Effects of Doped Elements (Si, Cr, W and Nb) on the Stability, Mechanical Properties and Electronic Structures of MoAlB Phase by the First-Principles Calculation



Abstract
1. Introduction
2. Methodology
3. Results and Discussion
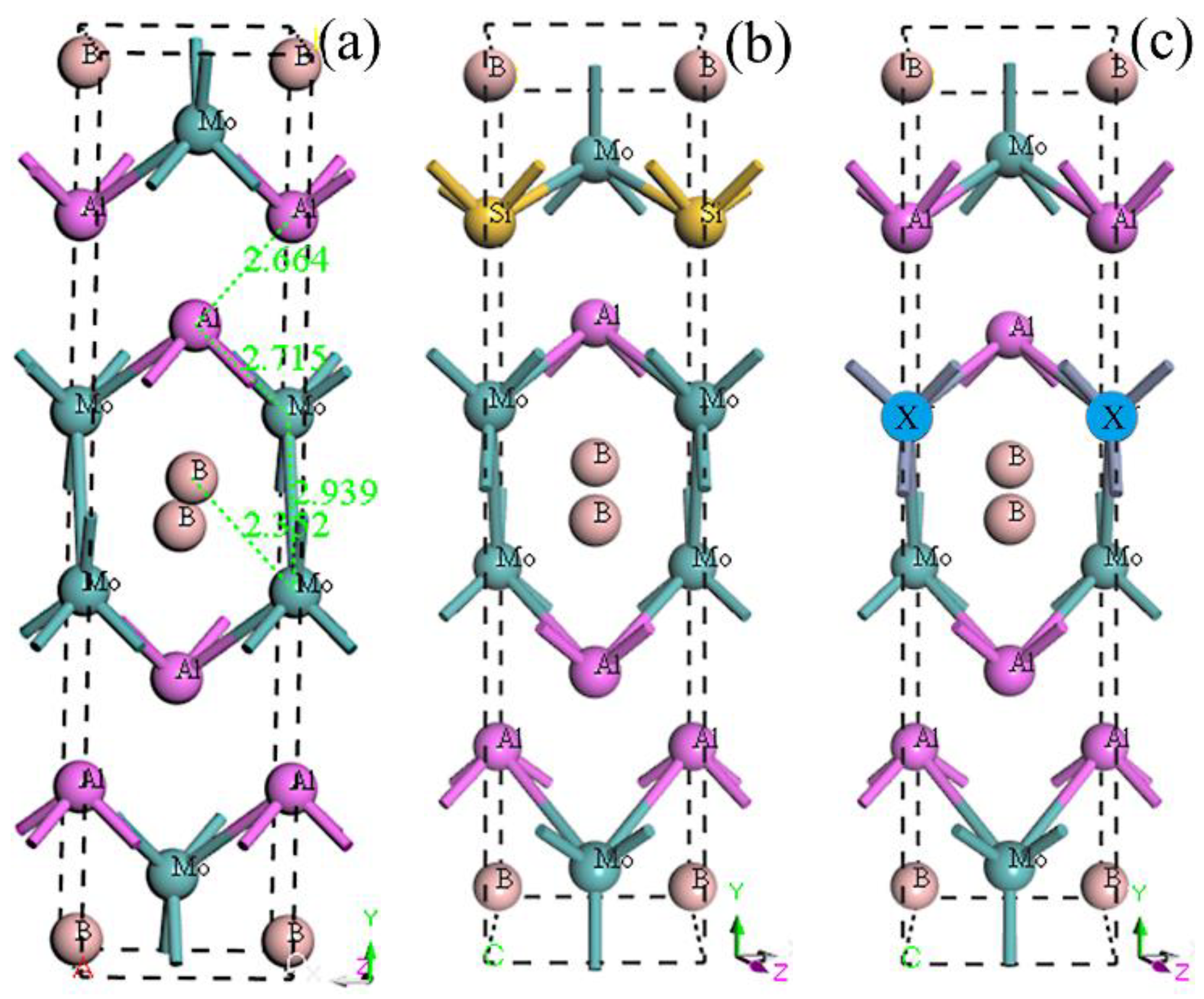
3.1. Equilibrium Structure and Stability
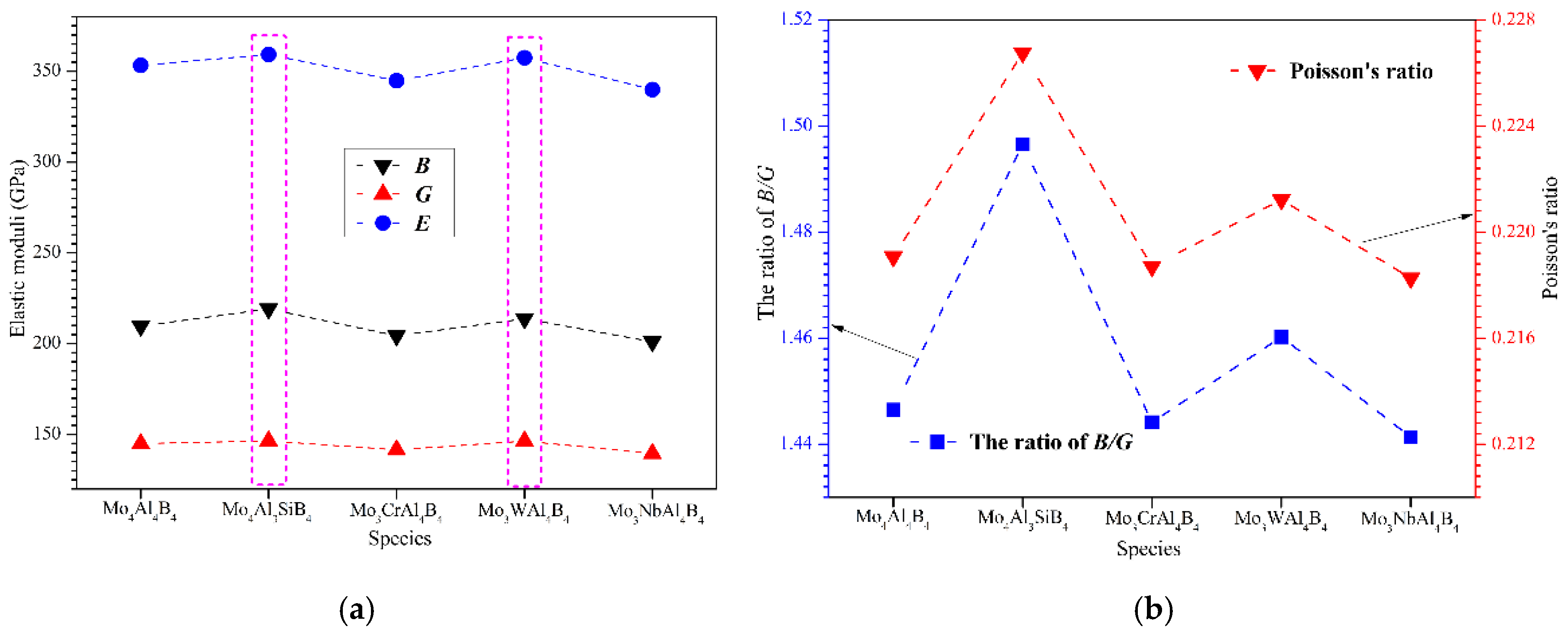
3.2. Elastic Properties
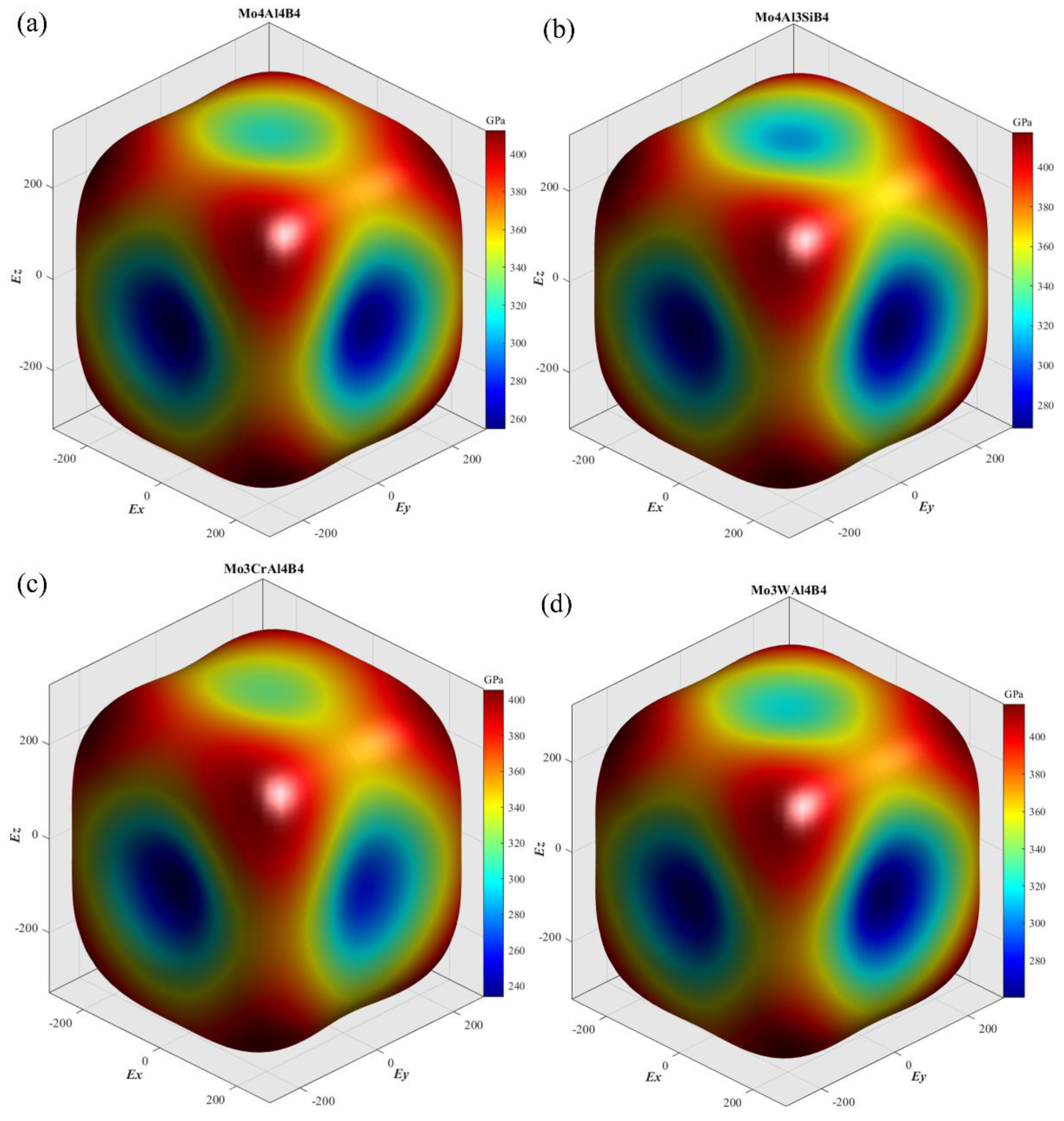
3.3. Elastic Anisotropy
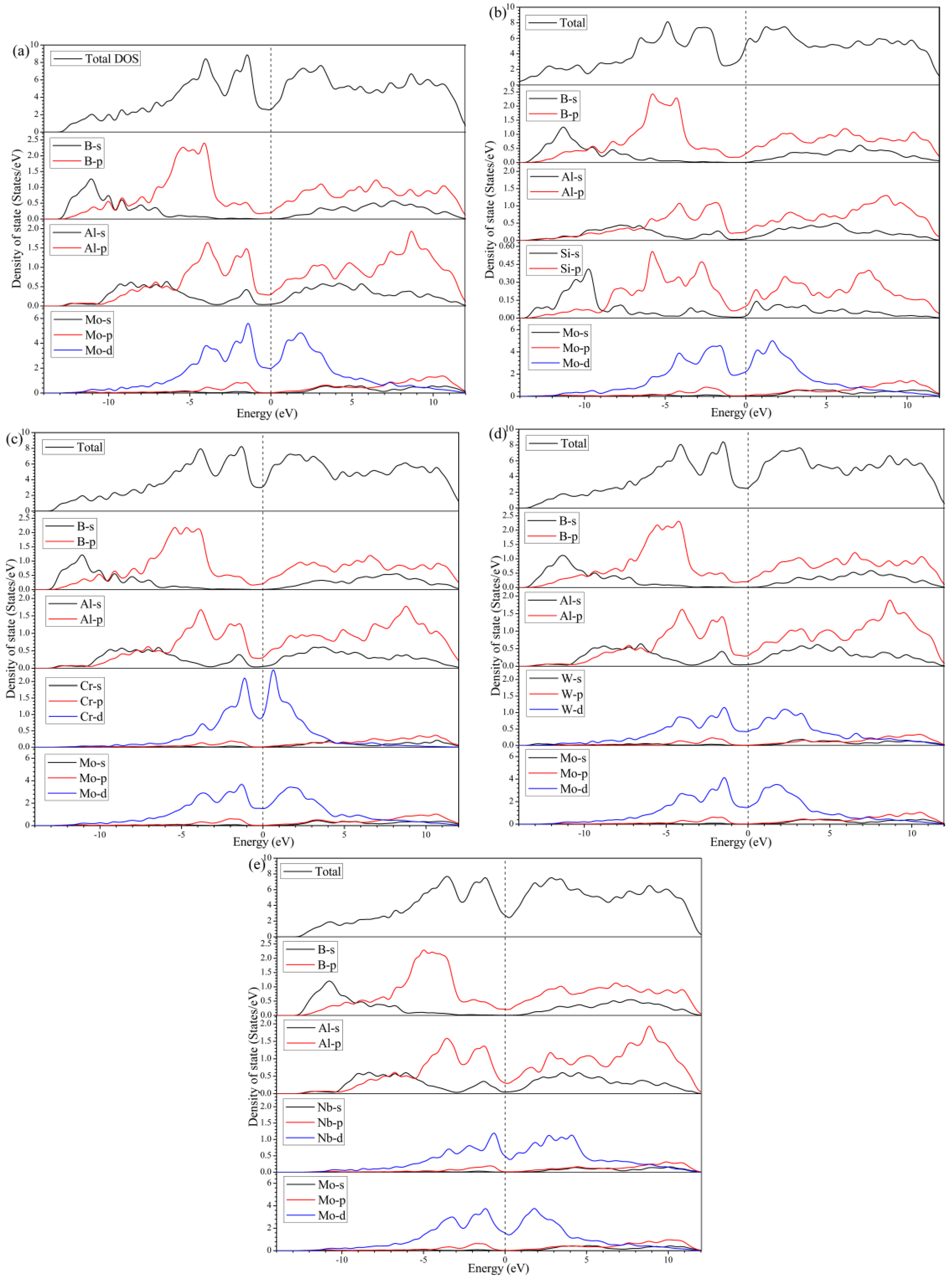
3.4. Electronic Structures
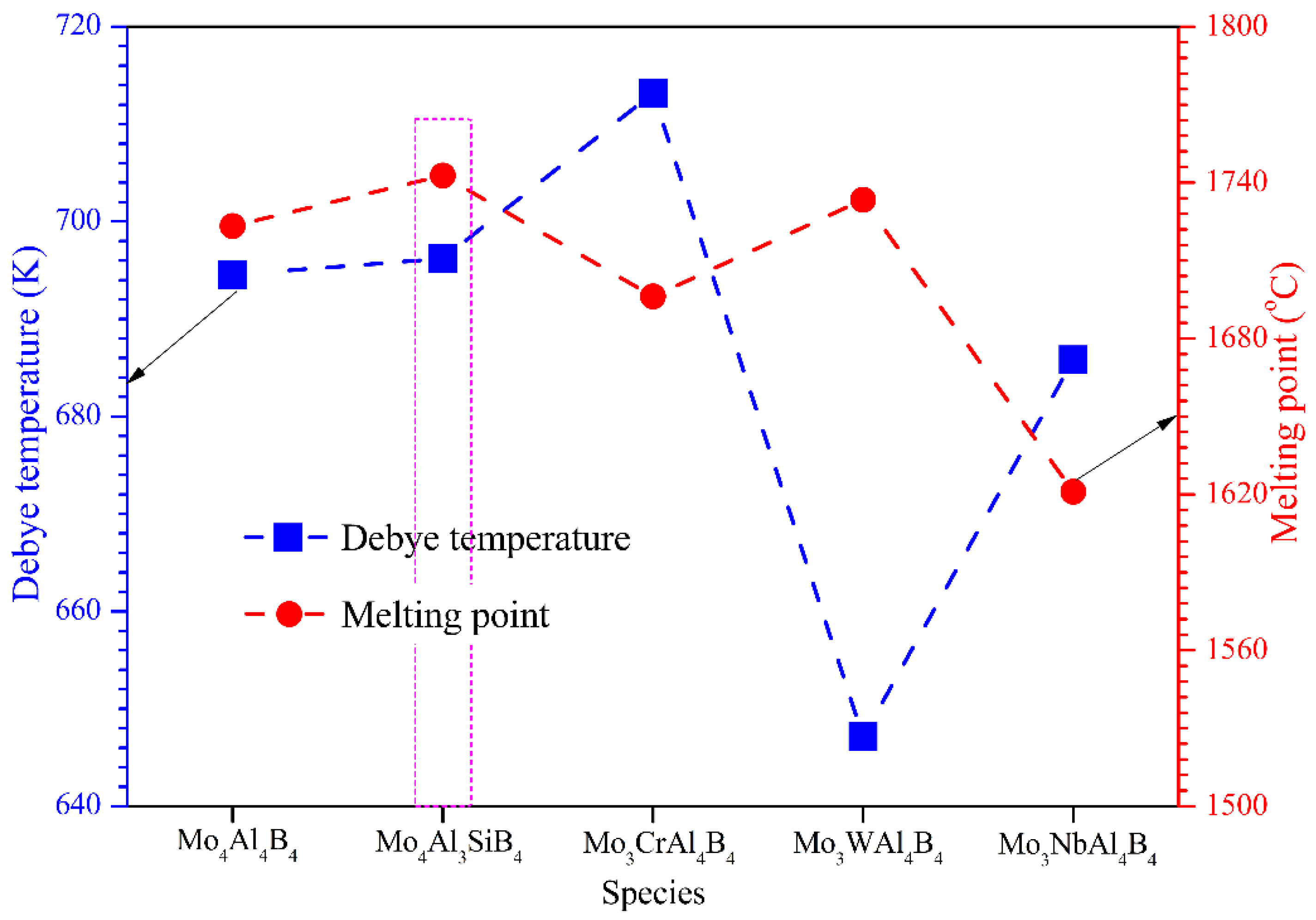
3.5. Debye Temperature
4. Conclusions
- The initial MoAlB, as well as the element (Si, Cr, W and Nb) doped crystals, are thermodynamically stable, confirmed by the negative cohesive energy and formation enthalpy.
- MoAlB possesses a considerably high Young’s modulus, but it is intrinsically brittle with a B/G ratio less than 1.75. Si and W doping can not only enhance the Young’s modulus of MoAlB but also improve its ductility. However, Cr and Nb play a negative role in the elastic moduli as well as the ductility.
- MoAlB exhibits significant elastic anisotropy: it shows the largest stiffness along the [1 1 1] direction, while it is the most compressible along [0 1 0]. Si and W doping benefit weakening of the anisotropy of Young’s modulus by strengthening the Young’s moduli along [0 1 0].
- According to the investigation of DOS, Mulliken populations, and electron density distribution, strong covalent bonding is supposed to exist in MoAlB, combined with ionic and metallic bonding. Si doping significantly intensifies the covalent bondings of Si-B, Si-Mo, and B-B, which accounts for the improved mechanical properties.
- Si doping can simultaneously increase the Debye temperature and melting point of MoAlB, which apparently shows superior ameliorating effects compared to the other elements. Overall, Si is supposed to be the best dopant in optimizing the comprehensive performance of MoAlB.
Author Contributions
Funding
Conflicts of Interest
References
- Türkmen, İ.; Yalamaç, E.; Keddam, M. Investigation of tribological behaviour and diffusion model of Fe2B layer formed by pack-boriding on SAE 1020 steel. Surf. Cost. Tech. 2019, 377, 124888. [Google Scholar] [CrossRef]
- Iyer, A.K.; Zhang, Y.; Scheifers, J.P.; Fokwa, B.P.T. Structural variations, relationships and properties of M2B metal borides. J. Solid State Chem. 2019, 270, 618–635. [Google Scholar] [CrossRef]
- Fahrenholtz, W.G.; Hilmas, G.E.; Talmy, I.G.; Zaykoski, J.A. Refractory Diborides of Zirconium and Hafnium. J. Am. Ceram. Soc. 2007, 90, 1347–1364. [Google Scholar] [CrossRef]
- Jeitschko, W. Die Kristallstruktur von MoAlB. Mon. Chem. 1966, 97, 1472–1476. [Google Scholar] [CrossRef]
- Kota, S.; Zapata-Solvas, E.; Ly, A.; Lu, J.; Elkassabany, O.; Huon, A.; Lee, W.E.; Hultman, L.; May, S.J.; Barsoum, M.W. Synthesis and Characterization of an Alumina Forming Nanolaminated Boride: MoAlB. Sci. Rep. 2016, 6, 26475. [Google Scholar] [CrossRef] [PubMed]
- Barsoum, M.W. The MN+1AXN phases: A new class of solids: Thermodynamically stable nanolaminates. Prog. Solid State Chem. 2000, 28, 201–281. [Google Scholar] [CrossRef]
- Radovic, M.; Barsoum, M.W. MAX phases: Bridging the gap between metals and ceramics. Am. Ceram. Soc. Bull. 2013, 92, 20–27. [Google Scholar]
- Wang, X.H.; Zhou, Y.C. Layered Machinable and Electrically Conductive Ti2AlC and Ti3AlC2 Ceramics: A Review. J. Mater. Sci. Technol. 2010, 26, 385–416. [Google Scholar] [CrossRef]
- Eklund, P.; Beckers, M.; Jansson, U.; Högberg, H.; Hultman, L. The Mn+1AXn phases: Materials science and thin-film processing. Thin Solid Films. 2010, 518, 1851–1878. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Zheng, L.; Ma, Y.; Lu, X.; Sun, Y.; Zhou, Y. Ti5Al2C3: A New Ternary Carbide Belonging to MAX Phases in the Ti–Al–C System. J. Am. Ceram. Soc. 2012, 95, 1508–1510. [Google Scholar] [CrossRef]
- Ade, M.; Hillebrecht, H. Ternary Borides Cr2AlB2, Cr3AlB4, and Cr4AlB6: The First Members of the Series (CrB2)nCrAl with n = 1, 2, 3 and a Unifying Concept for Ternary Borides as MAB-Phases. Inorg. Chem. 2015, 54, 6122–6135. [Google Scholar] [CrossRef] [PubMed]
- Kota, S.; Zapata-Solvas, E.; Chen, Y.; Radovic, M.; Lee, W.E.; Barsoum, M.W. Isothermal and Cyclic Oxidation of MoAlB in Air from 1100 °C to 1400 °C. J. Electrochem. Soc. 2017, 164, 930–938. [Google Scholar] [CrossRef]
- Lou, T. Microstructure and Properties of Spark Plasma Sintered MoAlB Ceramics. Master’s Thesis, University of Nebraska-Lincoln, Lincoln, NE, USA, 2016. [Google Scholar]
- Xu, L.; Shi, O.; Liu, C.; Zhu, D.; Grasso, S.; Hu, C. Synthesis, microstructure and properties of MoAlB ceramics. Ceram. Int. 2018, 44, 13396–13401. [Google Scholar] [CrossRef]
- Bei, G.; van der Zwaag, S.; Kota, S.; Barsoum, M.W.; Sloof, W.G. Ultra-high temperature ablation behavior of MoAlB ceramics under an oxyacetylene flame. J. Eur. Ceram. Soc. 2019, 39, 2010–2017. [Google Scholar] [CrossRef]
- Benamor, A.; Kota, S.; Chiker, N.; Haddad, A.; Hadji, Y.; Natu, V.; Abdi, S.; Yahi, M.; Benamar, M.E.; Sahraoui, T.; et al. Friction and wear properties of MoAlB against Al2O3 and 100Cr6 steel counterparts. J. Eur. Ceram. Soc. 2019, 39, 868–877. [Google Scholar] [CrossRef]
- Chen, Y.; Kota, S.; Barsoum, M.W.; Radovic, M. Compressive deformation of MoAlB up to 1100 °C. J. Alloy. Compd. 2019, 774, 1216–1222. [Google Scholar] [CrossRef]
- Kota, S.; Sokol, M.; Barsoum, M.W. A progress report on the MAB phases: Atomically laminated, ternary transition metal borides. Int. Mater. Rev. 2020, 65, 226–255. [Google Scholar] [CrossRef]
- Lu, X.; Li, S.; Zhang, W.; Yao, B.; Yu, W.; Zhou, Y. Crack healing behavior of a MAB phase: MoAlB. J. Eur. Ceram. Soc. 2019, 39, 4023–4028. [Google Scholar] [CrossRef]
- Lu, X.; Li, S.; Zhang, W.; Yu, W.; Zhou, Y. Thermal shock behavior of a nanolaminated ternary boride: MoAlB. Ceram. Int. 2019, 45, 9386–9389. [Google Scholar] [CrossRef]
- Shi, O.; Xu, L.; Jiang, A.; Xu, Q.; Xiao, Y.; Zhu, D.; Grasso, S.; Hu, C. Synthesis and oxidation resistance of MoAlB single crystals. Ceram. Int. 2019, 45, 2446–2450. [Google Scholar] [CrossRef]
- Wang, S.; Xu, Y.; Yu, Z.; Tan, H.; Du, S.; Zhang, Y.; Yang, J.; Liu, W. Synthesis, microstructure and mechanical properties of a MoAlB ceramic prepared by spark plasma sintering from elemental powders. Ceram. Int. 2019, 45, 23515–23521. [Google Scholar] [CrossRef]
- Su, X.; Dong, J.; Chu, L.; Sun, H.; Grasso, S.; Hu, C. Synthesis, microstructure and properties of MoAlB ceramics prepared by in situ reactive spark plasma sintering. Ceram. Int. 2020, 46, 15214–15221. [Google Scholar] [CrossRef]
- Yu, Z.; Tan, H.; Wang, S.; Cheng, J.; Sun, Q.; Yang, J.; Liu, W. High-temperature tribological behaviors of MoAlB ceramics sliding against Al2O3 and Inconel 718 alloy. Ceram. Int. 2020, 46, 14713–14720. [Google Scholar] [CrossRef]
- Wei, X.; Chen, Z.; Zhong, J.; Wang, L.; Yang, W.; Wang, Y. Effect of alloying elements on mechanical, electronic and magnetic properties of Fe2B by first-principles investigations. Comp. Mater. Sci. 2018, 147, 322–330. [Google Scholar]
- Rojacz, H.; Premauer, M.; Varga, M. Alloying and strain hardening effects in abrasive contacts on iron based alloys. Wear 2018, 410, 173–180. [Google Scholar] [CrossRef]
- Park, N.; Lee, S.-C.; Cha, P.-R. Effects of alloying elements on the stability and mechanical properties of Fe3Al from first-principles calculations. Comp. Mater. Sci. 2018, 146, 303–309. [Google Scholar] [CrossRef]
- Wang, C.; Yan, L.; Han, J.; Xu, W.; Deng, B.; Liu, X. Effects of alloying elements on the structural, elastic and thermodynamic properties of Co3Ta compounds from first-principles calculations. J. Alloy. Compd. 2017, 726, 490–497. [Google Scholar] [CrossRef]
- Jian, Y.; Huang, Z.; Xing, J.; Gao, Y. Effects of chromium on the morphology and mechanical properties of Fe2B intermetallic in Fe-3.0B alloy. J. Mater. Sci. 2018, 53, 5329–5338. [Google Scholar] [CrossRef]
- Jian, Y.; Huang, Z.; Liu, X.; Sun, J.; Xing, J. Microstructure, mechanical properties and toughening mechanism of directional Fe2B crystal in Fe-B alloy with trace Cr addition. J. Mater. Sci. Technol. 2020, 57, 172–179. [Google Scholar] [CrossRef]
- Jian, Y.; Huang, Z.; Xing, J.; Guo, X.; Wang, Y.; Lv, Z. Effects of Mn addition on the two-body abrasive wear behavior of Fe-3.0 wt% B alloy. Tribol. Int. 2016, 103, 243–251. [Google Scholar] [CrossRef]
- Shen, Y.; Huang, Z.; Xiao, P.; Zhang, L.; Li, K.; Cao, Z.; Jian, Y. Sintering mechanism, microstructure evolution and nanomechanical properties of Cr-added Mo2FeB2 based cermets. Ceram. Int. 2020, 46, 15482–15491. [Google Scholar] [CrossRef]
- Okada, S.; Iizumi, K.; Kudaka, K.; Kudou, K.; Miyamoto, M.; Yu, Y.; Lundström, T. Single Crystal Growth of (MoXCr1−X)AlB and (MoXW1−X)AlB by Metal Al Solutions and Properties of the Crystals. J. Solid State Chem. 1997, 133, 36–43. [Google Scholar] [CrossRef]
- Ma, P.; Li, S.; Hu, J.; Lu, X.; Yu, W.; Zhou, Y. Processing and characterization of MoAl1−xSixB solid solutions. J. Alloy. Compd. 2020, 814, 152290. [Google Scholar] [CrossRef]
- Li, X.; Cui, H.; Zhang, R. First-principles study of the electronic and optical properties of a new metallic MoAlB. Sci. Rep. 2016, 6, 39790. [Google Scholar] [CrossRef]
- Bai, Y.; Qi, X.; Duff, A.; Li, N.; Kong, F.; He, X.; Wang, R.; Lee, W.E. Density functional theory insights into ternary layered boride MoAlB. Acta Mater. 2017, 132, 69–81. [Google Scholar] [CrossRef]
- Xiang, H.; Feng, Z.; Li, Z.; Zhou, Y. Theoretical investigations on mechanical and dynamical properties of MAlB (M = Mo, W) nanolaminated borides at ground-states and elevated temperatures. J. Alloy. Compd. 2018, 738, 461–472. [Google Scholar] [CrossRef]
- Ali, M.M.; Hadi, M.A.; Rahman, M.L.; Haque, F.H.; Haider, A.F.M.Y.; Aftabuzzaman, M. DFT investigations into the physical properties of a MAB phase Cr4AlB4. J. Alloy. Compd. 2020, 821, 153547. [Google Scholar] [CrossRef]
- Payne, M.C.; Teter, M.P.; Allan, D.C.; Arias, T.A.; Joannopoulos, A.J. Iterative minimization techniques for ab initio total-energy calculations: Molecular dynamics and conjugate gradients. Rev. Mod. Phys. 1992, 64, 1045–1097. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.; Probert, M.A.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter. 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef] [PubMed]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of Crystals with the Quasi-Newton Method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Zhang, P.; Zhou, Y.; Yang, J.; Li, D.; Ren, X.; Yang, Y.; Yang, Q. Optimization on mechanical properties of Fe7−xCrxC3 carbides by first-principles investigation. J. Alloy. Compd. 2013, 560, 49–53. [Google Scholar] [CrossRef]
- Lu, J.; Kota, S.; Barsoum, M.W.; Hultman, L. Atomic structure and lattice defects in nanolaminated ternary transition metal borides. Mater. Res. Lett. 2016, 5, 235–241. [Google Scholar] [CrossRef]
- Dai, F.-Z.; Feng, Z.; Zhou, Y. First-principles investigation on the chemical bonding, elastic properties and ideal strengths of MoAlB and WAlB nanolaminated MAB phases. Comp. Mater. Sci. 2018, 147, 331–337. [Google Scholar] [CrossRef]
- Feng, J.; Xiao, B.; Chen, J.; Du, Y.; Yu, J.; Zhou, R. Stability, thermal and mechanical properties of PtxAly compounds. Mater. Design. 2011, 32, 3231–3239. [Google Scholar] [CrossRef]
- Wu, J.; Chong, X.; Jiang, Y.; Feng, J. Stability, electronic structure, mechanical and thermodynamic properties of Fe-Si binary compounds. J. Alloy. Compds. 2017, 693, 859–870. [Google Scholar] [CrossRef]
- Jian, Y.; Huang, Z.; Xing, J.; Sun, L.; Liu, Y.; Gao, P. Phase stability, mechanical properties and electronic structures of Ti Al binary compounds by first principles calculations. Mater. Chem. Phys. 2019, 221, 311–321. [Google Scholar] [CrossRef]
- Jian, Y.; Huang, Z.; Liu, X.; Xing, J. Comparative investigation on the stability, electronic structures and mechanical properties of Mo2FeB2 and Mo2NiB2 ternary borides by first-principles calculations. Results Phys. 2019, 15, 102698. [Google Scholar] [CrossRef]
- Patil, S.; Khare, S.; Tuttle, B.; Bording, J.; Kodambaka, S. Mechanical stability of possible structures of PtN investigated using first-principles calculations. Phys. Rev. B 2006, 73, 104118. [Google Scholar] [CrossRef]
- Born, M.; Huang, K. Dynamical Theory of Crystal Lattices; The Clarendon Press: Oxford, UK, 1954. [Google Scholar]
- Zhou, Y.; Xiang, H.; Zhang, H.; Dai, F.-Z. Theoretical prediction on the stability, electronic structure, room and elevated temperature properties of a new MAB phase Mo2AlB2. J. Mater. Sci. Technol. 2019, 35, 2926–2934. [Google Scholar] [CrossRef]
- Chung, D.; Buessem, W. The Voigt-Reuss-Hill Approximation and Elastic Moduli of Polycrystalline MgO, CaF2, β-ZnS, ZnSe, and CdTe. J. Appl. Phys. 1967, 38, 2535–2540. [Google Scholar] [CrossRef]
- Kota, S.; Agne, M.; Zapata-Solvas, E.; Dezellus, O.; Lopez, D.; Gardiola, B.; Radovic, M.; Barsoum, M.W. Elastic properties, thermal stability, and thermodynamic parameters of MoAlB. Phys. Rev. B 2017, 95. [Google Scholar] [CrossRef]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dubl. Phil. Mag. J. Sci. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- He, L.; Lin, Z.; Wang, J.; Bao, Y.; Li, M.; Zhou, Y. Synthesis and Characterization of Bulk Zr2Al3C4 Ceramic. J. Am. Ceram. Soc. 2007, 90, 3687–3689. [Google Scholar] [CrossRef]
- Bai, Y.; Duff, A.; Jayaseelan, D.D.; Wang, R.; He, X.; Lee, W.E. DFT Predictions of Crystal Structure, Electronic Structure, Compressibility, and Elastic Properties of Hf–Al–C Carbides. J. Am. Ceram. Soc. 2016, 99, 3449–3457. [Google Scholar] [CrossRef]
- Sun, Z.; Li, S.; Ahuja, R.; Schneider, J.M. Calculated elastic properties of M2AlC (M = Ti, V, Cr, Nb and Ta). Solid State Commun. 2004, 129, 589–592. [Google Scholar] [CrossRef]
- Feng, J.; Xiao, B.; Zhou, R.; Pan, W.; Clarke, D.R. Anisotropic elastic and thermal properties of the double perovskite slab–rock salt layer Ln2SrAl2O7 (Ln=La, Nd, Sm, Eu, Gd or Dy) natural superlattice structure. Acta Mater. 2012, 60, 3380–3392. [Google Scholar] [CrossRef]
- Chen, H.; Yang, L.; Long, J. First-principles investigation of the elastic, Vickers hardness and thermodynamic properties of Al–Cu intermetallic compounds. Superlattices Microst. 2015, 79, 156–165. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Formula | Lattice Parameters (Å) | Vol (Å3) | ρ (g/cm3) | Ecoh (eV/atom) | ∆Hr (eV/atom) | ||
|---|---|---|---|---|---|---|---|
| a | b | c | |||||
| Mo4Al4B4 | 3.214 | 14.038 | 3.110 | 140.292 | 6.332 | −7.628 | −0.449 |
| 3.210 [5] | 13.980 [5] | 3.100 [5] | ─ | ─ | ─ | ─ | |
| 3.200 [46] | 13.960 [46] | 3.100 [46] | ─ | ─ | ─ | ─ | |
| 3.216 [47] | 14.062 [47] | 3.103 [47] | ─ | ─ | ─ | ─ | |
| Mo4Al3SiB4 | 3.211 | 13.766 | 3.122 | 138.016 | 6.449 | −7.746 | −0.426 |
| Mo3CrAl4B4 | 3.172 | 13.977 | 3.080 | 136.586 | 5.969 | −7.503 | −0.416 |
| Mo3WAl4B4 | 3.216 | 14.024 | 3.112 | 140.334 | 7.370 | −7.633 | −0.412 |
| Mo3NbAl4B4 | 3.226 | 14.194 | 3.129 | 143.283 | 6.164 | −7.590 | −0.477 |
| Species | C11 | C12 | C13 | C22 | C23 | C33 | C44 | C55 | C66 |
|---|---|---|---|---|---|---|---|---|---|
| Mo4Al4B4 | 348.66 | 140.80 | 150.45 | 322.30 | 121.32 | 398.32 | 193.60 | 159.73 | 176.43 |
| Mo4Al3SiB4 | 357.48 | 150.87 | 149.71 | 351.81 | 134.92 | 393.61 | 191.41 | 162.74 | 176.75 |
| Mo3CrAl4B4 | 341.88 | 139.62 | 139.42 | 304.52 | 124.98 | 393.80 | 190.72 | 157.27 | 173.26 |
| Mo3WAl4B4 | 350.73 | 143.95 | 154.20 | 333.69 | 124.09 | 400.90 | 194.74 | 162.60 | 177.49 |
| Mo3NbAl4B4 | 326.66 | 138.54 | 145.38 | 297.68 | 125.97 | 374.18 | 185.88 | 162.37 | 186.67 |
| Species | BV | BR | BVRH | GV | GR | GVRH | E | ν | B/G |
|---|---|---|---|---|---|---|---|---|---|
| Mo4Al4B4 | 210.49 | 208.67 | 209.58 | 149.73 | 140.04 | 144.89 | 353.26 | 0.219 | 1.446 |
| 232.90 [56] | 151.20 [56] | 372.90 [56] | |||||||
| Mo4Al3SiB4 | 219.32 | 218.94 | 219.13 | 150.67 | 142.18 | 146.42 | 359.25 | 0.227 | 1.497 |
| Mo3CrAl4B4 | 205.36 | 203.29 | 204.33 | 146.66 | 136.31 | 141.49 | 344.86 | 0.219 | 1.444 |
| Mo3WAl4B4 | 214.42 | 212.99 | 213.71 | 151.17 | 141.54 | 146.35 | 357.46 | 0.221 | 1.460 |
| Mo3NbAl4B4 | 202.03 | 200.13 | 201.08 | 146.23 | 132.79 | 139.51 | 339.91 | 0.218 | 1.441 |
| Species | A1 | A2 | A3 | AU | AB | AG |
|---|---|---|---|---|---|---|
| Mo4Al4B4 | 1.736 | 1.337 | 1.813 | 0.355 | 0.435% | 3.344% |
| Mo4Al3SiB4 | 1.695 | 1.369 | 1.735 | 0.301 | 0.089% | 2.901% |
| Mo3CrAl4B4 | 1.670 | 1.403 | 1.887 | 0.390 | 0.505% | 3.658% |
| Mo3WAl4B4 | 1.757 | 1.337 | 1.790 | 0.347 | 0.334% | 3.291% |
| Mo3NbAl4B4 | 1.813 | 1.547 | 2.150 | 0.516 | 0.474% | 4.817% |
| Mo4Al4B4 | Mo4Al3SiB4 | Mo3CrAl4B4 | ||||||
| Bonds | P | L (Å) | Bonds | P | L (Å) | Bonds | P | L (Å) |
| B-B | 1.37 | 1.81 | B-B | 1.42 | 1.80 | B-B | 1.40 | 1.80 |
| Al-B | 0.16 | 2.32 | Al/Si-B | 0.20 | 2.29 | Al-B | 0.15 | 2.30 |
| Mo-B | −0.01 | 2.35 | Mo-B | 0.00 | 2.34 | Mo/Cr-B | −0.02 | 2.31 |
| Mo-B | 0.66 | 2.37 | Mo-B | 0.57 | 2.38 | Mo/Cr-B | 0.66 | 2.34 |
| Al-Al | 0.95 | 2.67 | Al/Si-Al | 0.95 | 2.64 | Al-Al | 0.97 | 2.66 |
| Al-Mo | 0.73 | 2.71 | Al/Si-Mo | 0.81 | 2.69 | Al-Mo/Cr | 0.71 | 2.69 |
| Al-Mo | −0.22 | 2.99 | Al-Mo | −0.23 | 2.87 | Al-Cr | −0.18 | 2.94 |
| Mo-Mo | −0.96 | 2.94 | Mo-Mo | −0.93 | 2.97 | Mo/Cr-Mo | −0.96 | 2.90 |
| Mo3WAl4B4 | Mo3NbAl4B4 | |||||||
| Bonds | P | L (Å) | Bonds | P | L (Å) | |||
| B | 1.33 | 1.82 | B-B | 1.39 | 1.82 | |||
| Al-B | 0.13 | 2.32 | Al-B | 0.15 | 2.36 | |||
| Mo/W-B | 0.00 | 2.35 | Mo/Nb-B | 0.00 | 2.35 | |||
| Mo/W-B | 0.72 | 2.37 | Mo/Nb-B | 0.65 | 2.38 | |||
| Al-Al | 0.92 | 2.66 | Al-Al | 0.96 | 2.68 | |||
| Al-Mo/W | 0.79 | 2.71 | Al-Mo/Nb | 0.73 | 2.74 | |||
| Al-Mo/W | −0.20 | 2.98 | Al-Mo | −0.22 | 2.98 | |||
| Mo-Mo/W | −0.81 | 2.94 | Mo/Nb-Mo | −1.01 | 2.95 | |||
| Species | Vl (Km/s) | Vt (Km/s) | Vm(Km/s) | θD (K) |
|---|---|---|---|---|
| Mo4Al4B4 | 7.98 | 4.78 | 5.29 | 694.54 |
| Mo4Al3SiB4 | 8.02 | 4.77 | 5.28 | 696.19 |
| Mo3CrAl4B4 | 8.11 | 4.87 | 5.39 | 713.18 |
| Mo3WAl4B4 | 7.45 | 4.46 | 4.93 | 647.12 |
| Mo3NbAl4B4 | 7.92 | 4.76 | 5.26 | 685.81 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jian, Y.; Huang, Z.; Wang, Y.; Xing, J. Effects of Doped Elements (Si, Cr, W and Nb) on the Stability, Mechanical Properties and Electronic Structures of MoAlB Phase by the First-Principles Calculation. Materials 2020, 13, 4221. https://doi.org/10.3390/ma13194221
Jian Y, Huang Z, Wang Y, Xing J. Effects of Doped Elements (Si, Cr, W and Nb) on the Stability, Mechanical Properties and Electronic Structures of MoAlB Phase by the First-Principles Calculation. Materials. 2020; 13(19):4221. https://doi.org/10.3390/ma13194221
Chicago/Turabian StyleJian, Yongxin, Zhifu Huang, Yu Wang, and Jiandong Xing. 2020. "Effects of Doped Elements (Si, Cr, W and Nb) on the Stability, Mechanical Properties and Electronic Structures of MoAlB Phase by the First-Principles Calculation" Materials 13, no. 19: 4221. https://doi.org/10.3390/ma13194221
APA StyleJian, Y., Huang, Z., Wang, Y., & Xing, J. (2020). Effects of Doped Elements (Si, Cr, W and Nb) on the Stability, Mechanical Properties and Electronic Structures of MoAlB Phase by the First-Principles Calculation. Materials, 13(19), 4221. https://doi.org/10.3390/ma13194221