CaCO3 as an Environmentally Friendly Renewable Material for Drug Delivery Systems: Uptake of HSA-CaCO3 Nanocrystals Conjugates in Cancer Cell Lines

 ,
,  ,
, 
 , , ,
, , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. CaCO3 Nanocrystals Synthesis and Bioconjugation
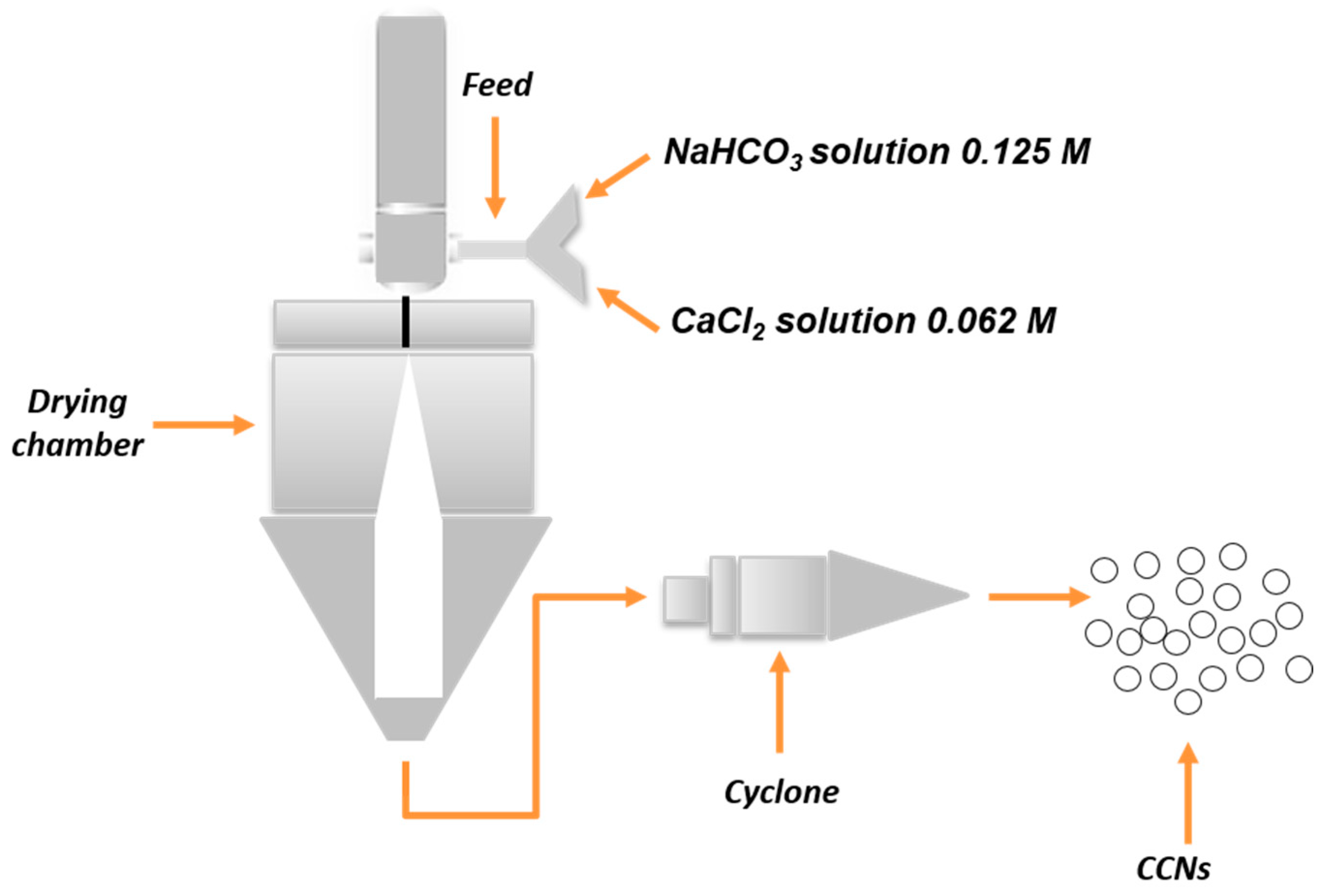
2.2.1. CaCO3 Nanocrystals Synthesis Procedure
2.2.2. Amino-Functionalization of CaCO3 Nanocrystals
2.2.3. Fluorescein Coupling of Amino-Functionalized CaCO3 Nanocrystals
2.2.4. Adsorption of HSA on CaCO3 nanocrystals
2.3. Material Characterization
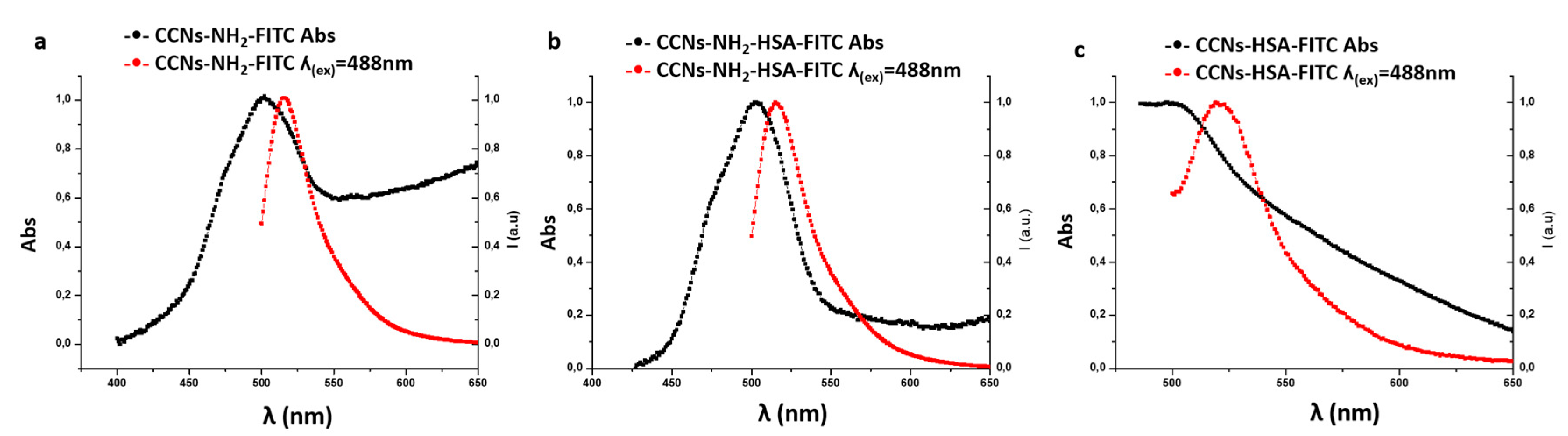
2.3.1. Fluorescence Stability of FITC-Coupled Nanocrystals
2.3.2. Evaluation of HSA Adsorbed on CaCO3 Nanocrystals
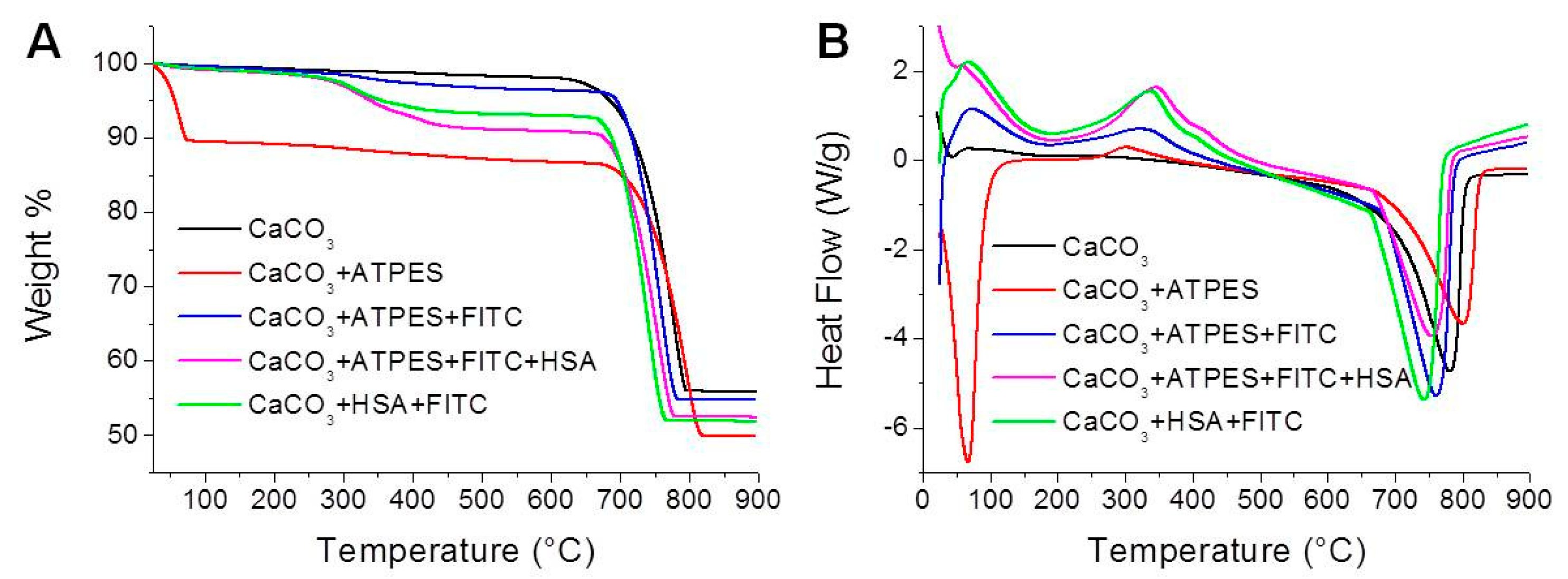
2.3.3. TGA/DSC Measurements
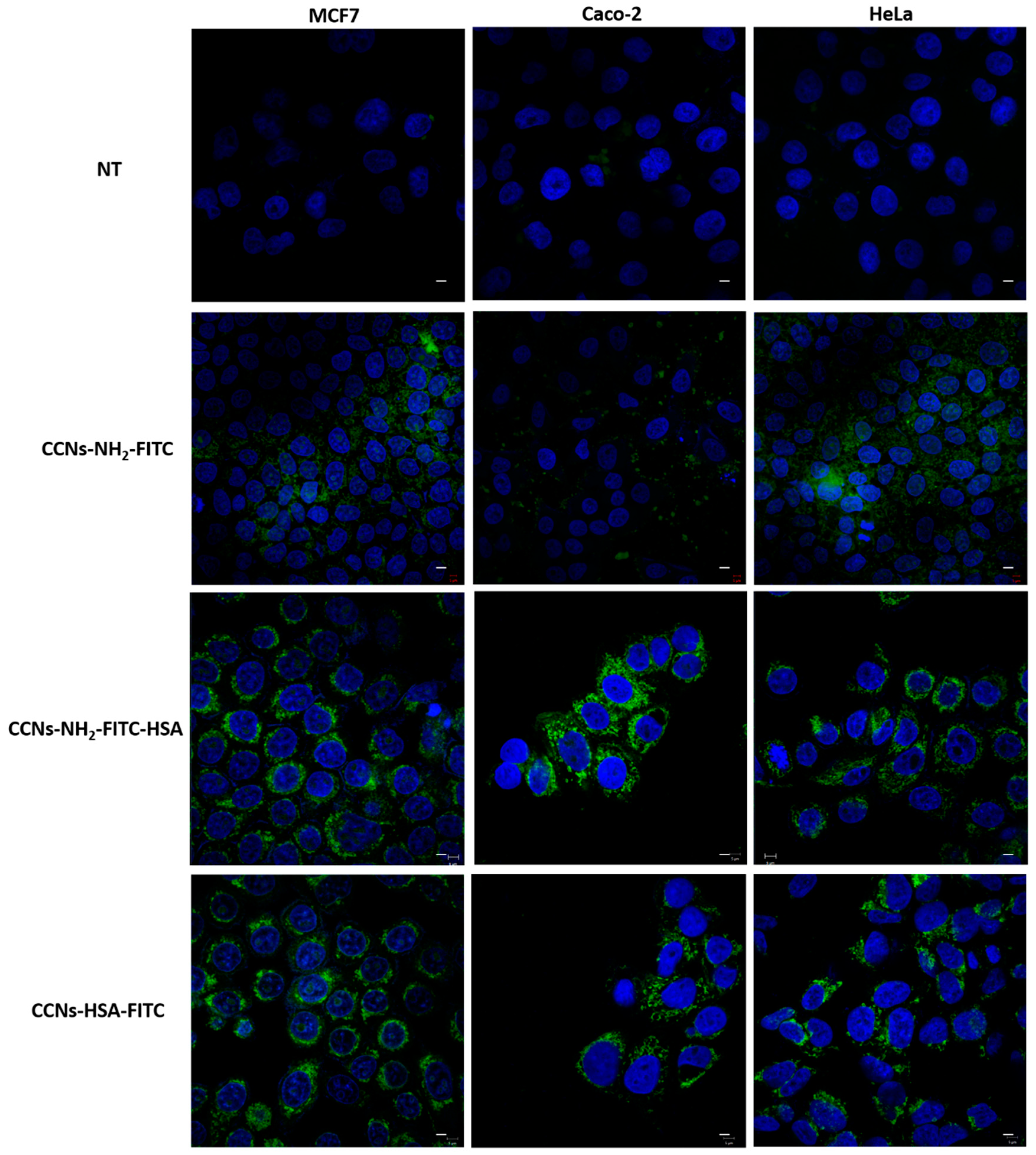
2.4. Internalization Assay
2.4.1. Cell Culture
2.4.2. Cell Proliferation Assay
2.4.3. Flow Cytometry
3. Results
3.1. Characterization of CaCO3 Nanocrystals
3.2. TGA–DSC Measurements
3.3. Characterization of FITC-Coupled Nanocrystals
3.3.1. Coupling Efficiency
3.3.2. Fluorescence Stability
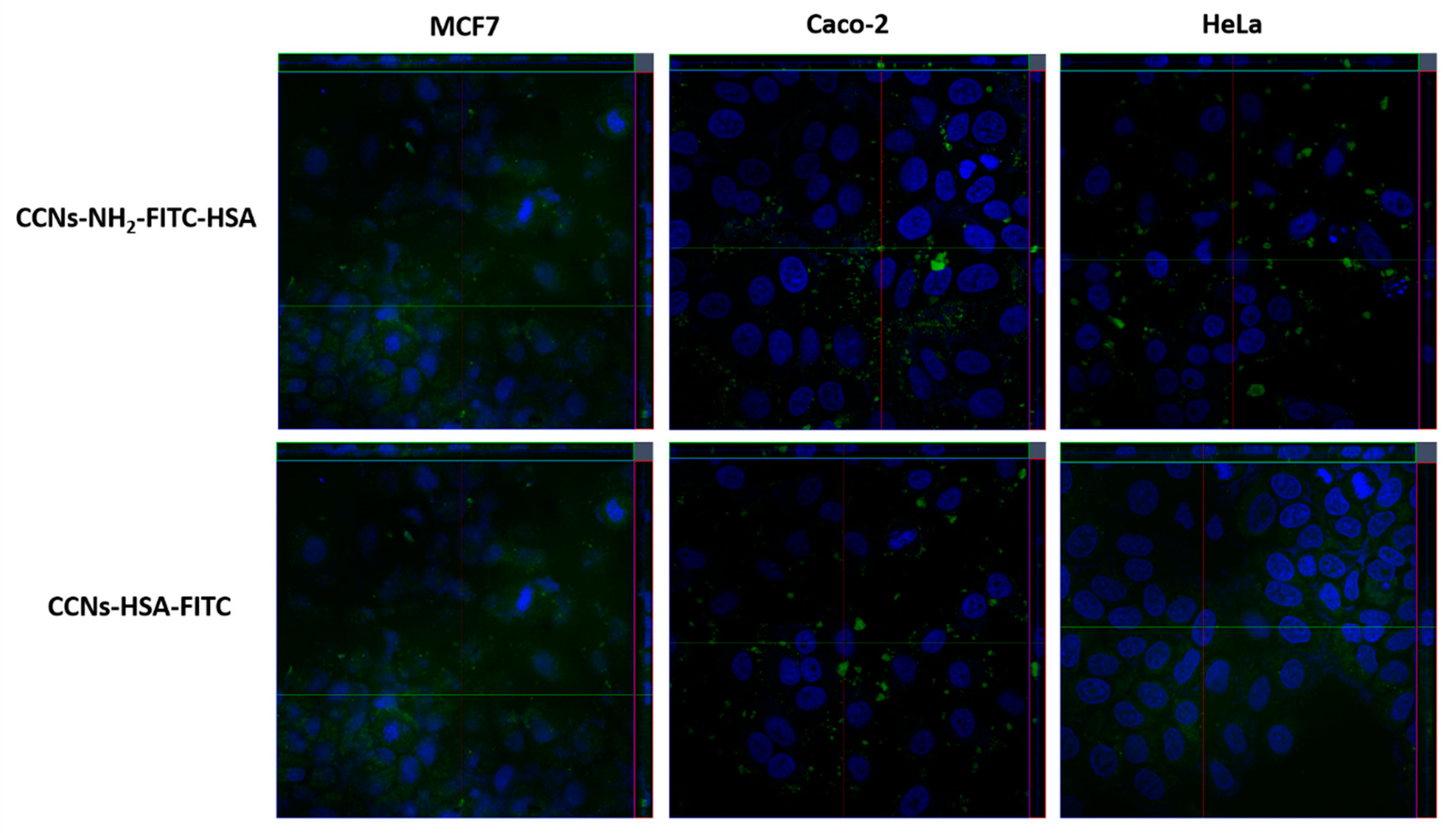
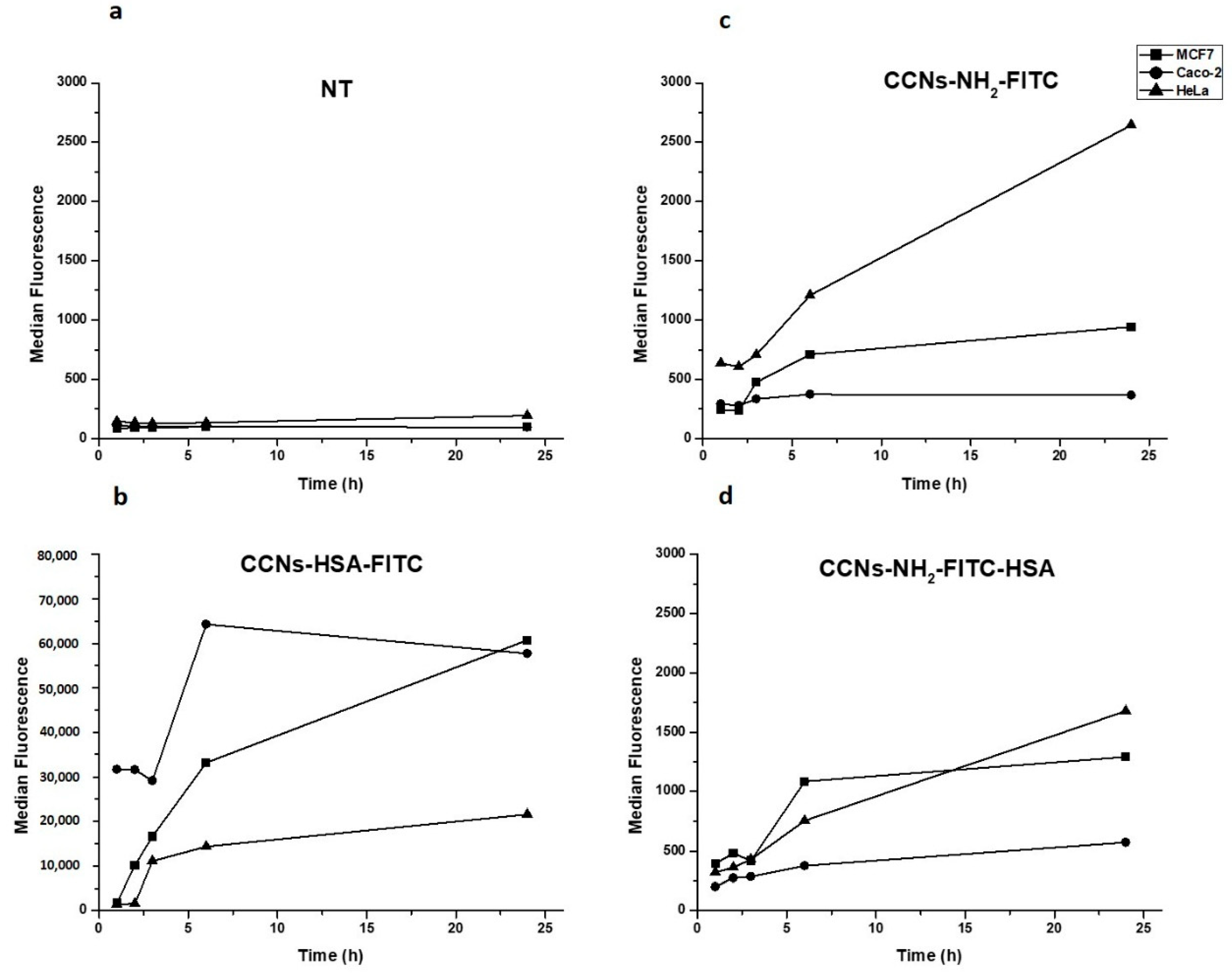
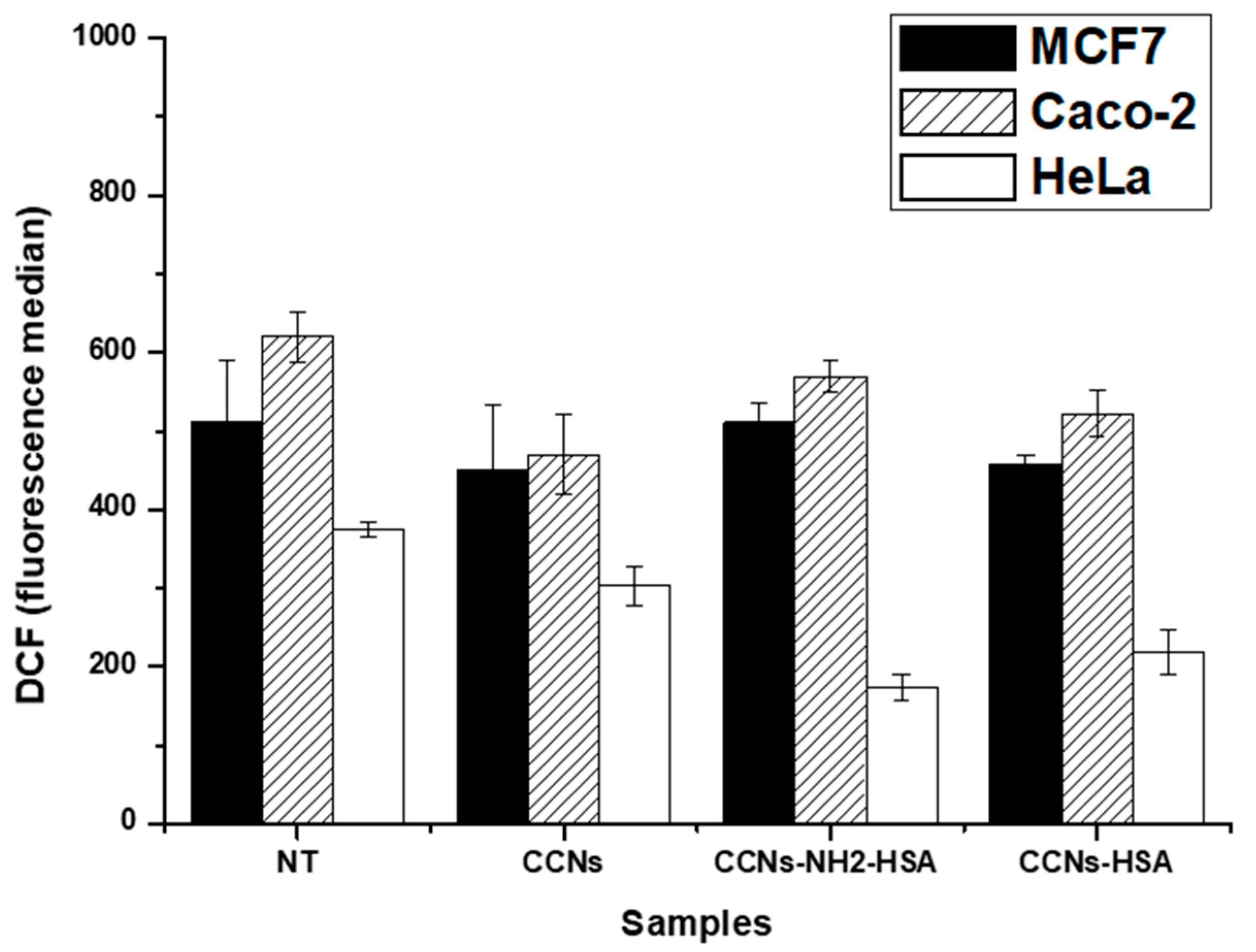
3.4. Internalization Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Valerio, V.; Giovanni, S.; Riccardo, N.; Fernanda, R.; Stefano, L.; Fabio, B. Smart Delivery and Controlled Drug Release with Gold Nanoparticles: New Frontiers in Nanomedicine. Recent Pat. Nanomed. 2012, 2, 34–44. [Google Scholar] [CrossRef]
- Conde, J.; Doria, G.; Baptista, P. Noble Metal Nanoparticles Applications in Cancer. J. Drug Deliv. 2012, 2012, 12. [Google Scholar] [CrossRef]
- Shroff, K. Polymer Nanoparticles: Newer Strategies towards Targeted Cancer Therapy. J. Phys. Chem. Biophys. 2013, 3, 1000125. [Google Scholar]
- Caldorera-Moore, M.; Guimard, N.; Shi, L.; Roy, K. Designer nanoparticles: Incorporating size, shape and triggered release into nanoscale drug carriers. Expert Opin. Drug Deliv. 2010, 7, 479–495. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, K.; Si, J.; Sui, M.; Shen, Y. Charge-Reversal Polymers for Biodelivery. In Bioinspired and Biomimetic Polymer Systems for Drug and Gene Delivery; Wiley-vch: Weinheim, Germany, 2013. [Google Scholar]
- David, R.K.; Stephen, J.F.; Alexandra, E.M.; Andrea, N.B. Recent Patents on Common Modifications Made to Traditional Micellar- Based Chemotherapeutics Designed to Improve Drug Delivery. Recent Pat. Nanomed. 2013, 3, 21–25. [Google Scholar] [CrossRef]
- Andresen, T.L.; Jensen, S.S.; Jorgensen, K. Advanced strategies in liposomal cancer therapy: Problems and prospects of active and tumor specific drug release. Prog. Lipid Res. 2005, 44, 68–97. [Google Scholar] [CrossRef] [PubMed]
- Dand, N.; Patel, P.; Ayre, A.; Kadam, V. Polymeric micelles as a drug carrier for tumor targeting. Chron. Young Sci. 2013, 4, 94–101. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, X.L.; Zhang, Y.F.; Gao, F.; Li, G.L.; He, Y.; Peng, M.L.; Fan, H.M. Magnetic nanoparticles based cancer therapy: Current status and applications. Sci. China Life Sci. 2018, 61, 400–414. [Google Scholar] [CrossRef]
- Mahmoudi, M.; Laurent, S.; Shokrgozar, M.A.; Hosseinkhani, M. Toxicity evaluations of superparamagnetic iron oxide nanoparticles: Cell “vision” versus physicochemical properties of nanoparticles. ACS Nano 2011, 5, 7263–7276. [Google Scholar] [CrossRef]
- Markides, H.; Rotherham, M.; El Haj, A.J. Biocompatibility and Toxicity of Magnetic Nanoparticles in Regenerative Medicine. J. Nanomater. 2012, 2012, 11. [Google Scholar] [CrossRef]
- Dey, N.S.; Rao, M.B. Quantum Dot: Novel Carrier for Drug Delivery; CBS: New Delhi, India, 2011; Volume 2. [Google Scholar]
- Fang, M.; Peng, C.-W.; Pang, D.-W.; Li, Y. Quantum dots for cancer research: Current status, remaining issues, and future perspectives. Cancer Biol. Med. 2012, 9, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.P.; Leong, K.W. Quantum dot-based theranostics. Nanoscale 2010, 2, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, Z.; Zhang, Y. The application of carbon nanotubes in target drug delivery systems for cancer therapies. Nanoscale Res. Lett. 2011, 6, 555. [Google Scholar] [CrossRef] [PubMed]
- Madani, S.Y.; Naderi, N.; Dissanayake, O.; Tan, A.; Seifalian, A.M. A new era of cancer treatment: Carbon nanotubes as drug delivery tools. Int. J. Nanomed. 2011, 6, 2963–2979. [Google Scholar] [CrossRef]
- Wang, K.; He, X.; Linthicum, W.; Mezan, R.; Wang, L.; Rojanasakul, Y.; Wen, Q.; Yang, Y. Carbon nanotubes induced fibrogenesis on nanostructured substrates. Environ. Sci. Nano 2017, 4, 689–699. [Google Scholar] [CrossRef]
- He, X.; Kiratipaiboon, C.; Porter, D.W.; Rojanasakul, L.W.; Dinu, C.Z.; Wang, K.; Yang, Y.; Rojanasakul, Y. Predicting Nanotube Fibrogenicity through Stem Cell-Mediated Fibroblast Focus and Spheroid Formation. Nano Lett. 2018, 18, 6500–6508. [Google Scholar] [CrossRef]
- Wang, L.; Mercer, R.R.; Rojanasakul, Y.; Qiu, A.; Lu, Y.; Scabilloni, J.F.; Wu, N.; Castranova, V. Direct Fibrogenic Effects of Dispersed Single-Walled Carbon Nanotubes on Human Lung Fibroblasts. J. Toxicol. Environ. Health Part A 2010, 73, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Schrand, A.M.; Dai, L.; Schlager, J.J.; Hussain, S.M. Toxicity Testing of Nanomaterials. In New Technologies for Toxicity Testing; Balls, M., Combes, R.D., Bhogal, N., Eds.; Springer: New York, NY, USA, 2012; pp. 58–75. [Google Scholar]
- Mercer, R.R.; Hubbs, A.F.; Scabilloni, J.F.; Wang, L.; Battelli, L.A.; Friend, S.; Castranova, V.; Porter, D.W. Pulmonary fibrotic response to aspiration of multi-walled carbon nanotubes. Part. Fibre Toxicol. 2011, 8, 21. [Google Scholar] [CrossRef]
- Krewski, D.; Andersen, M.E.; Mantus, E.; Zeise, L. Toxicity Testing in the 21st Century: Implications for Human Health Risk Assessment. Risk Anal. 2009, 29, 474–479. [Google Scholar] [CrossRef]
- Li, Y.; Wang, P.; Hu, C.; Wang, K.; Chang, Q.; Liu, L.; Han, Z.; Shao, Y.; Zhai, Y.; Zuo, Z.; et al. Protein corona of airborne nanoscale PM2.5 induces aberrant proliferation of human lung fibroblasts based on a 3D organotypic culture. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Baldassarre, F. Biocatalytic Synthesis of Phospholipids and Their Application as Coating Agents for CaCO Nano-crystals: Characterization and Intracellular Localization Analysis. Chem. Sel. 2016, 1, 6507–6514. [Google Scholar] [CrossRef]
- Vergaro, V.; Carata, E.; Baldassarre, F.; Panzarini, E.; Dini, L.; Carlucci, C.; Leporatti, S.; Scremin, B.F.; Altamura, D.; Giannini, C.; et al. Scalable production of calcite nanocrystals by atomization process: Synthesis, characterization and biological interactions study. Adv. Powder Technol. 2017, 28, 2445–2455. [Google Scholar] [CrossRef]
- Baldassarre, F. Polyelectrolyte Capsules as Carriers for Growth Factor Inhibitor Delivery to Hepatocellular Carcinoma. Macromol. Biosci. 2012, 12, 656–665. [Google Scholar] [CrossRef]
- Vergaro, V. Nanostructured polysaccharidic microcapsules for intracellular release of cisplatin. Int. J. Biol. Macromol. 2017, 99, 187–195. [Google Scholar] [CrossRef]
- Vergaro, V. Cell-Penetrating CaCO3 Nanocrystals for Improved Transport of NVP-BEZ235 across Membrane Barrier in T-Cell Lymphoma. Cancers 2018, 10, 31. [Google Scholar] [CrossRef]
- Vergaro, V. Synthesis of biocompatible polymeric nano-capsules based on calcium carbonate: A potential cisplatin delivery system. J. Inorg. Biochem. 2015, 153, 284–292. [Google Scholar] [CrossRef]
- Vergaro, V. TGF-Beta Inihibitor-loaded Polyelectrolyte Multilayers Capsules for Sustained Targeting of Hepatocarcinoma Cells. Curr. Pharm. Des. 2012, 18, 4155–4164. [Google Scholar] [CrossRef]
- Chatterjee, A. Novel synthesis with an atomized microemulsion technique and characterization of nano-calcium carbonate (CaCO3)/poly(methyl methacrylate) core–shell nanoparticles. Particuology 2013, 11, 760–767. [Google Scholar] [CrossRef]
- Mishra, S.; Sonawane, S.; Chitodkar, V. Comparative Study on Improvement in Mechanical and Flame Retarding Properties of Epoxy-CaCO3 Nano and Commercial Composites. Polym. Plast. Technol. Eng. 2005, 44, 463–473. [Google Scholar] [CrossRef]
- Mishra, S.; Sonawane, S.H.; Singh, R.P. Studies on characterization of nano CaCO3 prepared by thein situ deposition technique and its application in PP-nano CaCO3 composites. J. Polym. Sci. Part B Polym. Phys. 2005, 43, 107–113. [Google Scholar] [CrossRef]
- Wehrmeister, U.; Soldati, A.L.; Jacob, D.E.; Häger, T.; Hofmeister, W. Raman spectroscopy of synthetic, geological and biological vaterite: A Raman spectroscopic study. J. Raman Spectrosc. 2010, 41, 193–201. [Google Scholar] [CrossRef]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79. [Google Scholar] [CrossRef]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer Nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Gerlowski, L.E.; Jain, R.K. Microvascular permeability of normal and neoplastic tissues. Microvasc. Res. 1986, 31, 288–305. [Google Scholar] [CrossRef]
- Vergaro, V.; Carlucci, C.; Cascione, M.; Lorusso, C.; Conciauro, F. Interaction between Human Serum Albumin and Different Anatase TiO Nanoparticles: A Nano-bio Interface Study. Nanomater. Nanotechnol. 2015, 5, 30. [Google Scholar] [CrossRef]
- Aggarwal, P.; Hall, J.B.; McLeland, C.B.; Dobrovolskaia, M.A.; McNeil, S.E. Nanoparticle interaction with plasma proteins as it relates to particle biodistribution, biocompatibility and therapeutic efficacy. Adv. Drug Deliv. Rev. 2009, 61, 428–437. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, Y.; Zhang, Z.; Duan, Y. Targeted polymeric therapeutic nanoparticles: Design and interactions with hepatocellular carcinoma. Biomaterials 2015, 56, 229–240. [Google Scholar] [CrossRef]
- Owens, D.E., 3rd; Peppas, N.A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef]
- Lemarchand, C.; Gref, R.; Lesieur, S.; Hommel, H.; Vacher, B.; Besheer, A.; Maeder, K.; Couvreur, P. Physico-chemical characterization of polysaccharide-coated nanoparticles. J. Controll. Release Off. J. Controll. Release Soc. 2005, 108, 97–111. [Google Scholar] [CrossRef]
- Muller, J.; Bauer, K.N.; Prozeller, D.; Simon, J.; Mailander, V.; Wurm, F.R.; Winzen, S.; Landfester, K. Coating nanoparticles with tunable surfactants facilitates control over the protein corona. Biomaterials 2017, 115, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gillich, T.; Acikgoz, C.; Isa, L.; Schluter, A.D.; Spencer, N.D.; Textor, M. PEG-stabilized core-shell nanoparticles: Impact of linear versus dendritic polymer shell architecture on colloidal properties and the reversibility of temperature-induced aggregation. ACS Nano 2013, 7, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Salvati, A.; Pitek, A.S.; Monopoli, M.P.; Prapainop, K.; Bombelli, F.B.; Hristov, D.R.; Kelly, P.M.; Åberg, C.; Mahon, E.; Dawson, K.A. Transferrin-Functionalized Nanoparticles Lose Their Targeting Capabilities When a Biomolecule Corona Adsorbs on the Surface. Nat. Nanotechnol. 2013, 8, 137. [Google Scholar] [CrossRef] [PubMed]
- Ehrenberg, M.S.; Friedman, A.E.; Finkelstein, J.N.; Oberdorster, G.; McGrath, J.L. The influence of protein adsorption on nanoparticle association with cultured endothelial cells. Biomaterials 2009, 30, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Chithrani, B.D.; Chan, W.C.W. Elucidating the Mechanism of Cellular Uptake and Removal of Protein-Coated Gold Nanoparticles of Different Sizes and Shapes. Nano Lett. 2007, 7, 1542–1550. [Google Scholar] [CrossRef]
- Chithrani, B.D.; Ghazani, A.A.; Chan, W.C.W. Determining the Size and Shape Dependence of Gold Nanoparticle Uptake into Mammalian Cells. Nano Lett. 2006, 6, 662–668. [Google Scholar] [CrossRef]
- Cho, E.C.; Xie, J.; Wurm, P.A.; Xia, Y. Understanding the role of surface charges in cellular adsorption versus int+ernalization by selectively removing gold nanoparticles on the cell surface with a I2/KI etchant. Nano Lett. 2009, 9, 1080–1084. [Google Scholar] [CrossRef]
- Ritz, S.; Schottler, S.; Kotman, N.; Baier, G.; Musyanovych, A.; Kuharev, J.; Landfester, K.; Schild, H.; Jahn, O.; Tenzer, S.; et al. Protein corona of nanoparticles: Distinct proteins regulate the cellular uptake. Biomacromolecules 2015, 16, 1311–1321. [Google Scholar] [CrossRef]
- Lee, Y.K.; Choi, E.J.; Webster, T.J.; Kim, S.H.; Khang, D. Effect of the protein corona on nanoparticles for modulating cytotoxicity and immunotoxicity. Int. J. Nanomed. 2015, 10, 97–113. [Google Scholar] [CrossRef]
- Pilkington, E.H.; Xing, Y.; Wang, B. Effects of Protein Corona on IAPP Amyloid Aggregation, Fibril Remodelling, and Cytotoxicity. Sci. Rep. 2017, 7, 2455. [Google Scholar] [CrossRef]
- Yan, Y.; Gause, K.T.; Kamphuis, M.M.; Ang, C.S.; O’Brien-Simpson, N.M.; Lenzo, J.C.; Reynolds, E.C.; Nice, E.C.; Caruso, F. Differential roles of the protein corona in the cellular uptake of nanoporous polymer particles by monocyte and macrophage cell lines. ACS Nano 2013, 7, 10960–10970. [Google Scholar] [CrossRef] [PubMed]
- Panzarini, E.; Mariano, S.; Vergallo, C.; Carata, E.; Fimia, G.M. Glucose capped silver nanoparticles induce cell cycle arrest in HeLa cells. Toxicol. In Vitro 2017, 41, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolskaia, M.A.; Patri, A.K.; Zheng, J.; Clogston, J.D.; Ayub, N.; Aggarwal, P.; Neun, B.W.; Hall, J.B.; McNeil, S.E. Interaction of colloidal gold nanoparticles with human blood: Effects on particle size and analysis of plasma protein binding profiles. Nanomed. Nanotechnol. Biol. Med. 2009, 5, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Jambhrunkar, S.; Thorn, P.; Chen, J.; Gu, W.; Yu, C. Hyaluronic acid modified mesoporous silica nanoparticles for targeted drug delivery to CD44-overexpressing cancer cells. Nanoscale 2013, 5, 178–183. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Grisorio, R.; Allegretta, G.; Suranna, G.P.; Mastrorilli, P.; Loiudice, A.; Rizzo, A.; Mazzeo, M.; Gigli, G. Monodispersed vs. polydispersed systems for bulk heterojunction solar cells: The case of dithienopyrrole/anthracene based materials. J. Mater. Chem. 2012, 22, 19752–19760. [Google Scholar] [CrossRef]
- Grisorio, R.; Piliego, C.; Striccoli, M.; Cosma, P.; Fini, P.; Gigli, G.; Mastrorilli, P.; Suranna, G.P.; Nobile, C.F. Influence of Keto Groups on the Optical, Electronic, and Electroluminescent Properties of Random Fluorenone-Containing Poly(fluorenylene-vinylene)s. J. Mater. Chem. C 2008, 112, 20076–20087. [Google Scholar] [CrossRef]
- Xu, W.; Riikonen, J.; Nissinen, T.; Suvanto, M.; Rilla, K.; Li, B.; Wang, Q.; Deng, F.; Lehto, V.P. Amine surface modifications and fluorescent labeling of thermally stabilized mesoporous silicon nanoparticles. J. Mater. Chem. C 2012, 116, 22307–22314. [Google Scholar] [CrossRef]
- Nairi, V.; Medda, S.; Piludu, M.; Casula, M.F.; Vallet-Regì, M.; Monduzzi, M.; Salis, A. Interactions between bovine serum albumin and mesoporous silica nanoparticles functionalized with biopolymers. Chem. Eng. J. 2018, 340, 42–50. [Google Scholar] [CrossRef]
- Grisorio, R.; Debellis, D.; Suranna, G.P.; Gigli, G.; Giansante, C. The Dynamic Organic/Inorganic Interface of Colloidal PbS Quantum Dots. Angew. Chem. Int. Ed. 2016, 55, 6628–6633. [Google Scholar] [CrossRef]
- Shu, Y.; Qiu, F.; Zhang, Y.; Cao, W.; Wu, Z.; Nian, S.; Zhou, N. Novel vaterite-containing tricalcium silicate bone cement by surface functionalization using 3-aminopropyltriethoxysilane: Setting behavior, in vitro bioactivity and cytocompatibility. Biomed. Mater. 2017, 12, 065007. [Google Scholar] [CrossRef]
- Zeng, X.; Xu, G.; Gao, Y.; An, Y. Surface Wettability of (3-Aminopropyl)triethoxysilane Self-Assembled Monolayers. J. Mater. Chem. B 2011, 115, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.A.; Chen, W. How to prevent the loss of surface functionality derived from aminosilanes. Langmuir ACS J. Surf. Colloids 2008, 24, 12405–12409. [Google Scholar] [CrossRef]
- Avnir, D.; Levy, D.; Reisfeld, R. The nature of the silica cage as reflected by spectral changes and enhanced photostability of trapped Rhodamine 6G. J. Mater. Chem. 1984, 88, 5956–5959. [Google Scholar] [CrossRef]
- Lunova, M.; Prokhorov, A.; Jirsa, M.; Hof, M.; Olzyńska, A.; Jurkiewicz, P.; Kubinová, Š.; Lunov, O.; Dejneka, A. Nanoparticle core stability and surface functionalization drive the mTOR signaling pathway in hepatocellular cell lines. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.; Xie, Q.; Wei, W.; Zhao, J. Revealing the immune perturbation of black phosphorus nanomaterials to macrophages by understanding the protein corona. Nat. Commun. 2018, 9, 2480. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.; Du, J.; Zhao, L.; Wang, L.; Liu, Y.; Li, D.; Yang, Y.; Zhou, R.; Zhao, Y.; Chai, Z.; et al. Binding of blood proteins to carbon nanotubes reduces cytotoxicity. Proc. Natl. Acad. Sci. USA 2011, 108, 16968–16973. [Google Scholar] [CrossRef] [PubMed]
- MiclǍuş, T.; Beer, C.; Chevallier, J.; Scavenius, C.; Bochenkov, V.E.; Enghild, J.J.; Sutherland, D.S. Dynamic protein coronas revealed as a modulator of silver nanoparticle sulphidation in vitro. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Monopoli, M.P.; Walczyk, D.; Campbell, A.; Elia, G.; Lynch, I.; Baldelli Bombelli, F.; Dawson, K.A. Physical-Chemical aspects of protein corona: Relevance to in vitro and in vivo biological impacts of nanoparticles. J. Am. Chem. Soc. 2011, 133, 2525–2534. [Google Scholar] [CrossRef]
- Göppert, T.M.; Müller, R.H. Polysorbate-stabilized solid lipid nanoparticles as colloidal carriers for intravenous targeting of drugs to the brain: Comparison of plasma protein adsorption patterns. J. Drug Target. 2005, 13, 179–187. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A.; McNeil, S.E. Immunological properties of engineered nanomaterials. Nat. Nanotechnol. 2007, 2, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Schöttler, S.; Landfester, K.; Mailänder, V. Controlling the Stealth Effect of Nanocarriers through Understanding the Protein Corona. Angew. Chem. Int. Ed. 2016, 55, 8806–8815. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Size (nm) (dH) | PDI Poly Dispersion Index | ζ-Potential (mV) |
|---|---|---|---|
| CCNs | 800.1 | 0.501 | −12.5 ± 1.23 |
| CCNs-NH2 | 587.2 | 0.313 | +14.8 ± 0.70 |
| CCNs-HSA | 497.2 | 0.467 | −19.4 ± 1.80 |
| CCNs-NH2-HSA | 285.4 | 0.105 | −24.9 ± 0.62 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vergaro, V.; Pisano, I.; Grisorio, R.; Baldassarre, F.; Mallamaci, R.; Santoro, A.; Suranna, G.P.; Papadia, P.; Fanizzi, F.P.; Ciccarella, G. CaCO3 as an Environmentally Friendly Renewable Material for Drug Delivery Systems: Uptake of HSA-CaCO3 Nanocrystals Conjugates in Cancer Cell Lines. Materials 2019, 12, 1481. https://doi.org/10.3390/ma12091481
Vergaro V, Pisano I, Grisorio R, Baldassarre F, Mallamaci R, Santoro A, Suranna GP, Papadia P, Fanizzi FP, Ciccarella G. CaCO3 as an Environmentally Friendly Renewable Material for Drug Delivery Systems: Uptake of HSA-CaCO3 Nanocrystals Conjugates in Cancer Cell Lines. Materials. 2019; 12(9):1481. https://doi.org/10.3390/ma12091481
Chicago/Turabian StyleVergaro, Viviana, Isabella Pisano, Roberto Grisorio, Francesca Baldassarre, Rosanna Mallamaci, Antonella Santoro, Gian Paolo Suranna, Paride Papadia, Francesco Paolo Fanizzi, and Giuseppe Ciccarella. 2019. "CaCO3 as an Environmentally Friendly Renewable Material for Drug Delivery Systems: Uptake of HSA-CaCO3 Nanocrystals Conjugates in Cancer Cell Lines" Materials 12, no. 9: 1481. https://doi.org/10.3390/ma12091481
APA StyleVergaro, V., Pisano, I., Grisorio, R., Baldassarre, F., Mallamaci, R., Santoro, A., Suranna, G. P., Papadia, P., Fanizzi, F. P., & Ciccarella, G. (2019). CaCO3 as an Environmentally Friendly Renewable Material for Drug Delivery Systems: Uptake of HSA-CaCO3 Nanocrystals Conjugates in Cancer Cell Lines. Materials, 12(9), 1481. https://doi.org/10.3390/ma12091481