-C and N-6 (pyridine-N), (iv) C-N-C and N-H, (v) C-N-C and N-X (pyridinic-N-oxide), and (vi) C-O-C (ethoxy). These groups will be called A, B, C, D, E, and F, respectively. These arrangements are shown in
Figure 11 and the results in
Table 4.
This analysis was performed to give a general idea of the interaction between the studied molecules (CO
2 and CH
4) and each of the functional groups theoretically present in the CMFs, considering a single carbon sheet; confinement effects and multiple interactions will be addressed elsewhere. CO
2 and CH
4 molecules are nonpolar, capable of forming weak interactions with the functional groups in CMFs. All binding energy values in
Table 4 show low affinity energies; however, among all functional groups, the oxygen functional groups stand out as having the most negative and highest absolutes. Nitrogen groups (B, C, D, and E) also have an effect, though one much more modest. Finally, A (pure carbon) and B (quaternary-N) are the configurations with lower reactivity. This fact means that a chemical surface rich in oxygen is preferred to enhance CO
2 adsorption. For CH
4, a similar behavior occurs; the F configuration shows the highest reactivity (C-O-C bonds). After the functional groups with oxygen, the most reactive species are N-6 (pyridine-N) and N-H. These calculations agree with experimental results; for instance, the CMF-600 material presents a higher amount of oxygen (
Figure 1B and
Table 1) and, therefore, a higher binding energy (
Table 4). In
Figure 10 this material has the highest isosteric enthalpy of adsorption, i.e., the higher interaction energy between the adsorbates (CO
2 or CH
4) and adsorbent (CMFs). At this point, N
2 adsorption was not calculated due to the very low uptake and adsorbate–adsorbent interaction. Moreover, CH
4 and CO
2 provides the specific reactivity trends of positive (H end) and negative (O end) molecules, respectively.
In this context, some studies have already focused on comparing the π-π type Van der Waals interactions of CO
2 with: (i) benzene, pyridine, and pyrrole [
44,
45] or (ii) carbonyl compounds [
46]. And the CH/π Interactions of CH
4 with (i) benzene [
47,
48,
49] or (ii) pyridine [
50]. In general, they have shown an interaction between CO
2 and CH
4 molecules with the different functional groups, and in some cases, a weak interaction, consistent with the results shown here. It is worth highlighting that the simulations presented herein consider a single layer of grapheme. While other effects, such as confinement, multiple sites and functional groups, could have further impact on the results, they are beyond the scope of the present manuscript and are currently subject of follow up work.
Although all the binding energy values (calculated in this work) are relatively small, they allowed concluding that the presence of these functional groups favors the interaction with molecules such as CO
2 or CH
4. Furthermore, the amount of these species has a crucial role, which likely caused the samples CMF-600 and CMF-700 to show the following behaviors: (i) the N
2 isotherms (at −196.15 °C) did not present a closed hysteresis cycle and (ii) the CO
2 and CH
4 adsorption uptakes were lower as compared to CMF-800 (which had the highest C and lowest N/O content of all MCF samples). An unclosed hysteresis loop in N
2 adsorption for carbonized materials at low temperatures may be a synergistic behavior between chemical composition and textural properties because these materials are the ones that present a lower degree of graphitization and more significant disorder in the arrangement of carbon sheets (behavior observed by Raman and XRD). Another conclusion that can be drawn is that the increase in carbonization temperature leads to a higher degree of graphitization, this means, for example, that temperatures above 2000 °C are only going to present C-C and C=C bonds (configuration A), and based on the results in
Table 3, these materials are going to adsorb a lower amount of CO
2, due to the low or null interaction between the CO
2 molecule and the sp
2 and sp
3 carbons (
Table 3 and A arrangement in
Figure 11); for this reason, it is imperative to choose a temperature that allows preserving a concentration of functional groups for an efficient adsorption of CO
2 or CH
4, because an increase in carbonization temperature also raises the specific surface area. Still, surface chemistry dominates the adsorption phenomena in these materials.


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}