Docosane-Organosilica Microcapsules for Structural Composites with Thermal Energy Storage/Release Capability

 ,
,  ,
,  , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of the Microcapsules
2.3. Preparation of the Epoxy-Based Matrices and Laminates
2.4. Characterization of the Microcapsules
2.5. Characterization of the Epoxy-Based Matrices and Laminates
3. Results and Discussion
3.1. Characterization of the PCM Microcapsules
3.1.1. Sample Preparation and Quality of the Docosane o/w Emulsion
3.1.2. SEM Micrography
3.1.3. FTIR Spectroscopy on the Microcapsules
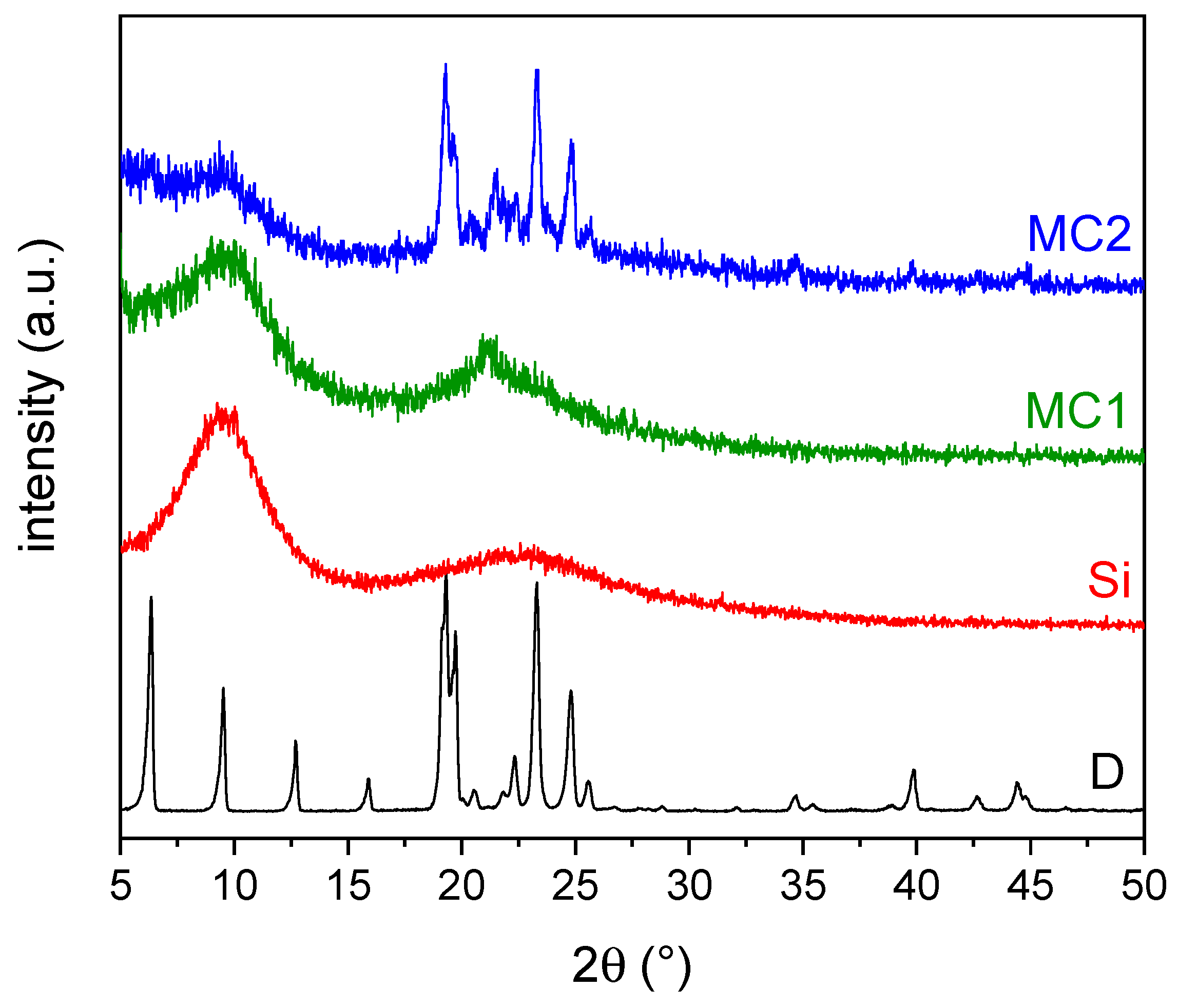
3.1.4. XRD Analysis
3.1.5. Solid-State NMR Analysis
3.1.6. DSC Analysis of the Microcapsules
3.1.7. TGA Analysis of the Microcapsules
3.2. Characterization of the Epoxy-Based Matrices and Laminates
3.2.1. SEM Microscopy on the Matrices and Laminates
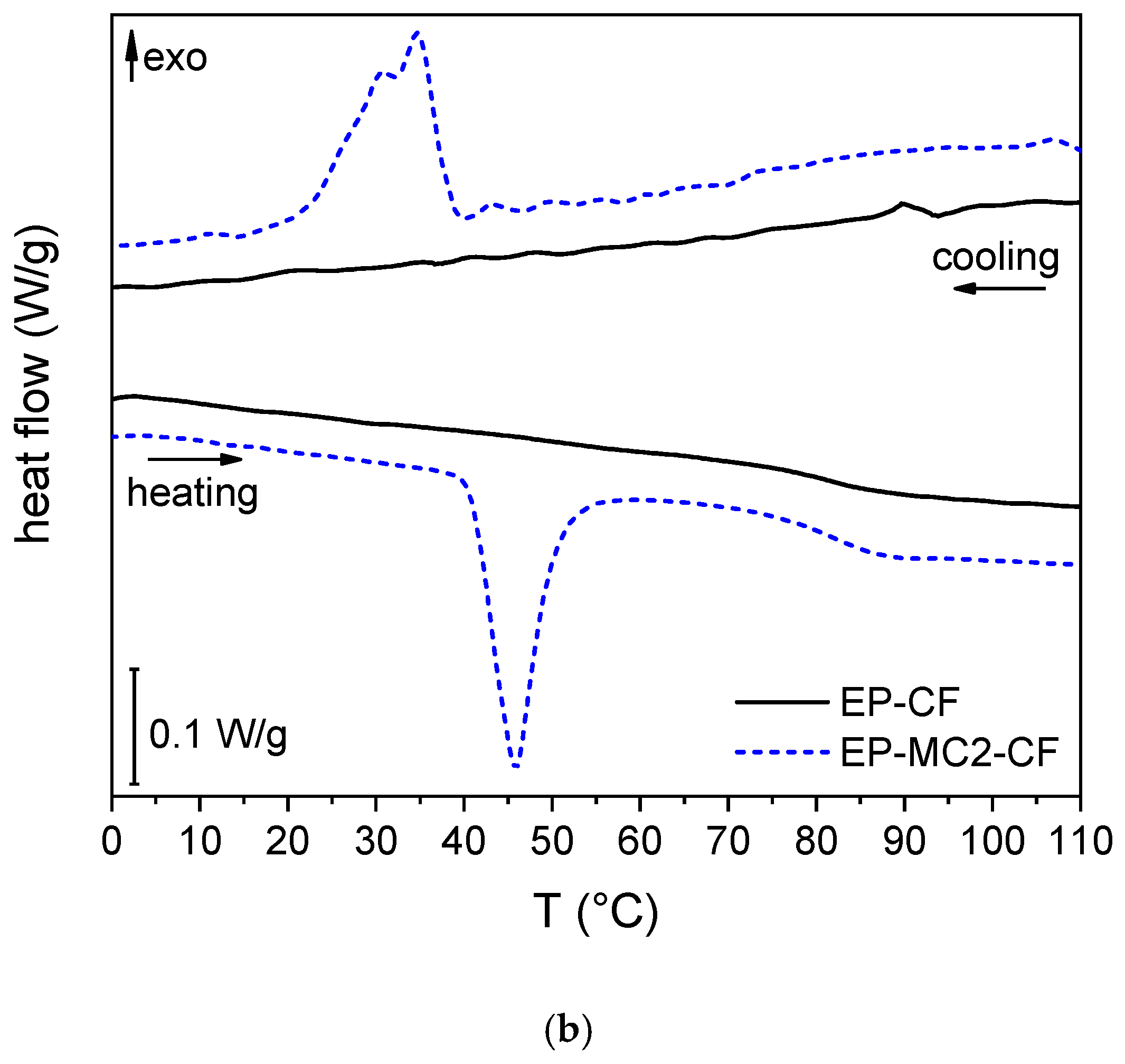
3.2.2. DSC on the Matrices and Laminates
3.2.3. TGA Analysis and Fraction of the Constituents
3.2.4. Three-Point Bending Test
3.2.5. DMA Analysis
3.2.6. Thermal Camera Imaging
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fleischer, A.S. Thermal Energy Storage Using Phase Change Materials—Fundamentals and Applications; Springer Briefs in Applied Science and Technology, Thermal Engineering and Applied Science; Springer: Minneapolis, MN, USA, 2015. [Google Scholar]
- Kenisarin, M.M.; Kenisarina, K.M. Form-stable phase change materials for thermal energy storage. Renew. Sustain. Energy Rev. 2012, 16, 1999–2040. [Google Scholar] [CrossRef]
- Xie, N.; Luo, J.; Li, Z.; Huang, Z.; Gao, X.; Fang, Y.; Zhang, Z. Salt hydrate/expanded vermiculite composite as a form-stable phase change material for building energy storage. Sol. Energy Mater. Sol. Cells 2019, 189, 33–42. [Google Scholar] [CrossRef]
- Kuznik, F.; David, D.; Johannes, K.; Roux, J.J. A review on phase change materials integrated in building walls. Renew. Sustain. Energy Rev. 2011, 15, 379–391. [Google Scholar] [CrossRef]
- Abhat, A. Low temperature latent heat thermal energy storage: Heat storage materials. Sol. Energy 1983, 30, 313–332. [Google Scholar] [CrossRef]
- Agyenim, F.; Hewitt, N.; Eames, P.; Smyth, M. A review of materials, heat transfer and phase change problem formulation for latent heat thermal energy storage systems (LHTESS). Renew. Sustain. Energy Rev. 2010, 14, 615–628. [Google Scholar] [CrossRef]
- Khadiran, T.; Hussein, M.Z.; Zainal, Z.; Rusli, R. Encapsulation techniques for organic phase change materials as thermal energy storage medium: A review. Sol. Energy Mater. Sol. Cells 2015, 143, 78–98. [Google Scholar] [CrossRef]
- Fan, L.; Khodadadi, J.M. Thermal conductivity enhancement of phase change materials for thermal energy storage: A review. Renew. Sustain. Energy Rev. 2011, 15, 24–46. [Google Scholar] [CrossRef]
- Pielichowska, K.; Pielichowski, K. Phase change materials for thermal energy storage. Prog. Mater. Sci. 2014, 65, 67–123. [Google Scholar] [CrossRef]
- Chen, F.; Wolcott, M. Polyethylene/paraffin binary composites for phase change material energy storage in building: A morphology, thermal properties, and paraffin leakage study. Sol. Energy Mater. Sol. Cells 2015, 137, 79–85. [Google Scholar] [CrossRef]
- Chen, F.; Wolcott, M.P. Miscibility studies of paraffin/polyethylene blends as form-stable phase change materials. Eur. Polym. J. 2014, 52, 44–52. [Google Scholar] [CrossRef]
- Lin, Y.; Zhu, C.; Fang, G. Synthesis and properties of microencapsulated stearic acid/silica composites with graphene oxide for improving thermal conductivity as novel solar thermal storage materials. Sol. Energy Mater. Sol. Cells 2019, 189, 197–205. [Google Scholar] [CrossRef]
- Jamekhorshid, A.; Sadrameli, S.M.; Farid, M. A review of microencapsulation methods of phase change materials (PCMs) as a thermal energy storage (TES) medium. Renew. Sustain. Energy Rev. 2014, 31, 531–542. [Google Scholar] [CrossRef]
- Hassan, A.; Shakeel Laghari, M.; Rashid, Y. Micro-encapsulated phase change materials: A review of encapsulation, safety and thermal characteristics. Sustainability 2016, 8, 1046. [Google Scholar] [CrossRef]
- Sarı, A.; Alkan, C.; Karaipekli, A. Preparation, characterization and thermal properties of PMMA/n-heptadecane microcapsules as novel solid–liquid microPCM for thermal energy storage. Appl. Energy 2010, 87, 1529–1534. [Google Scholar] [CrossRef]
- Brown, E.N.; White, S.R.; Sottos, N.R. Microcapsule induced toughening in a self-healing polymer composite. J. Mater. Sci. 2004, 39, 1703–1710. [Google Scholar] [CrossRef]
- Fortuniak, W.; Slomkowski, S.; Chojnowski, J.; Kurjata, J.; Tracz, A.; Mizerska, U. Synthesis of a paraffin phase change material microencapsulated in a siloxane polymer. Colloid Polym. Sci. 2013, 291, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yang, Y.; Li, Y.; Zhao, L.; Wang, H.; Song, G.; Tang, G. Microencapsulated phase change materials with TiO2-doped PMMA shell for thermal energy storage and UV-shielding. Sol. Energy Mater. Sol. Cells 2017, 168, 62–68. [Google Scholar] [CrossRef]
- Peng, H.; Zhang, D.; Ling, X.; Li, Y.; Wang, Y.; Yu, Q.; She, X.; Li, Y.; Ding, Y. N-alkanes phase change materials and their microencapsulation for thermal energy storage: A critical review. Energy Fuels 2018, 32, 7262–7293. [Google Scholar] [CrossRef]
- Yu, S.; Wang, X.; Wu, D. Microencapsulation of n-octadecane phase change material with calcium carbonate shell for enhancement of thermal conductivity and serving durability: Synthesis, microstructure, and performance evaluation. Appl. Energy 2014, 114, 632–643. [Google Scholar] [CrossRef]
- Fang, G.; Chen, Z.; Li, H. Synthesis and properties of microencapsulated paraffin composites with SiO2 shell as thermal energy storage materials. Chem. Eng. J. 2010, 163, 154–159. [Google Scholar] [CrossRef]
- Cao, L.; Tang, F.; Fang, G. Synthesis and characterization of microencapsulated paraffin with titanium dioxide shell as shape-stabilized thermal energy storage materials in buildings. Energy Build. 2014, 72, 31–37. [Google Scholar] [CrossRef]
- Cao, L.; Tang, F.; Fang, G. Preparation and characteristics of microencapsulated palmitic acid with TiO2 shell as shape-stabilized thermal energy storage materials. Sol. Energy Mater. Sol. Cells 2014, 123, 183–188. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, S.; Wang, X.; Wu, D. Fabrication of microencapsulated phase change materials based on n-octadecane core and silica shell through interfacial polycondensation. Colloids Surf. A Physicochem. Eng. Asp. 2011, 389, 104–117. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, X.; Wu, D. Silica encapsulation of n-octadecane via sol-gel process: A novel microencapsulated phase-change material with enhanced thermal conductivity and performance. J. Colloid Interface Sci. 2010, 343, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Ciriminna, R.; Sciortino, M.; Alonzo, G.; de Schrijver, A.; Pagliaro, M. From molecules to systems: Sol-gel microencapsulation in silica-based materials. Chem. Rev. 2011, 111, 765–789. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Liu, H.; Wang, X.; Wu, D. Innovative design of superhydrophobic thermal energy-storage materials by microencapsulation of n-docosane with nanostructured ZnO/SiO2 shell. Appl. Energy 2019, 237, 549–565. [Google Scholar] [CrossRef]
- Chen, Z.; Cao, L.; Shan, F.; Fang, G. Preparation and characteristics of microencapsulated stearic acid as composite thermal energy storage material in buildings. Energy Build. 2013, 62, 469–474. [Google Scholar] [CrossRef]
- Wang, L.Y.; Tsai, P.S.; Yang, Y.M. Preparation of silica microspheres encapsulating phase-change material by sol-gel method in o/w emulsion. J. Microencapsul. 2006, 23, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Cao, L.; Fang, G.; Shan, F. Synthesis and characterization of microencapsulated paraffin microcapsules as shape-stabilized thermal energy storage materials. Nanoscale Microscale Thermophys. Eng. 2013, 17, 112–123. [Google Scholar] [CrossRef]
- Tang, F.; Liu, L.; Alva, G.; Jia, Y.; Fang, G. Synthesis and properties of microencapsulated octadecane with silica shell as shape–stabilized thermal energy storage materials. Sol. Energy Mater. Sol. Cells 2017, 160, 1–6. [Google Scholar] [CrossRef]
- Alkan, C.; Sarı, A.; Karaipekli, A.; Uzun, O. Preparation, characterization, and thermal properties of microencapsulated phase change material for thermal energy storage. Sol. Energy Mater. Sol. Cells 2009, 93, 143–147. [Google Scholar] [CrossRef]
- Felix De Castro, P.; Ahmed, A.; Shchukin, D.G. Confined-volume effect on the thermal properties of encapsulated phase change materials for thermal energy storage. Chemistry 2016, 22, 4389–4394. [Google Scholar] [CrossRef]
- Felix De Castro, P.; Shchukin, D.G. New polyurethane/docosane microcapsules as phase-change materials for thermal energy storage. Chemistry 2015, 21, 11174–11179. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, K. Routes for Achieving Multifunctionality in Reinforced Polymers and Composite Structures. In Multifunctionality of Polymer Composites: Challenges and New Solutions; Friedrich, K., Breuer, U., Eds.; Elsevier: Waltham, MA, USA, 2015; pp. 3–41. [Google Scholar]
- Fredi, G.; Dorigato, A.; Fambri, L.; Pegoretti, A. Wax confinement with carbon nanotubes for phase changing epoxy blends. Polymers 2017, 9, 405. [Google Scholar] [CrossRef] [PubMed]
- Fredi, G.; Dorigato, A.; Fambri, L.; Pegoretti, A. Multifunctional epoxy/carbon fiber laminates for thermal energy storage and release. Compos. Sci. Technol. 2018, 158, 101–111. [Google Scholar] [CrossRef]
- Fredi, G.; Dorigato, A.; Pegoretti, A. Multifunctional glass fiber/polyamide composites with thermal energy storage/release capability. eXPRESS Polym. Lett. 2018, 12, 349–364. [Google Scholar] [CrossRef]
- Dorigato, A.; Fredi, G.; Pegoretti, A. Novel phase change materials using thermoplastic composites. In Proceedings of the 9th International Conference on “Times of Polymers and Composites”, Ischia, Italy, 17–21 June 2018; pp. 020044-1–020044-4. [Google Scholar]
- Fredi, G.; Dorigato, A.; Pegoretti, A. Novel reactive thermoplastic resin as a matrix for laminates containing phase change microcapsules. Polym. Compos. 2019. [Google Scholar] [CrossRef]
- Fredi, G.; Dorigato, A.; Unterberger, S.; Artuso, N.; Pegoretti, A. Discontinuous carbon fiber/polyamide composites with microencapsulated paraffin for thermal energy storage. J. Appl. Polym. Sci. 2019, 136, 47408. [Google Scholar] [CrossRef]
- Dorigato, A.; Fredi, G.; Pegoretti, A. Application of the thermal energy storage concept to novel epoxy-short carbon fiber composites. J. Appl. Polym. Sci. 2019, 136, 47434. [Google Scholar] [CrossRef]
- Dirè, S.; Tagliazucca, V.; Callone, E.; Quaranta, A. Effect of functional groups on condensation and properties of sol-gel silica nanoparticles prepared by direct synthesis from organoalkoxysilanes. Mater. Chem. Phys. 2011, 126, 909–917. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, C.; Liu, J.; Fang, X.; Zhang, Z. Highly stable graphite nanoparticle-dispersed phase change emulsions with little supercooling and high thermal conductivity for cold energy storage. Appl. Energy 2017, 188, 97–106. [Google Scholar] [CrossRef]
- Shao, J.; Darkwa, J.; Kokogiannakis, G. Review of phase change emulsions (PCMEs) and their applications in HVAC systems. Energy Build. 2015, 94, 200–217. [Google Scholar] [CrossRef]
- Wang, F.; Lin, W.; Ling, Z.; Fang, X. A comprehensive review on phase change material emulsions: Fabrication, characteristics, and heat transfer performance. Sol. Energy Mater. Sol. Cells 2019, 191, 218–234. [Google Scholar] [CrossRef]
- Park, J.-H.; Oh, C.; Shin, S.-I.; Moon, S.-K.; Oh, S.-G. Preparation of hollow silica microspheres in w/o emulsions with polymers. J. Colloid Interface Sci. 2003, 266, 107–114. [Google Scholar] [CrossRef]
- Proverbio, Z.E.; Bardavid, S.M.; Arancibi, E.L.; Schulz, P.C. Hydrophile-lipophile balance and solubility parameter of cationic surfactants. Colloids Surf. A Physicochem. Eng. Asp. 2003, 214, 167–171. [Google Scholar] [CrossRef]
- Cardoso, K.P.; Ferrão, L.F.A.; Kawachi, E.Y. Preparation of paraffin-based solid combustible for hybrid propulsion rocket motor. J. Propuls. Power 2017, 33, 448–455. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts; Wiley: Oxford, UK, 2001. [Google Scholar]
- Dirè, S.; Borovin, E.; Ribot, F. Architecture of Silsesquioxanes. In Handbook of Sol-Gel Science and Technology: Processing, Characterization and Applications; Klein, L., Aparicio, M., Jitanu, A., Eds.; Springer: New York, NY, USA, 2018. [Google Scholar]
- Nouar, H.; Petitjean, D.; Bouroukba, M.; Dirand, M. Binary phase diagram of the system: N-docosane-n-tricosane. J. Mol. Struct. 1998, 443, 197–204. [Google Scholar] [CrossRef]
- Lüth, H.; Nyburg, S.C.; Robinson, P.M.; Scott, H.G. Crystallographic and calorimetric phase studies of the n-eicosane, C20H42: N-docosane, C22H46 system. Mol. Cryst. Liq. Cryst. 1974, 27, 337–357. [Google Scholar] [CrossRef]
- Wang, D.; Sui, J.; Qi, D.; Wei, Y.; Wang, X.; Lan, X.Z. Phase transition of docosane in nanopores. J. Therm. Anal. Calorim. 2019, 135, 2869–2877. [Google Scholar] [CrossRef]
- Speight, R.J.; Rourke, J.P.; Wong, A.; Barrow, N.S.; Ellis, P.R.; Bishop, P.T.; Smith, M.E. 1H and 13C solution- and solid-state NMR investigation into wax products from the Fischer-Tropsch process. Solid State Nucl. Magn. Reason. 2011, 39, 58–64. [Google Scholar] [CrossRef]
- Okazaki, M.; Toriyama, K. Alternation of spin-lattice relaxation-times for even and odd linear alkane crystals—A high-resolution solid-state NMR study. J. Phys. Chem. 1989, 93, 2883–2885. [Google Scholar] [CrossRef]
- Möller, M.; Cantow, H.-J.; Drotloff, H.; Emeis, D.; Lee, K.-S.; Wegner, G. Phase transitions and defect structures in the lamellar surface of polyethylene and n-alkane crystallites. Magic angle spinning 13C NMR studies. Makromol. Chem. 1987, 187, 1237–1252. [Google Scholar] [CrossRef]
- Inoue, D.; Kurosu, H.; Chen, Q.; Ando, I. Structural and dynamical studies of 13C-labeled polyethylene adsorbed on the surface of silica gel by high-resolution solid-state 13C NMR spectroscopy. Acta Polym. 1995, 46, 420–423. [Google Scholar] [CrossRef]
- Liu, X.; Lou, Y. Preparation of microencapsulated phase change materials by the sol-gel process and its application on textiles. Fibres Text. East. Eur. 2015, 23, 63–67. [Google Scholar]
- Li, B.; Liu, T.; Hu, L.; Wang, Y.; Gao, L. Fabrication and properties of microencapsulated paraffin/SiO2 phase change composite for thermal energy storage. ACS Sustain. Chem. Eng. 2013, 1, 374–380. [Google Scholar] [CrossRef]
- Sun, N.; Xiao, Z. Synthesis and performances of phase change materials microcapsules with a polymer/bn/tio2 hybrid shell for thermal energy storage. Energy Fuels 2017, 31, 10186–10195. [Google Scholar] [CrossRef]
- Wang, S.; Tozaki, K.-i.; Hayashi, H.; Hosaka, S.; Inaba, H. Observation of multiple phase transitions in n-c22h46 using a high resolution and super-sensitive DSC. Thermochim. Acta 2003, 408, 31–38. [Google Scholar] [CrossRef]
- Anghel, E.M.; Georgiev, A.; Petrescu, S.; Popov, R.; Constantinescu, M. Thermo-physical characterization of some paraffins used as phase change materials for thermal energy storage. J. Therm. Anal. Calorim. 2014, 117, 557–566. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Docosane o/w Emulsion | MTES Solution | ||||
|---|---|---|---|---|---|---|
| Docosane (g) | CTAB (g) | Water (mL) | MTES (mL) | HCl 10−2 M (mL) | ethanol (mL) | |
| MC1 | 5 | 0.05 | 50 | 4.2 | 6.7 | 5.1 |
| MC2 | 25.5 | |||||
| Si | - | - | 5.1 | |||
| Sample | Composition |
|---|---|
| EP | Neat epoxy |
| EP-MC2 | Epoxy + MC2 (10 wt%) |
| EP-CF | Neat epoxy/carbon fiber laminate |
| EP-MC2-CF | Epoxy/carbon fiber laminate + MC2 (10 wt% with respect to the epoxy) |
| Sample | (%) | ||||
|---|---|---|---|---|---|
| D | 46.2 | 33.5 | 234.6 | −234.2 | 100 |
| MC1 | 41.2 | 25.1 | 33.0 | −32.9 | 14.1 |
| MC2 | 43.8 | 34.5 | 142.7 | −141.4 | 60.6 |
| Sample | (wt%) | (wt%) | (g/cm3) | (g/cm3) | (vol%) | (wt%) | (wt%) |
|---|---|---|---|---|---|---|---|
| EP | 6.0 | - | 1.179 ± 0.001 | - | - | 97.4 | 0 |
| EP-MC2 | 9.4 | - | 1.089 ± 0.013 | - | - | 90.1 | 7.6 |
| EP-CF | 66.8 | 64.7 | 1.454 ± 0.006 | 1.508 | 3.8 | 98.7 | 0 |
| EP-MC2-CF | 48.2 | 42.8 | 1.269 ± 0.008 | 1.306 | 2.7 | 95.9 | 4.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fredi, G.; Dirè, S.; Callone, E.; Ceccato, R.; Mondadori, F.; Pegoretti, A. Docosane-Organosilica Microcapsules for Structural Composites with Thermal Energy Storage/Release Capability. Materials 2019, 12, 1286. https://doi.org/10.3390/ma12081286
Fredi G, Dirè S, Callone E, Ceccato R, Mondadori F, Pegoretti A. Docosane-Organosilica Microcapsules for Structural Composites with Thermal Energy Storage/Release Capability. Materials. 2019; 12(8):1286. https://doi.org/10.3390/ma12081286
Chicago/Turabian StyleFredi, Giulia, Sandra Dirè, Emanuela Callone, Riccardo Ceccato, Francesco Mondadori, and Alessandro Pegoretti. 2019. "Docosane-Organosilica Microcapsules for Structural Composites with Thermal Energy Storage/Release Capability" Materials 12, no. 8: 1286. https://doi.org/10.3390/ma12081286
APA StyleFredi, G., Dirè, S., Callone, E., Ceccato, R., Mondadori, F., & Pegoretti, A. (2019). Docosane-Organosilica Microcapsules for Structural Composites with Thermal Energy Storage/Release Capability. Materials, 12(8), 1286. https://doi.org/10.3390/ma12081286