Effect of Aromatic System Expansion on Crystal Structures of 1,2,5-Thia- and 1,2,5-Selenadiazoles and Their Quaternary Salts: Synthesis, Structure, and Spectroscopic Properties

 ,
,  , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. X-ray Measurements and Refinements
2.3. DFT Calculations
3. Results and Discussion
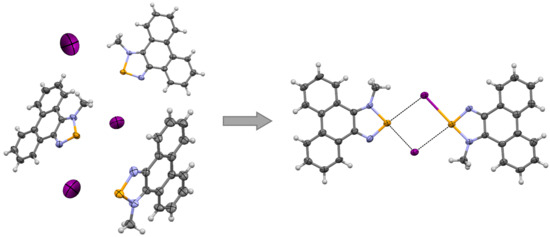
3.1. X-ray Crystallography
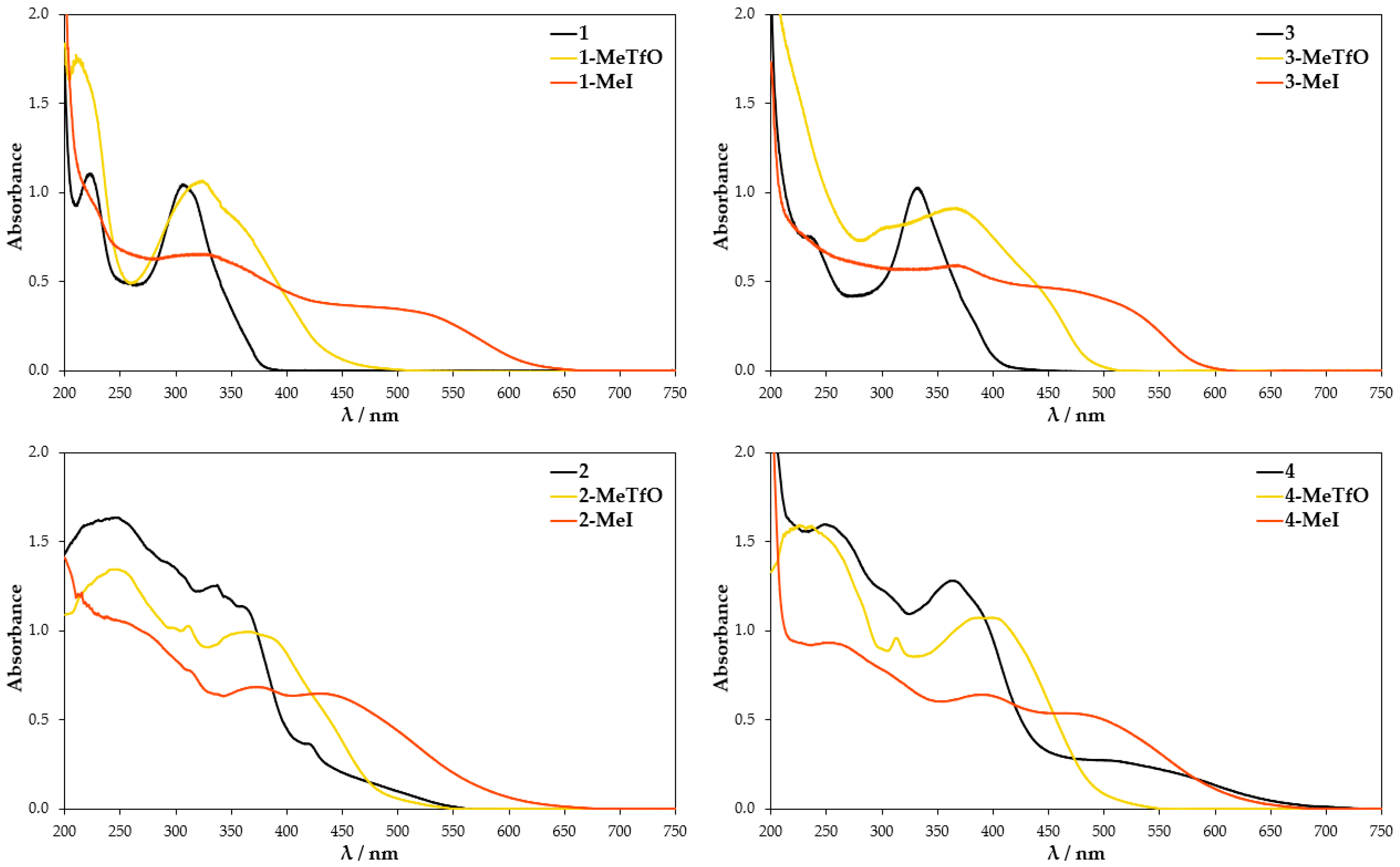
3.2. UV-Vis Spectra
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Da Cruz, E.H.G.; Carvalho, P.H.P.R.; Corrêa, J.R.; Silva, D.A.C.; Diogo, E.B.T.; De Souza Filho, J.D.; Cavalcanti, B.C.; Pessoa, C.; De Oliveira, H.C.B.; Guido, B.C.; et al. Design, synthesis and application of fluorescent 2,1,3-benzothiadiazole- triazole-linked biologically active lapachone derivatives. New J. Chem. 2014, 38, 2569–2580. [Google Scholar] [CrossRef]
- Neto, B.A.D.; Lapis, A.A.M.; Da Silva Júnior, E.N.; Dupont, J. 2,1,3-benzothiadiazole and derivatives: Synthesis, properties, reactions, and applications in light technology of small molecules. Eur. J. Org. Chem. 2013, 2013, 228–255. [Google Scholar] [CrossRef]
- Xia, D.; Guo, X.; Chen, L.; Baumgarten, M.; Keerthi, A.; Müllen, K. Layered electron acceptors by dimerization of acenes end- capped with 1,2,5-thiadiazoles. Angew. Chem. Int. Ed. 2016, 55, 941–944. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Wang, X.Y.; Guo, X.; Baumgarten, M.; Li, M.; Müllen, K. Fused Bis-Benzothiadiazoles as Electron Acceptors. Cryst. Growth Des. 2016, 16, 7124–7129. [Google Scholar] [CrossRef]
- Jung, J.W.; Jo, J.W.; Jung, E.H.; Jo, W.H. Recent progress in high efficiency polymer solar cells by rational design and energy level tuning of low bandgap copolymers with various electron-withdrawing units. Org. Electron. 2016, 31, 149–170. [Google Scholar] [CrossRef]
- Konstantinova, L.S.; Baranovsky, I.V.; Pritchina, E.A.; Mikhailov, M.S.; Bagryanskaya, I.Y.; Semenov, N.A.; Irtegova, I.G.; Salnikov, G.E.; Lyssenko, K.A.; Gritsan, N.P.; et al. Fused 1,2,3-Thiaselenazoles Synthesized from 1,2,3-Dithiazoles through Selective Chalcogen Exchange. Chem. Eur. J. 2017, 23, 17037–17047. [Google Scholar] [CrossRef]
- Shuku, Y.; Hirai, Y.; Semenov, N.A.; Kadilenko, E.; Gritsan, N.P.; Zibarev, A.V.; Rakitin, O.A.; Awaga, K. 3D molecular network and magnetic ordering, formed by multi-dentate magnetic couplers, bis(benzene)chromium(i) and [1,2,5]thiadiazolo[3,4-: C] [1,2,5]thiadiazolidyl. Dalton Trans. 2018, 47, 9897–9902. [Google Scholar] [CrossRef]
- Ams, M.R.; Trapp, N.; Schwab, A.; Milić, J.V.; Diederich, F. Chalcogen Bonding “2S–2N Squares” versus Competing Interactions: Exploring the Recognition Properties of Sulfur. Chem. Eur. J. 2019, 25, 323–333. [Google Scholar] [CrossRef]
- Riwar, L.J.; Trapp, N.; Root, K.; Zenobi, R.; Diederich, F. Supramolecular Capsules: Strong versus Weak Chalcogen Bonding. Angew. Chem. Int. Ed. 2018, 57, 17259–17264. [Google Scholar] [CrossRef] [PubMed]
- Rahman, F.; Tzeli, D.; Petsalakis, I.D.; Ballester, P.; Rebek, J., Jr.; Yu, Y. Chalcogen Bonding and Hydrophobic Effects Force Molecules into Small Spaces Chalcogen Bonding and Hydrophobic Effects Force Molecules into Small Spaces. J. Am. Chem. Soc. 2020, 142, 5876–5883. [Google Scholar] [CrossRef] [PubMed]
- Langis-Barsetti, S.; Maris, T.; Wuest, J.D. Molecular Organization of 2,1,3-Benzothiadiazoles in the Solid State. J. Org. Chem. 2017, 82, 5034–5045. [Google Scholar] [CrossRef] [PubMed]
- Vogel, L.; Wonner, P.; Huber, S.M. Chalcogen Bonding: An Overview. Angew. Chem. Int. Ed. 2019, 58, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Leroy, C.; Bryce, D.L. Halide ion recognition: Via chalcogen bonding in the solid state and in solution. Directionality and linearity. CrystEngComm 2018, 20, 6406–6411. [Google Scholar] [CrossRef]
- Scilabra, P.; Murray, J.S.; Terraneo, G.; Resnati, G. Chalcogen Bonds in Crystals of Bis(o-anilinium)diselenide Salts. Cryst. Growth Des. 2019, 19, 1149–1154. [Google Scholar] [CrossRef]
- Fourmigué, M.; Dhaka, A. Chalcogen bonding in crystalline diselenides and selenocyanates: From molecules of pharmaceutical interest to conducting materials. Coord. Chem. Rev. 2020, 403, 213084. [Google Scholar] [CrossRef]
- Benz, S.; Poblador-Bahamonde, A.I.; Low-Ders, N.; Matile, S. Catalysis with Pnictogen, Chalcogen, and Halogen Bonds. Angew. Chem. Int. Ed. 2018, 57, 5408–5412. [Google Scholar] [CrossRef] [PubMed]
- Benz, S.; López-Andarias, J.; Mareda, J.; Sakai, N.; Matile, S. Catalysis with Chalcogen Bonds. Angew. Chem. 2017, 129, 830–833. [Google Scholar] [CrossRef]
- Taylor, M.S. Anion recognition based on halogen, chalcogen, pnictogen and tetrel bonding. Coord. Chem. Rev. 2020, 413, 213270. [Google Scholar] [CrossRef]
- Biot, N.; Bonifazi, D. Chalcogen-bond driven molecular recognition at work. Coord. Chem. Rev. 2020, 413, 213243. [Google Scholar] [CrossRef]
- Milios, C.J.; Ioannou, P.V.; Raptopoulou, C.P.; Papaefstathiou, G.S. Crystal engineering with 2,1,3-benzoselenadiazole and mercury(II) chloride. Polyhedron 2009, 28, 3199–3202. [Google Scholar] [CrossRef]
- Mukherjee, G.; Singh, P.; Ganguri, C.; Sharma, S.; Singh, H.B.; Goel, N.; Singh, U.P.; Butcher, R.J. Selenadiazolopyridine: A synthon for supramolecular assembly and complexes with metallophilic interactions. Inorg. Chem. 2012, 51, 8128–8140. [Google Scholar] [CrossRef]
- Risto, M.; Reed, R.W.; Robertson, C.M.; Oilunkaniemi, R.; Laitinen, R.S.; Oakley, R.T. Self-association of the N-methyl benzotellurodiazolylium cation: Implications for the generation of super-heavy atom radicals. Chem. Commun. 2008, 28, 3278–3280. [Google Scholar] [CrossRef]
- Chivers, T.; Gao, X.; Parvez, M. Preparation, Crystal Structures, and Isomerization of the Tellurium Diimide Dimers RNTe(μ-NR’)2TeNR (R = R’ = tBu; R = PPh2NSiMe3, R’ = tBu, tOct): X-ray Structure of the Telluradiazole Dimer [tBu2C6H2N2Te] 2. Inorg. Chem. 1996, 35, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, A.F.; Britten, J.F.; Vargas-Baca, I. The effect of steric hindrance on the association of telluradiazoles through Te-N secondary bonding interactions. Cryst. Growth Des. 2006, 6, 181–186. [Google Scholar] [CrossRef]
- Cozzolino, A.F.; Vargas-Baca, I.; Mansour, S.; Mahmoudkhani, A.H. The nature of the supramolecular association of 1,2,5-chalcogenadiazoles. J. Am. Chem. Soc. 2005, 127, 3184–3190. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.M.; Corless, V.B.; Tran, M.; Jenkins, H.; Britten, J.F.; Vargas-Baca, I. Synthetic, structural, and computational investigations of N-alkyl benzo-2,1,3-selenadiazolium iodides and their supramolecular aggregates. Dalton Trans. 2016, 45, 3285–3293. [Google Scholar] [CrossRef]
- Mancilha, F.S.; DaSilveira Neto, B.A.; Lopes, A.S.; Moreira, P.F.; Quina, F.H.; Gonçalves, R.S.; Dupont, J. Are molecular 5,8-π-extended quinoxaline derivatives good chromophores for photoluminescence applications? Eur. J. Org. Chem. 2006, 4924–4933. [Google Scholar] [CrossRef]
- Nunn, A.J.; Ralph, J.T. Quaternisation of 2,1,3-Benzothiadiazole and 2,1,3-Benzoselenadiazole. Part I. Preparation of Methyl- and Ethyl-2,1,3-benzothiadiazolium and -benzoselenadiazolium Salts. J. Chem. Soc. 1965, 1254, 6769–6777. [Google Scholar] [CrossRef]
- Buu-Hoï, N.P. The Chemistry of Carcinogenic Nitrogen-compounds. Part 111. Polysubstituted Pyrroles and Indoles as Potential Cocarcinogens. J. Chem. Soc. 1949, 2882–2888. [Google Scholar] [CrossRef]
- Oxford Diffraction Ltd. CrysAlis CCD and CrysAlis RED, Version 1.171.36.24; Oxford Diffraction Ltd.: Yarnton, UK, 2012. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.K. ORTEP II, Report ORNL-5138; Oak Ridge National Laboratory: OakRidge, TN, USA, 1976.
- Motherwell, S.; Clegg, S. PLUTO-78, Program for Drawing and Molecular Structure; University of Cambridge: Cambridge, UK, 1978. [Google Scholar]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Van De Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Labanowski, J.K.; Andzelm, J.K. Density Functional Methods in Chemistry; Springer: New York, NY, USA, 1991. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Scalmani, G.; Frisch, M.J.; Mennucci, B.; Tomasi, J.; Cammi, R.; Barone, V. Geometries and properties of excited states in the gas phase and in solution: Theory and application of a time-dependent density functional theory polarizable continuum model. J. Chem. Phys. 2006, 124, 094107. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular interactions in solution: An overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. A new definition of cavities for the computation of solvation free energies by the polarizable continuum model. J. Chem. Phys. 1997, 107, 3210–3221. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 (Revision C.01); Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Chemcraft—Graphical Software for Visualization of Quantum Chemistry Computations. Available online: https://www.chemcraftprog.com (accessed on 20 April 2020).
- Suzuki, T.; Tsuji, T.; Okubo, T.; Okada, A.; Obana, Y.; Fukushima, T.; Miyashi, T.; Yamashita, Y. Preparation, structure, and amphoteric redox properties of p-phenylenediamine-type dyes fused with a chalcogenadiazole unit. J. Org. Chem. 2001, 66, 8954–8960. [Google Scholar] [CrossRef]
- Gomes, A.C.; Biswas, G.; Banerjee, A.; Duax, W.L. Structure of a planar organic compound: 2,1,3-benzoselenadiazole (piaselenole). Acta Crystallogr. Sect. C 1989, 45, 73–75. [Google Scholar] [CrossRef]
- Linder, T.; Badiola, E.; Baumgartner, T.; Sutherland, T.C. Synthesis of π-extended thiadiazole (oxides) and their electronic properties. Org. Lett. 2010, 12, 4520–4523. [Google Scholar] [CrossRef] [PubMed]
- Clark, T. Halogen bonds and σ-holes. Faraday Discuss. 2017, 203, 9–27. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The σ-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef]
- Shukla, S.; Bishnoi, A.; Fatma, S.; Verma, A.K.; Devi, P. Computational and experimental FT-IR, NMR, UV-Vis spectral studies of 5,5′-((4-chlorophenyl)methylene)bis(1,3-dimethyl-6-(methylamino)pyrimidine-2,4(1H,3H)-dione). Chem. Sel. 2018, 3, 7800–7808. [Google Scholar] [CrossRef]
- Ullah, H.; Shah, A.-H.A.; Bilal, S.; Ayub, K. Doping and dedoping processes of polypyrrole: DFT study with hybrid functionals. J. Phys. Chem. C 2014, 118, 17819–17830. [Google Scholar] [CrossRef]
- Bibi, S.; Ullah, H.; Ahmad, S.M.; Ali Shah, A.-H.; Bilal, S.; Tahir, A.A.; Ayub, K. Molecular and electronic structure elucidation of polypyrrole gas sensors. J. Phys. Chem. C 2015, 119, 15994–16003. [Google Scholar] [CrossRef]
- Rad, A.S.; Ayub, K. Adsorption of thiophene on the surfaces of X 12 Y 12 (X = Al, B, and Y = N, P) nanoclusters; A DFT study. J. Mol. Liq. 2017, 238, 303–309. [Google Scholar] [CrossRef]
- Cano Ordaz, J.; Chigo Anota, E.; Salazar Villanueva, M.; Castro, M. Possibility of a magnetic [BN fullerene: B 6 cluster] − nanocomposite as a vehicle for the delivery of dapsone. New J. Chem. 2017, 41, 8045–8052. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| (Å) | 3.35 | 3.35 | 3.45 | 3.45 |
| (Å) | 3.22 | 3.09 | 3.16 | 2.91; 2.97 |
| (°) | 168 | 174 | 169 | 168; 167 |
| δ%a | 96% | 92% | 92% | 84%; 86% |
| 1-MeTfO | 2-MeTfO | 3-MeTfO a | 4-MeTfO | |
|---|---|---|---|---|
| (Å) | 3.32 | 3.32 | 3.42 | 3.42 |
| (Å) | 2.82 | 2.88; 3.11 | 2.82; 3.24 | 2.65; 3.03 |
| (°) | 177 | 164; 158 | 148; 123 | 168; 162 |
| δ% | 85% | 87%; 94% | 82%; 95% | 77%; 89% |
| 1-MeIs a | 2-MeI | 3-MeI | 4-MeI | |
|---|---|---|---|---|
| (Å) | 3.78 | 3.78 | 3.88 | 3.88 |
| (Å) | 3.48; 3.39; 3.31 | 3.27 | 3.18; 3.61 | 3.067; 3.070; 3.55; 3.62 |
| (°) | 160; 171; 170 | 174 | 174; 176 | 178; 176; 175; 167 |
| δ% | 92%; 90%; 88% | 87% | 82%; 93% | 79%; 79%; 91%; 93% |
| 1 | 2 | 3 | 4 | [1-Me]+ | [2-Me]+ | [3-Me]+ | [4-Me]+ | |
|---|---|---|---|---|---|---|---|---|
| Dipole moment | 1.99 | 2.45 | 1.36 | 1.85 | 3.23 | 4.66 | 3.59 | 4.16 |
| EHOMO (eV) | −6.89 | −6.55 | −6.75 | −6.46 | −11.48 | −9.94 | −11.30 | −9.86 |
| ELUMO (eV) | −2.67 | −2.34 | −2.76 | −2.44 | −7.78 | −6.93 | −7.77 | −6.93 |
| ΔEH–L gap (eV) | 4.22 | 4.21 | 4.00 | 4.02 | 3.69 | 3.01 | 3.54 | 2.94 |
| Ionization potential IP (eV) | 6.89 | 6.55 | 6.75 | 6.46 | 11.48 | 9.94 | 11.30 | 9.86 |
| Electron affinity EA (eV) | 2.67 | 2.34 | 2.76 | 2.44 | 7.78 | 6.93 | 7.77 | 6.93 |
| Hardness η (eV) | 2.11 | 2.10 | 2.00 | 2.01 | 1.85 | 1.50 | 1.77 | 1.47 |
| Softness ζ (eV–1) | 0.24 | 0.24 | 0.25 | 0.25 | 0.27 | 0.33 | 0.28 | 0.34 |
| Electronegativity χ (eV) | 4.78 | 4.45 | 4.76 | 4.45 | 9.63 | 8.44 | 9.54 | 8.40 |
| Electrophilicity index ψ (eV) | 5.41 | 4.70 | 5.66 | 4.92 | 25.10 | 23.65 | 25.70 | 24.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfuth, J.; Zadykowicz, B.; Sikorski, A.; Połoński, T.; Eichstaedt, K.; Olszewska, T. Effect of Aromatic System Expansion on Crystal Structures of 1,2,5-Thia- and 1,2,5-Selenadiazoles and Their Quaternary Salts: Synthesis, Structure, and Spectroscopic Properties. Materials 2020, 13, 4908. https://doi.org/10.3390/ma13214908
Alfuth J, Zadykowicz B, Sikorski A, Połoński T, Eichstaedt K, Olszewska T. Effect of Aromatic System Expansion on Crystal Structures of 1,2,5-Thia- and 1,2,5-Selenadiazoles and Their Quaternary Salts: Synthesis, Structure, and Spectroscopic Properties. Materials. 2020; 13(21):4908. https://doi.org/10.3390/ma13214908
Chicago/Turabian StyleAlfuth, Jan, Beata Zadykowicz, Artur Sikorski, Tadeusz Połoński, Katarzyna Eichstaedt, and Teresa Olszewska. 2020. "Effect of Aromatic System Expansion on Crystal Structures of 1,2,5-Thia- and 1,2,5-Selenadiazoles and Their Quaternary Salts: Synthesis, Structure, and Spectroscopic Properties" Materials 13, no. 21: 4908. https://doi.org/10.3390/ma13214908
APA StyleAlfuth, J., Zadykowicz, B., Sikorski, A., Połoński, T., Eichstaedt, K., & Olszewska, T. (2020). Effect of Aromatic System Expansion on Crystal Structures of 1,2,5-Thia- and 1,2,5-Selenadiazoles and Their Quaternary Salts: Synthesis, Structure, and Spectroscopic Properties. Materials, 13(21), 4908. https://doi.org/10.3390/ma13214908