N-Lipidated Amino Acids and Peptides Immobilized on Cellulose Able to Split Amide Bonds


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
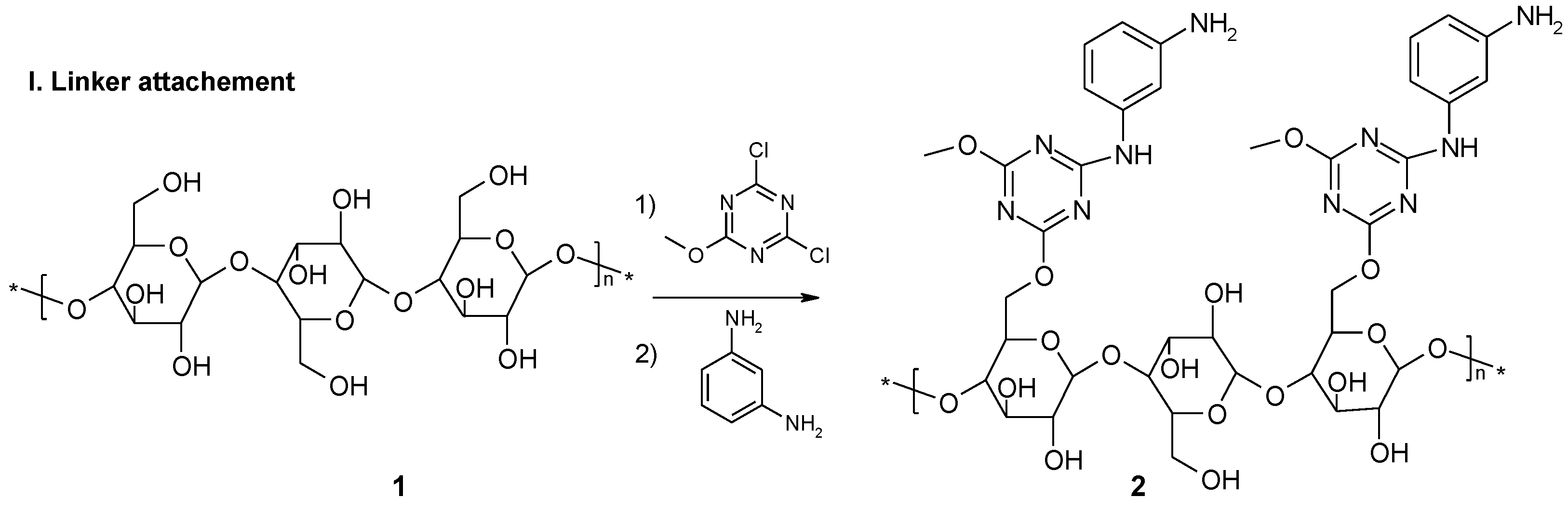
2.1. Functionalization of Whatman 7 Cellulose Sheets Using 2,4-Dichloro-6-Methoxy-1,3,5-Triazine (DCMT)
- for starting cellulose: %N 0.00–0.05%; Cl 0.01–0.05.
- for cellulose with attached chloro-dimethoxy-1,3,5-triazine: %N 3.64, %Cl 2.66.
2.2. Immobilization of M-Fenylenediamine
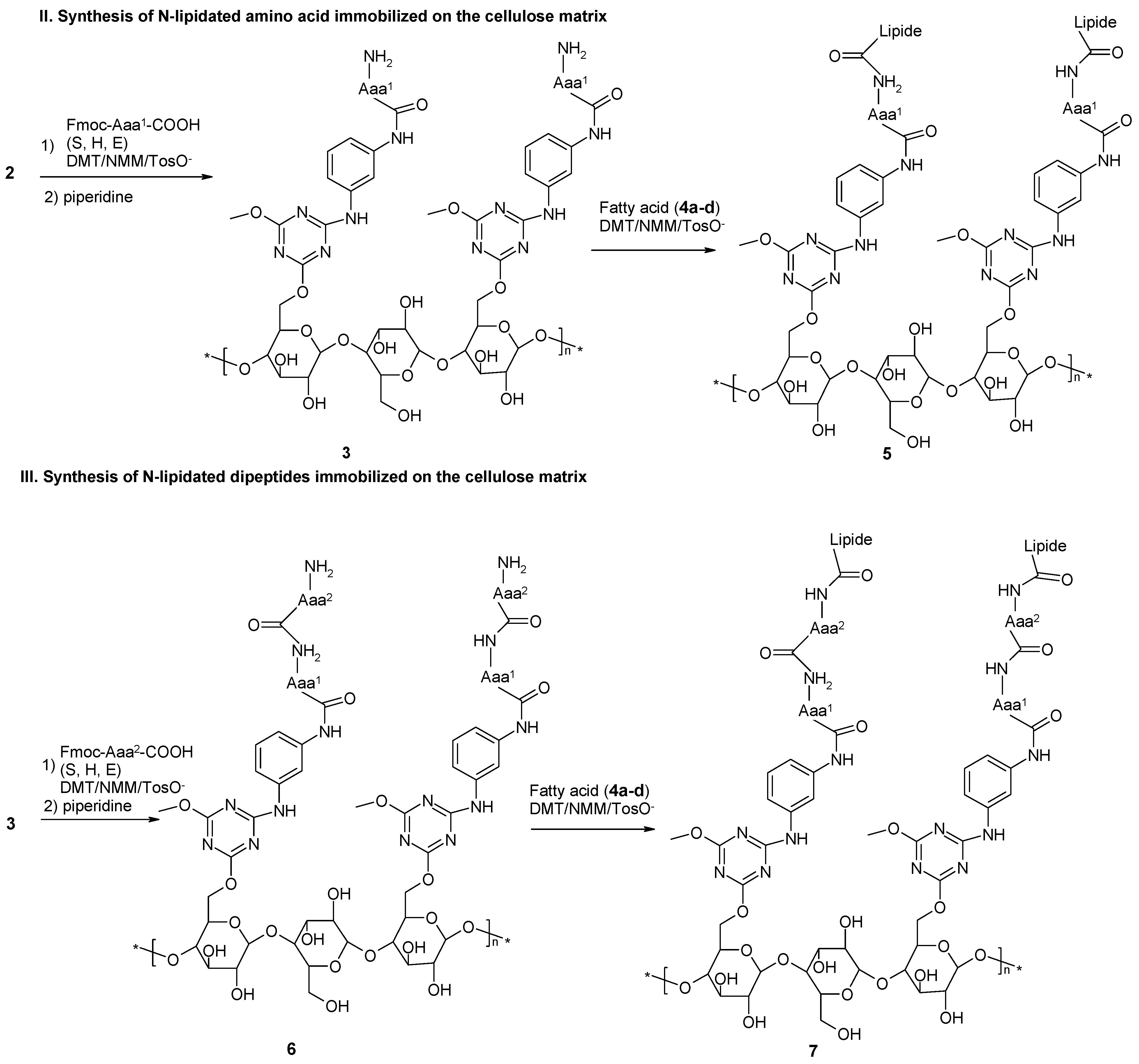
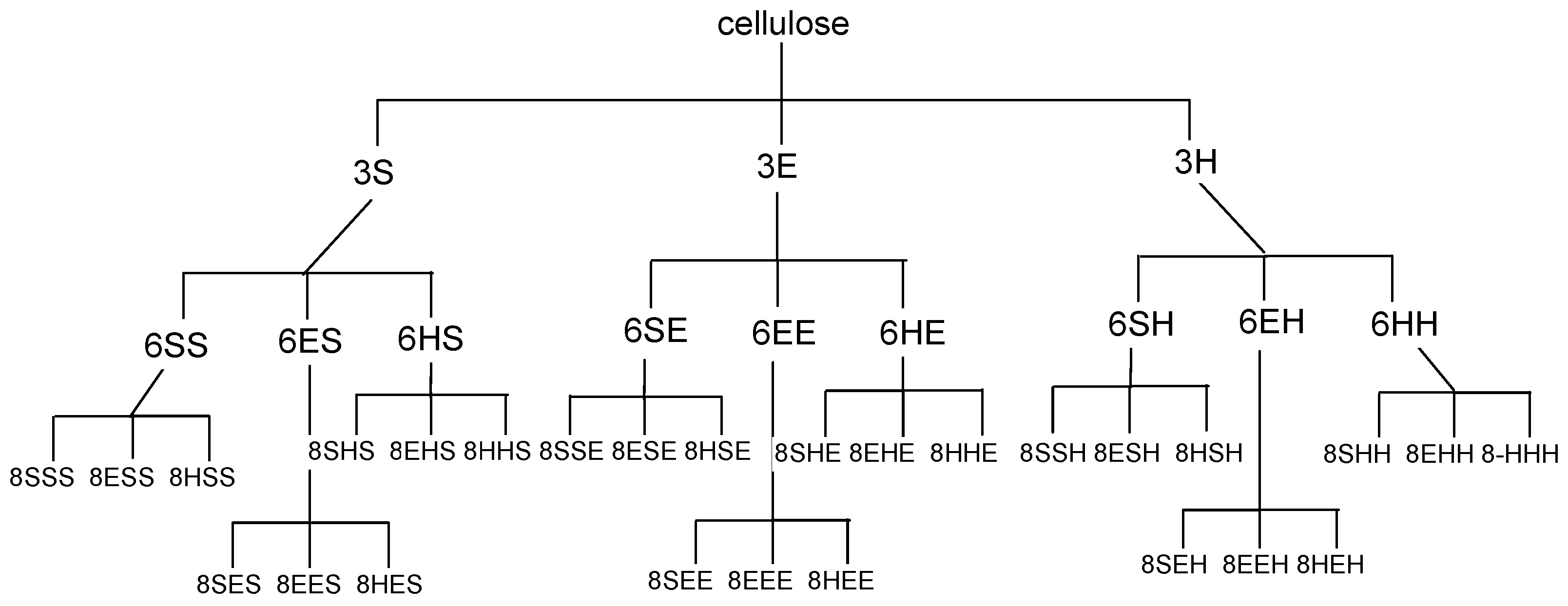
2.3. Loading of C-Terminal Amino Acid. Sub-Libraries {E},{S} and {H}
2.4. Removal of Fmoc-Protecting Group
2.5. Incorporation of the Second Amino Acid
2.6. Removal of Fmoc-Protecting Group
2.7. Incorporation of the N-Terminal Amino Acid
2.8. Removal of Fmoc-Protecting Group
2.9. Incorporation of Lipid Fragment
2.10. Removal of Side Chain Protecting Groups
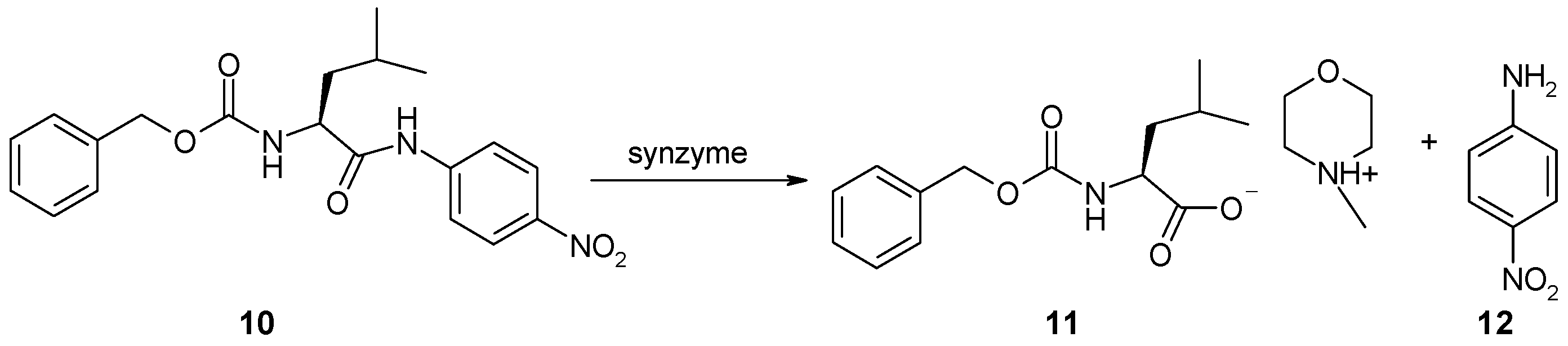
2.11. Synthesis of Z-Leu-NA (10)
2.12. Hydrolysis of Z-Leu-NA in the Presence of N-Lipidated Amino Acids, Di- and Tripeptides Immobilized on Cellulose
2.12.1. Hydrolysis of Z-Leu-NA in Solution at pH 7
2.12.2. Hydrolysis of Z-Leu-NA in Solution at pH 8.5
2.13. Docking of Metal Iions into the Catalytic Pockets of a Synzymes Library Immobilized on Cellulose
2.14. Hydrolysis of Z-Leu-NA in the Presence of N-Lipidated Amino Acids, Di- and Tripeptides Immobilized on Cellulose and Metal Ions
2.15. Blank Experiments
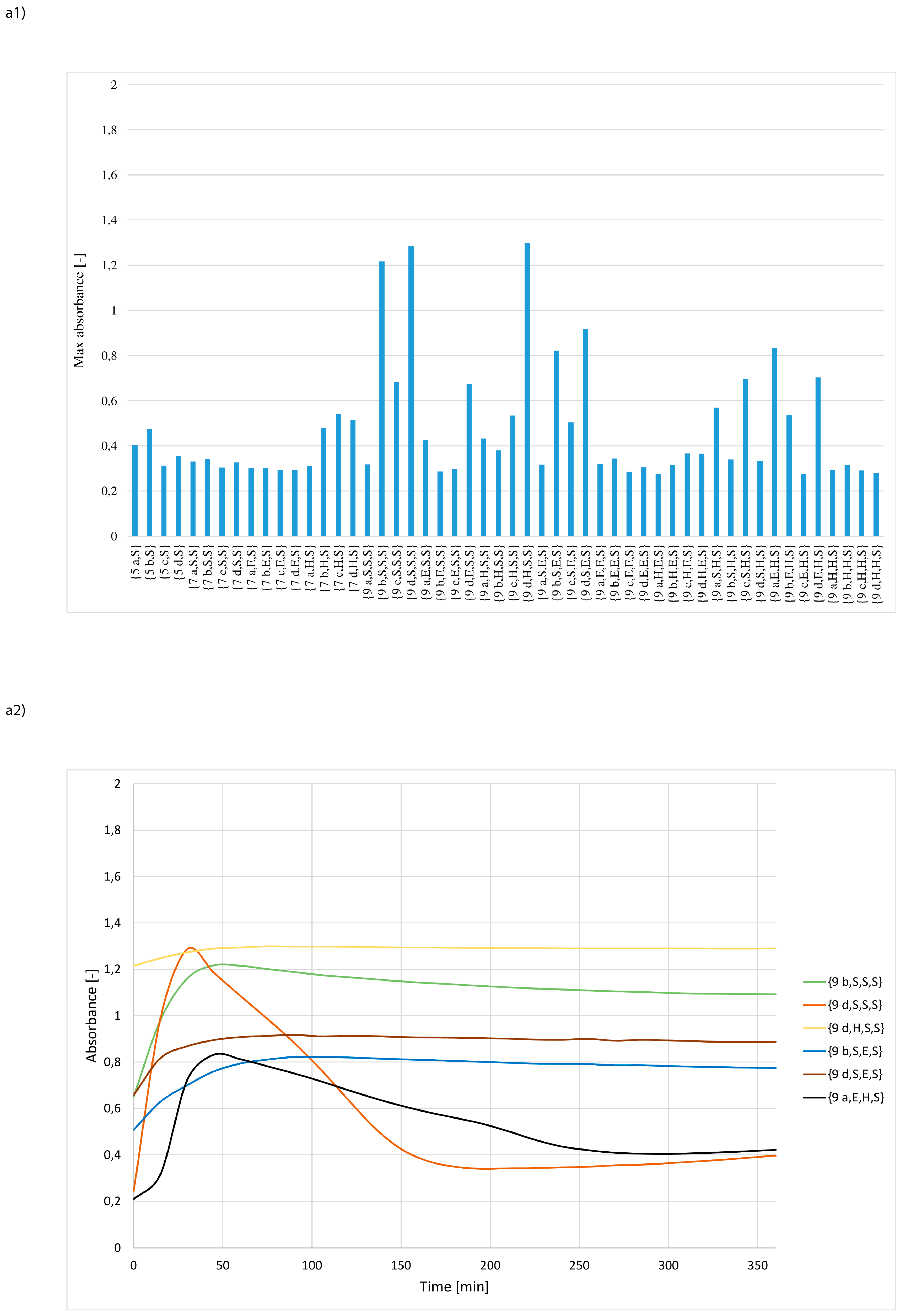
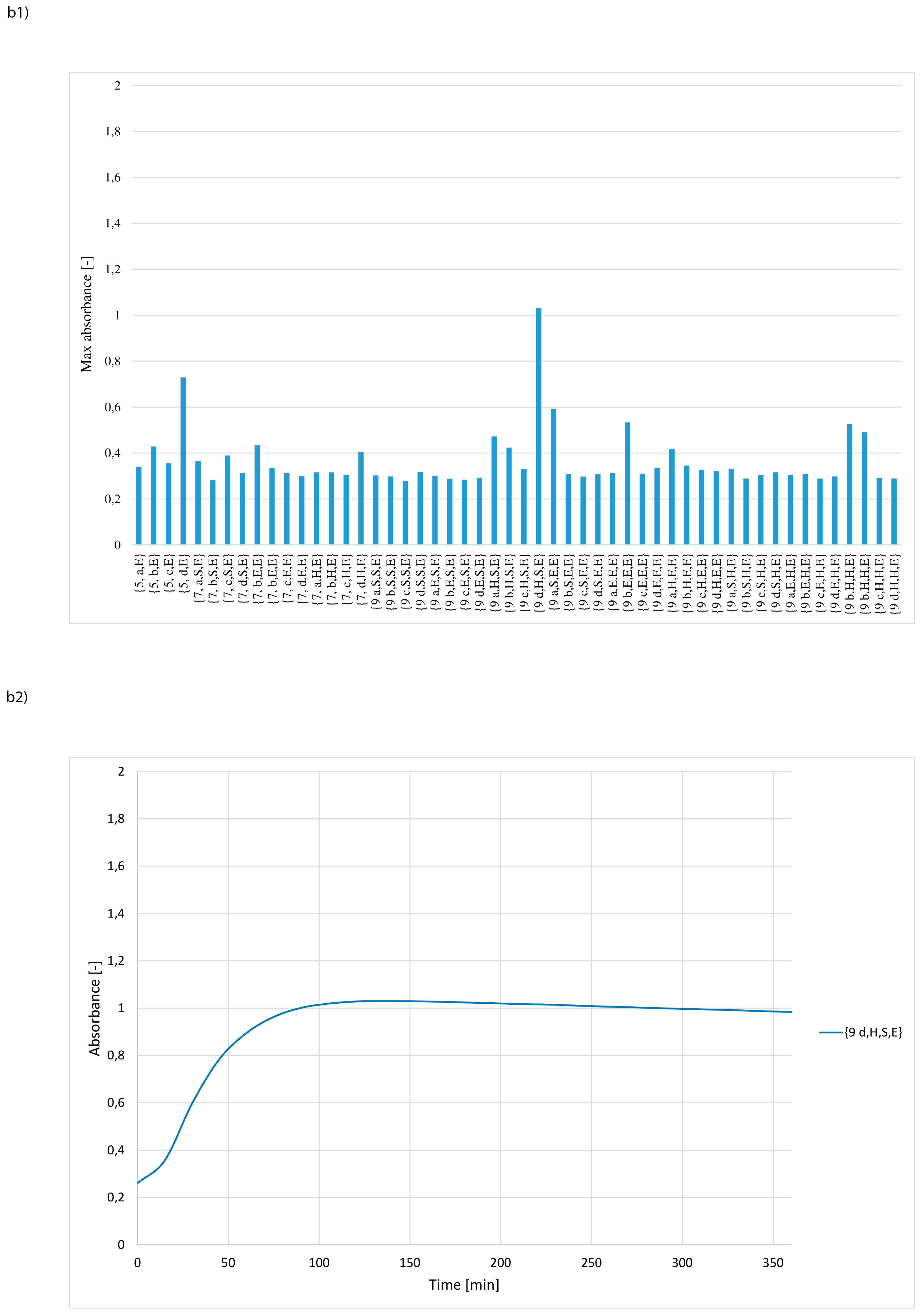
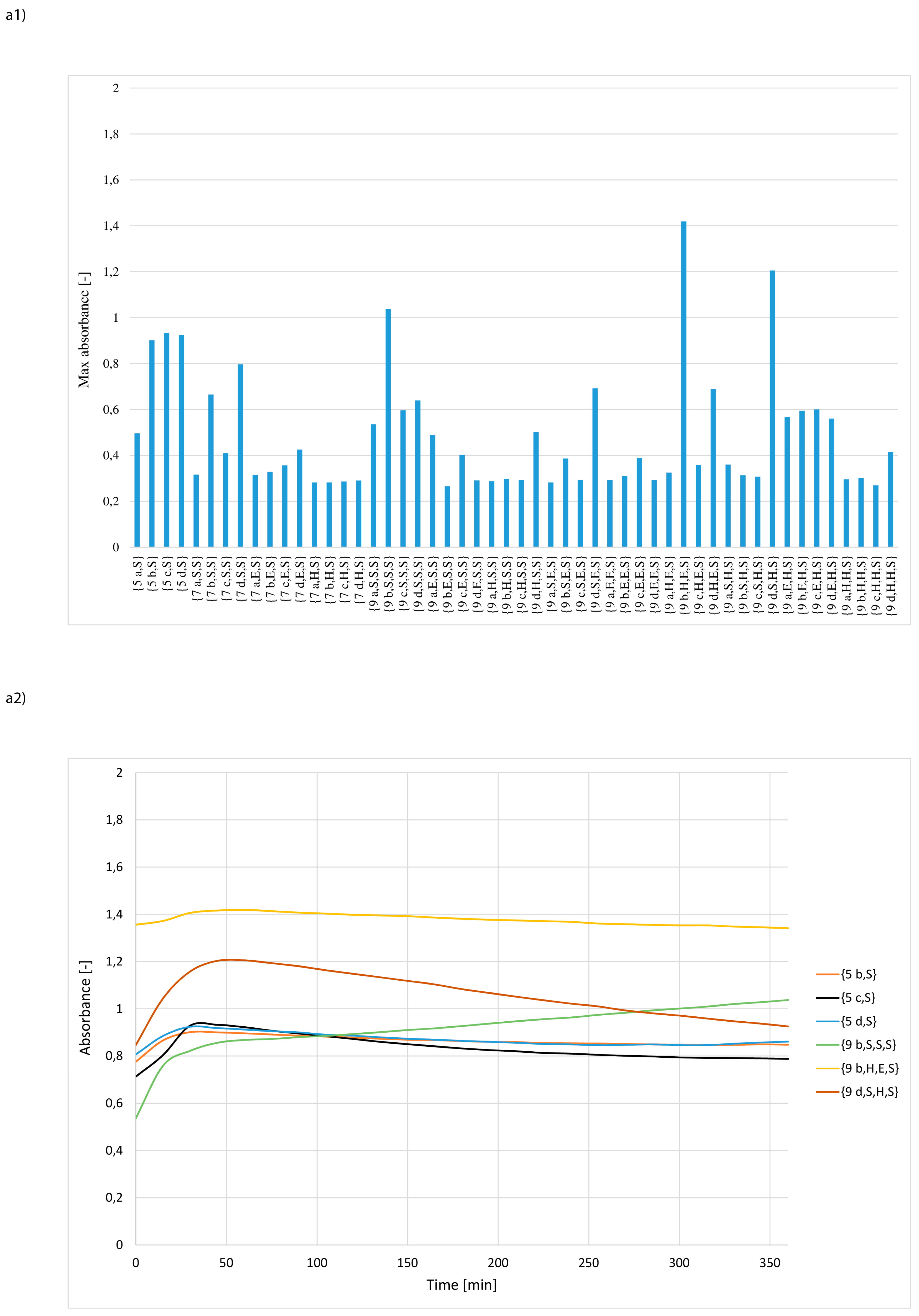
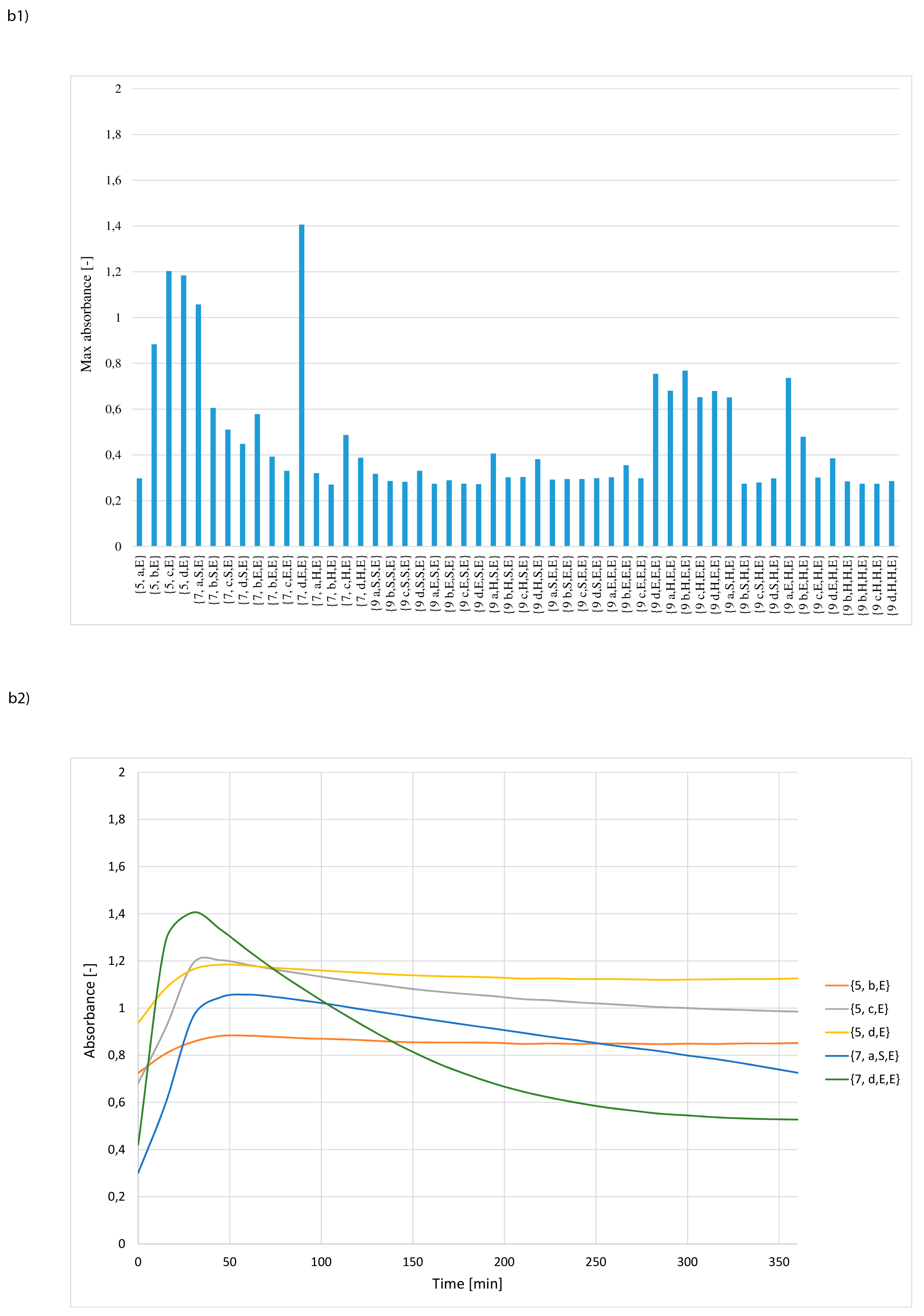
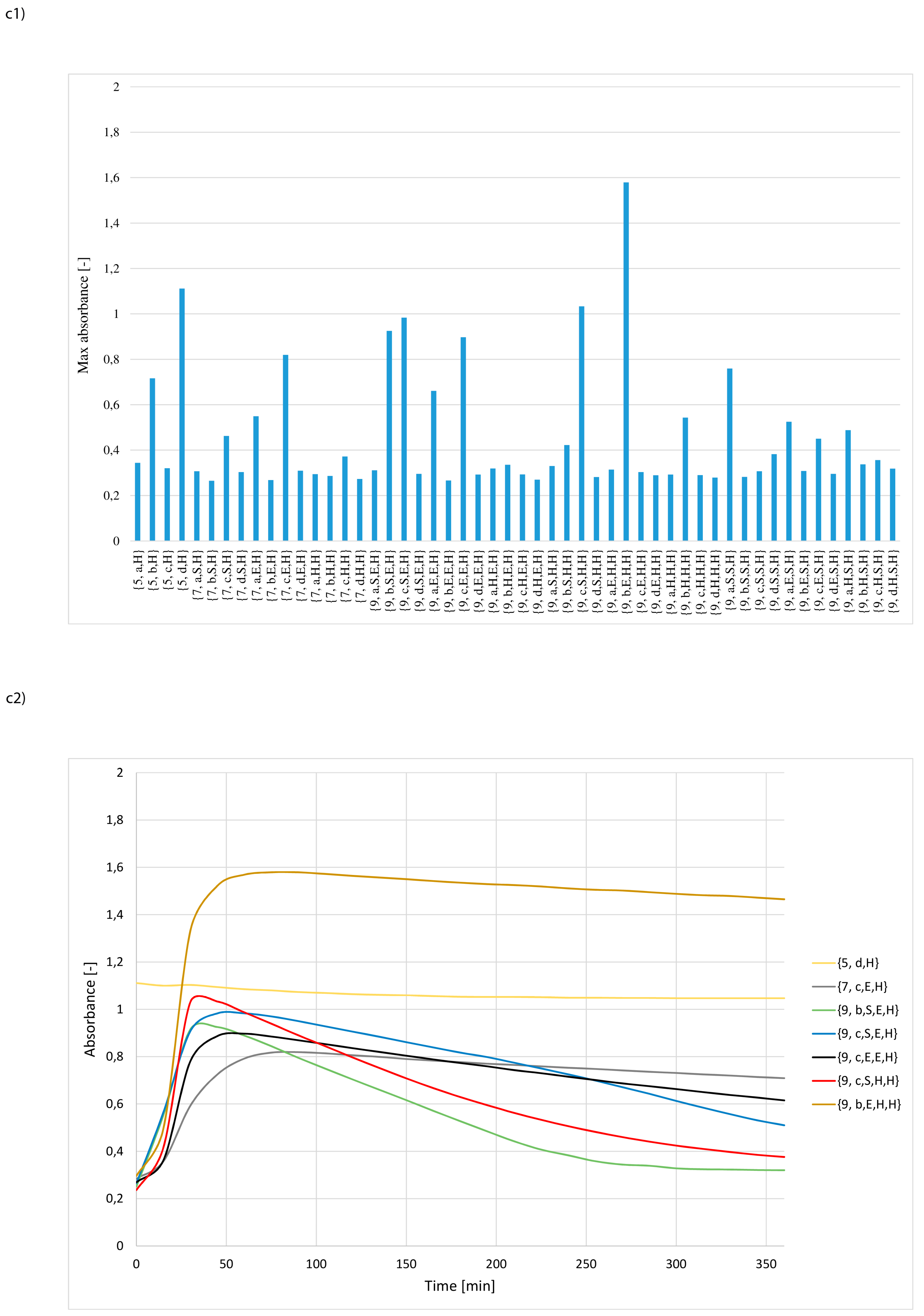
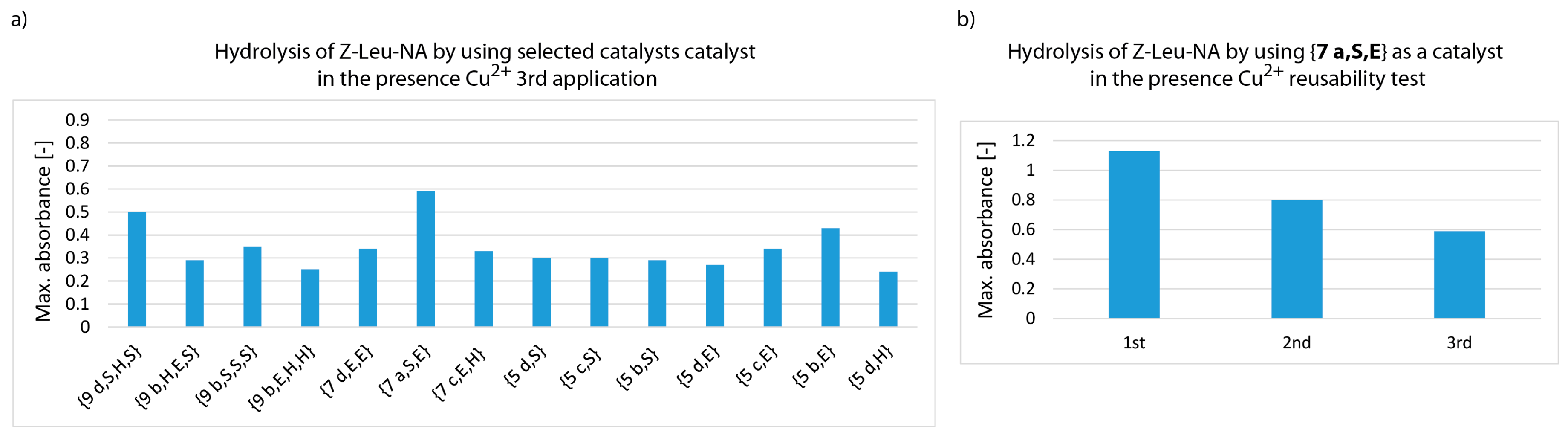
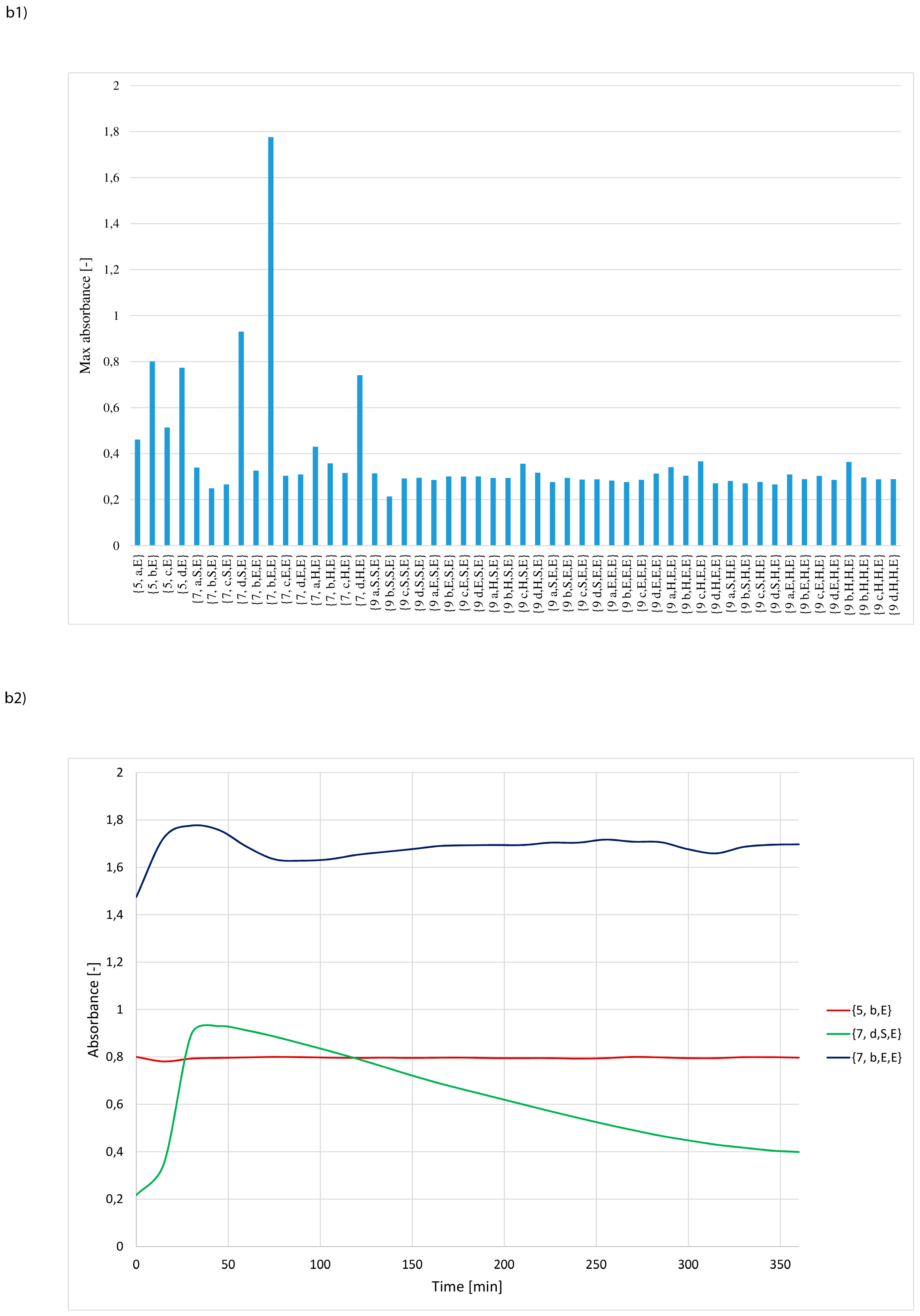
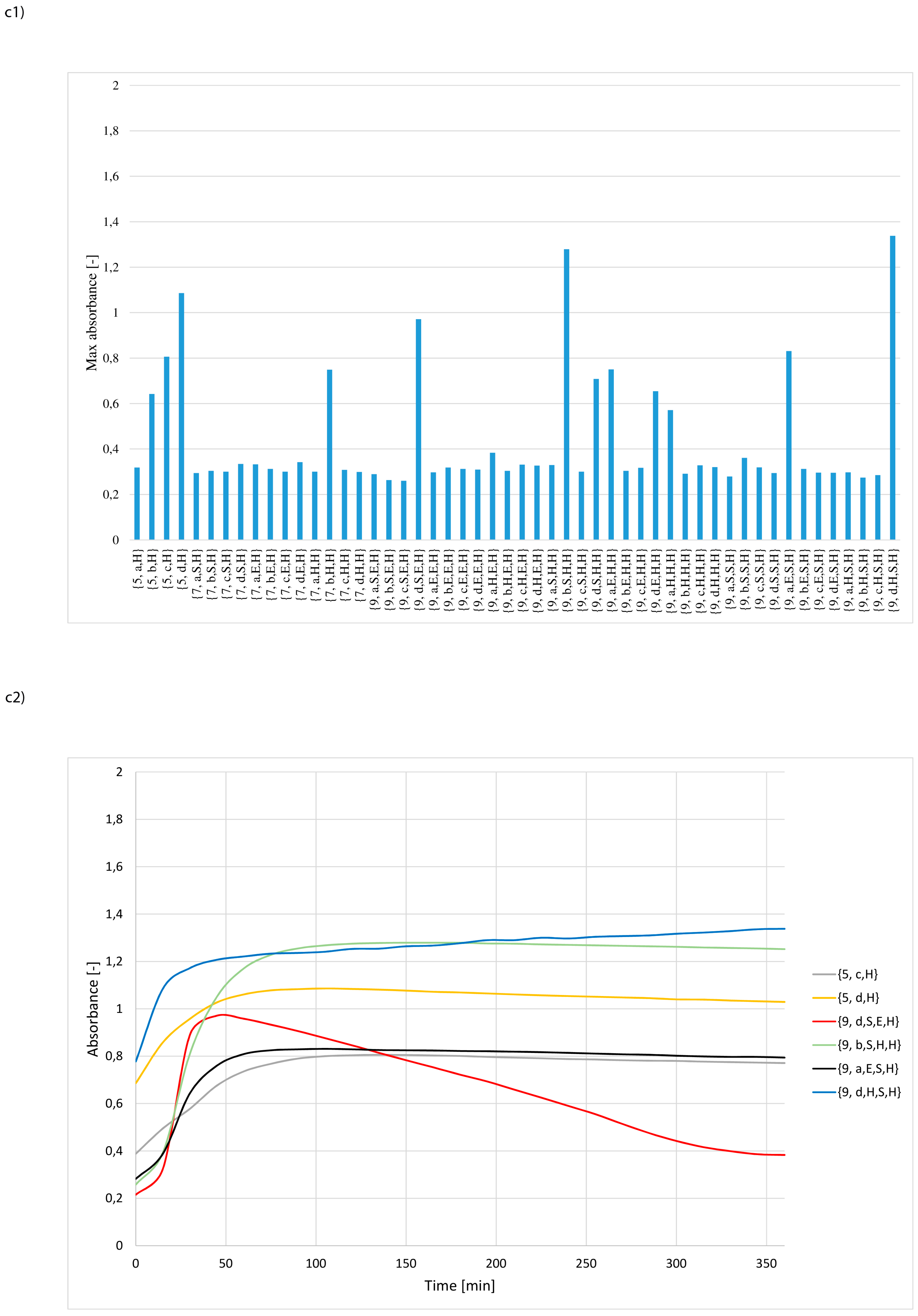
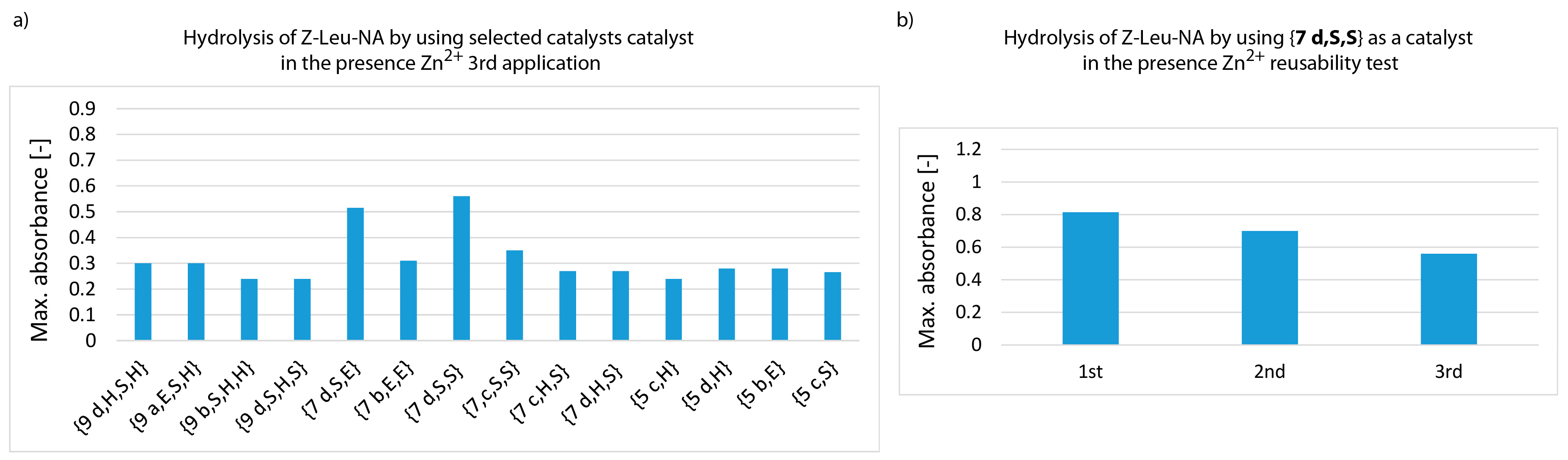
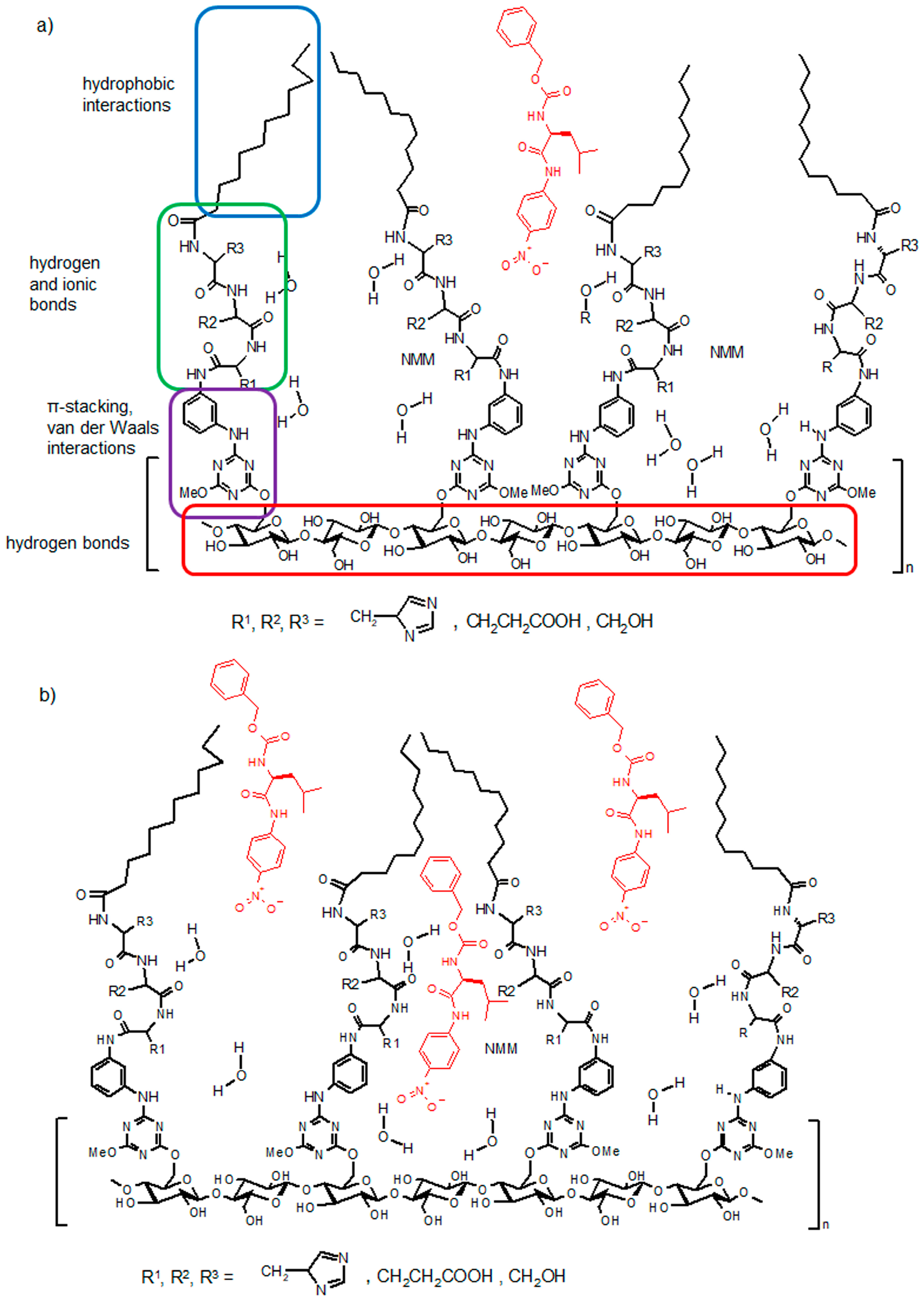
3. Results and Discussion
- whether structures composed of S, H and E residues would promote the hydrolysis of the amide/peptide bond (protease activity),
- the optimal synzyme structure for hydrolysis of the amide/peptide bond,
- if the synzymes could be used many times, giving the possibility of their use in many tests.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Matsuo, T.; Hirota, S. Artificial enzymes with protein scaffolds: Structural design and modification. Bioorg. Med. Chem. 2014, 22, 5638–5656. [Google Scholar] [CrossRef] [PubMed]
- Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P.W.N.M. Supramolecular catalysis. Part 2: Artificial enzyme mimics. Chem. Soc. Rev. 2014, 43, 1734–1787. [Google Scholar] [CrossRef] [PubMed]
- Schwizer, F.; Okamoto, Y.; Heinisch, T.; Gu, Y.; Pellizzoni, M.M.; Lebrun, V.; Reuter, R.; Kohler, V.; Lewis, J.C.; Ward, T.R. Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev. 2018, 118, 142–231. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Ren, J.; Qu, X. Catalytically active nanomaterials: A promising candidate for artificial enzymes. Acc. Chem. Res. 2014, 47, 1097–1105. [Google Scholar] [CrossRef]
- Wei, H.; Wang, E. Nanomaterials with enzyme-like characteristics (nanozymes): Next-generation artificial enzymes. Chem. Soc. Rev. 2013, 42, 6060–6093. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.A.; Gallivan, J.P. A flow cytometry-based screen for synthetic riboswitches. Nucleic Acids Res. 2009, 37, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Mathew, D.; Thomas, B.; Devaky, K.S. Biomimetic recognition and peptidase activities of transition state analogue imprinted chymotrypsin mimics. React. Funct. Polym. 2018, 124, 121–128. [Google Scholar] [CrossRef]
- Cao, L.; van Langen, L.; Roger, A.; Sheldon, R.A. Immobilised enzymes: Carrier-bond or carrier-free? Curr. Opin. Biotechnol. 2003, 14, 387–394. [Google Scholar] [CrossRef]
- Tischer, W.; Kasche, V. Immobilized enzymes: Crystals or carriers? Trends Biotechnol. 1999, 17, 326–335. [Google Scholar] [CrossRef]
- Janssen, M.H.A.; van Langen, L.M.; Pereira, S.R.M.; van Rantwijk, F.; Sheldon, R.A. Evaluation of the performance of immobilized penicillin G acylase using active-site titration. Biotechnol. Bioeng. 2002, 78, 425–432. [Google Scholar] [CrossRef]
- Dong, Z.; Luo, Q.; Liu, J. Artificial enzymes based on supramolecular scaffolds. Chem. Soc. Rev. 2012, 41, 7890–7908. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.T.; Freemont, P.S. Computational protein design with backbone plasticity. Biochem. Soc. Trans. 2016, 44, 1523–1529. [Google Scholar] [CrossRef] [PubMed]
- Kirby, A. Effective Molarities for Intramolecular Reactions. Adv. Phys. Org. Chem. 1980, 17, 183–278. [Google Scholar] [CrossRef]
- Kluger, R.; Lam, C.H. Carboxylic acid participation in amide hydrolysis. External general base catalysis and general acid catalysis in reactions of norbornenylanilic acids. J. Am. Chem. Soc. 1978, 100, 2191–2197. [Google Scholar] [CrossRef]
- Suh, J. Artificial Peptidases and Nucleases Using Macromolecular Catalytic System. Acc. Chem. Res. 2003, 36, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Suh, J. Synthesis of polymeric enzyme-like catalysts. Synlett 2001, 9, 1343–1363. [Google Scholar] [CrossRef]
- Suh, J.; Hah, S.S. Organic artificial proteinase with active site comprising three salicylate residues. J. Am. Chem. Soc. 1998, 120, 10088–10093. [Google Scholar] [CrossRef]
- Oh, S.; Chang, W.; Suh, J. An aspartic protease analogue: Intermolecular catalysis of peptide hydrolysis by carboxyl groups. Bioorg. Med. Chem. Lett. 2001, 11, 1469–1472. [Google Scholar] [CrossRef]
- Suh, J.; Oh, S. Remarkable proteolytic activity of imidazoles attached to cross-linked polystyrene. J. Org. Chem. 2000, 65, 7534–7540. [Google Scholar] [CrossRef]
- Wong, Y.-M.; Hoshino, Y.; Sudesh, K.; Miura, Y.; Numata, K. Optimization of Poly(N-isopropylacrylamide) as an Artificial Amidase. Biomacromolecules 2015, 16, 411–421. [Google Scholar] [CrossRef]
- Zhu, L.; Kostic, N.M. Toward artificial metallopeptidases: mechanisms by which platinum(II) and palladium(II) complexes promote selective, fast hydrolysis of unactivated amide bonds in peptides. Inorg. Chem. 1992, 31, 3994–4001. [Google Scholar] [CrossRef]
- Zozulia, O.; Dolan, M.A.; Korendovych, I.V. Catalytic peptide assemblies. Chem. Soc. Rev. 2018, 47, 3621–3639. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Tena-Solsona, M.; Miravet, J.F.; Escuder, B. Towards Supramolecular Catalysis with Small Self-Assembled Peptides. Isr. J. Chem. 2015, 55, 711–723. [Google Scholar] [CrossRef]
- Duncan, K.L.; Ulijn, R.V. Short Peptides in Minimalistic Biocatalyst Design. Biocatalysis 2015, 1, 67–81. [Google Scholar] [CrossRef]
- Rasale, D.B.; Das, A.K. Chemical Reactions Directed Peptide Self-Assembly. Int. J. Mol. Sci. 2015, 16, 10797–10820. [Google Scholar] [CrossRef] [PubMed]
- Mikolajczak, D.J.; Heier, J.L.; Schade, B.; Koksch, B. Catalytic Activity of Peptide-Nanoparticle Conjugates Regulated by a Conformational Change. Biomacromolecules 2017, 18, 3557–3562. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Lv, Y.; Liu, X.; Qi, W.; Su, R.; He, Z. Enhancing the Activity of Peptide-Based Artificial Hydrolase with Catalytic Ser/His/Asp Triad and Molecular Imprinting. ACS Appl. Mater. Interfaces 2016, 8, 14133–14141. [Google Scholar] [CrossRef]
- Fraczyk, J.; Malawska, B.; Kaminski, Z.J. The application of a library of artificial receptors formed by the self-organization of N-lipidated peptides immobilized on cellulose in studying the effects of the incorporation of a fluorine atom. J. Comb. Chem. 2009, 11, 446–451. [Google Scholar] [CrossRef]
- Fraczyk, J.; Kolesinska, B.; Czarnecka, A.; Malawska, B.; Wieckowska, A.; Bajda, M.; Kaminski, Z.J. The application of a library of artificial receptors formed by the self-organization of N-lipidated peptides immobilized on cellulose for preliminary studies of binding of N-phenylpiperazines. QSAR Comb. Sci. 2009, 28, 728–736. [Google Scholar] [CrossRef]
- Eichhorn, S.J.; Rahatekar, S.S.; Vignolini, S.; Windle, A.H. New horizons for cellulose nanotechnology. Philos. Trans. A Math. Phys. Eng. Sci. 2018, 376, 20170200. [Google Scholar] [CrossRef]
- Nishiyama, Y. Structure and properties of the cellulose microfibril. J. Wood Sci. 2009, 55, 241–249. [Google Scholar] [CrossRef]
- Ding, S.Y.; Himmel, M.E. The maize primary cell wall microfibril: A new model derived from direct visualization. J. Agric. Food Chem. 2006, 54, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.; Bansal, P.; Lee, J.H.; Realff, M.J.; Bommarius, A.S. Cellulose crystallinity—A key predictor of the enzymatic hydrolysis rate. FEBS J. 2010, 277, 1571–1582. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Siesler, H.W.; Mitsui, K.; Tsuchikawa, S. Difference of the Crystal Structure of Cellulose in Wood after Hydrothermal and Aging Degradation: A NIR Spectroscopy and XRD Study. Biomacromolecules 2010, 11, 2300–2305. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, M.C. Structure of native cellulose microfibrils, the starting point for nanocellulose manufacture. Philos. Trans. A Math. Phys. Eng. Sci. 2018, 376, 20170045. [Google Scholar] [CrossRef] [PubMed]
- Nugmanov, O.K.; Pertsin, A.I.; Zabelin, L.V.; Marchenko, G.N. The Molecular-crystal Structure of Cellulose. Russ. Chem. Rev. 1987, 56, 764–776. [Google Scholar] [CrossRef]
- Andersson, S.; Serimaa, R.; Paakkari, T.; Saranpä, P.; Pesonen, E. Crystallinity of wood and the size of cellulose crystallites in Norway spruce (Picea abies). J. Wood Sci. 2003, 49, 531–537. [Google Scholar] [CrossRef]
- Fraczyk, J.; Mrozek, A.; Kaminski, Z.J. Structure-activity relationship in binding ligands to library of artificial receptors: The search for biocompatible sensor. Bioelectrochemistry 2010, 80, 2–9. [Google Scholar] [CrossRef]
- Fraczyk, J.; Kaminski, Z.J. Designing, synthesis and application of a library of supramolecular structures formed by N-lipidated peptides immobilized on cellulose. Artificial receptors. J. Comb. Chem. 2008, 10, 934–940. [Google Scholar] [CrossRef]
- Fraczyk, J.; Kolesinska, B.; Kaminski, Z.J. The new approach to organocatalysts. Synthesis of a library of N-lipidated oligopeptides immobilized on cellulose and screening of their catalytic activity. ARKIVOC 2012, 4, 186–195. [Google Scholar] [CrossRef]
- Fraczyk, J.; Kolesinska, B.; Kaminski, Z.J. N-lipidated oligopeptides immobilized on cellulose as new type of organocatalysts. Comb. Chem. High Throughput Screen. 2013, 16, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Oyamada, H.; Saito, T.; Inaba, S.; Ueki, M. Reinvestigation of the phosphazo method and synthesis of N-(t-butoxycarbonyl)-l-arginine p-nitroanilide and a chromogenic enzyme substrate for the factor Xa. Bull. Chem. Soc. Jpn. 1991, 64, 1422–1424. [Google Scholar] [CrossRef]
- Kolesinska, B.; Rozniakowski, K.K.; Fraczyk, J.; Relich, I.; Papini, A.M.; Kaminski, Z.J. The Effect of Counterion and Tertiary Amine on the Efficiency of N-Triazinylammonium Sulfonates in Solution and Solid-Phase Peptide Synthesis. EurJOC 2015, 401–408. [Google Scholar] [CrossRef]
- Fraczyk, J.; Kaminski, Z.J.; Katarzynska, J.; Kolesinska, B. 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium Toluene-4-sulfonate (DMT/NMM/TsO-) Universal Coupling Reagent for Synthesis in Solution. Helv. Chim. Acta 2017, 100, e1700187. [Google Scholar] [CrossRef]
- Mohamad, N.R.; Marzuki, N.H.C.; Buang, N.A.; Huyop, F.; Wahab, R.A. An overview of technologies for immobilization of enzymes and surface analysis techniques for immobilized enzymes. Biotechnol. Biotechnol. Equip. 2015, 29, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Jannat, M.; Yang, K.-L. Immobilization of Enzymes on Flexible Tubing Surfaces for Continuous Bioassays. Langmuir 2018, 34, 14226–14233. [Google Scholar] [CrossRef]
- Bavaro, T.; Cattaneo, G.; Serra, I.; Benucci, I.; Pregnolato, M.; Terreni, M. Immobilization of Neutral Protease from Bacillus subtilis for Regioselective Hydrolysis of Acetylated Nucleosides: Application to Capecitabine Synthesis. Molecules 2016, 21, 1621. [Google Scholar] [CrossRef]
- Rehm, F.B.H.; Chen, S.; Rehm, B.H.A. Enzyme Engineering for In Situ Immobilization. Molecules 2016, 21, 1370. [Google Scholar] [CrossRef]
- Divya, M.; Benny, T.; Devaky, K.S. Amidase activity of phosphonate TSA-built polymer catalysts derived from organic monomers in the amidolysis of amino acid p-nitroanilides. Appl. Catal. A-Gen. 2016, 528, 93–103. [Google Scholar] [CrossRef]
- Divya, M.; Benny, T.; Christy, P.; Aparna, E.P.; Devaky, K.S. Catalytic amidolysis of amino acid p-nitroanilides using transition state analogue imprinted artificial enzymes: Cooperative effect of pyridine moiety. Bioorg. Chem. 2017, 74, 91–103. [Google Scholar] [CrossRef]
- Bond, J.S.; Butler, E. Intracellular Proteases. Ann. Rev. Biochem. 1987, 56, 333–364. [Google Scholar] [CrossRef] [PubMed]
- Birkedal-Hansen, H.; Moore, W.G.I.; Bodden, M.K.; Windsor, L.J.; Birkedal-Hansen, B.; DeCarlo, A.; Engler, J.A. Matrix Metalloproteinases: A Review. Crit. Rev. Oral. Biol. Med. 1993, 4, 197–250. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.E.; Vallee, B.L. Metallocarboxypeptidase-substrate complexes. Biochemistry 1962, 1, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, J.A.; Major Jourden, J.L.; Miller, M.T.; Cohen., S.M. To bind zinc or not to bind zinc: An examination of innovative approaches to improved metalloproteinase inhibition. BBA Mol. Cell Res. 2010, 803, 72–94. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fraczyk, J.; Kaminski, Z.J. N-Lipidated Amino Acids and Peptides Immobilized on Cellulose Able to Split Amide Bonds. Materials 2019, 12, 578. https://doi.org/10.3390/ma12040578
Fraczyk J, Kaminski ZJ. N-Lipidated Amino Acids and Peptides Immobilized on Cellulose Able to Split Amide Bonds. Materials. 2019; 12(4):578. https://doi.org/10.3390/ma12040578
Chicago/Turabian StyleFraczyk, Justyna, and Zbigniew J. Kaminski. 2019. "N-Lipidated Amino Acids and Peptides Immobilized on Cellulose Able to Split Amide Bonds" Materials 12, no. 4: 578. https://doi.org/10.3390/ma12040578
APA StyleFraczyk, J., & Kaminski, Z. J. (2019). N-Lipidated Amino Acids and Peptides Immobilized on Cellulose Able to Split Amide Bonds. Materials, 12(4), 578. https://doi.org/10.3390/ma12040578