On the Corrosion Mechanism of CO2 Transport Pipeline Steel Caused by Condensate: Synergistic Effects of NO2 and SO2




Abstract
:
1. Introduction
2. Materials and Methods
2.1. Exposure and Electrochemical Test Setup
2.2. Analyses
3. Results and Discussion
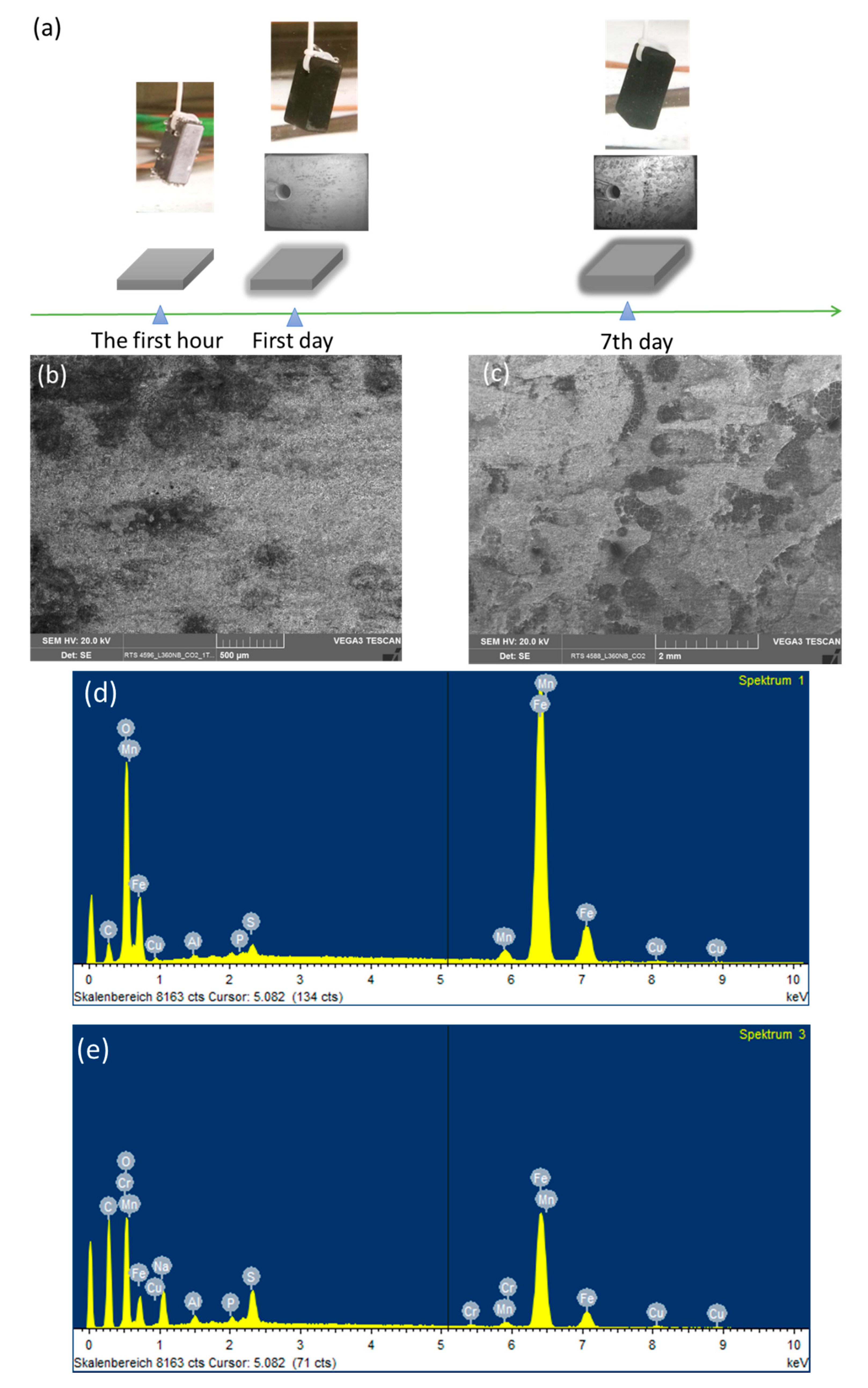
3.1. Corrosion Behaviors of L360NB in Condensate and the Role of CO2
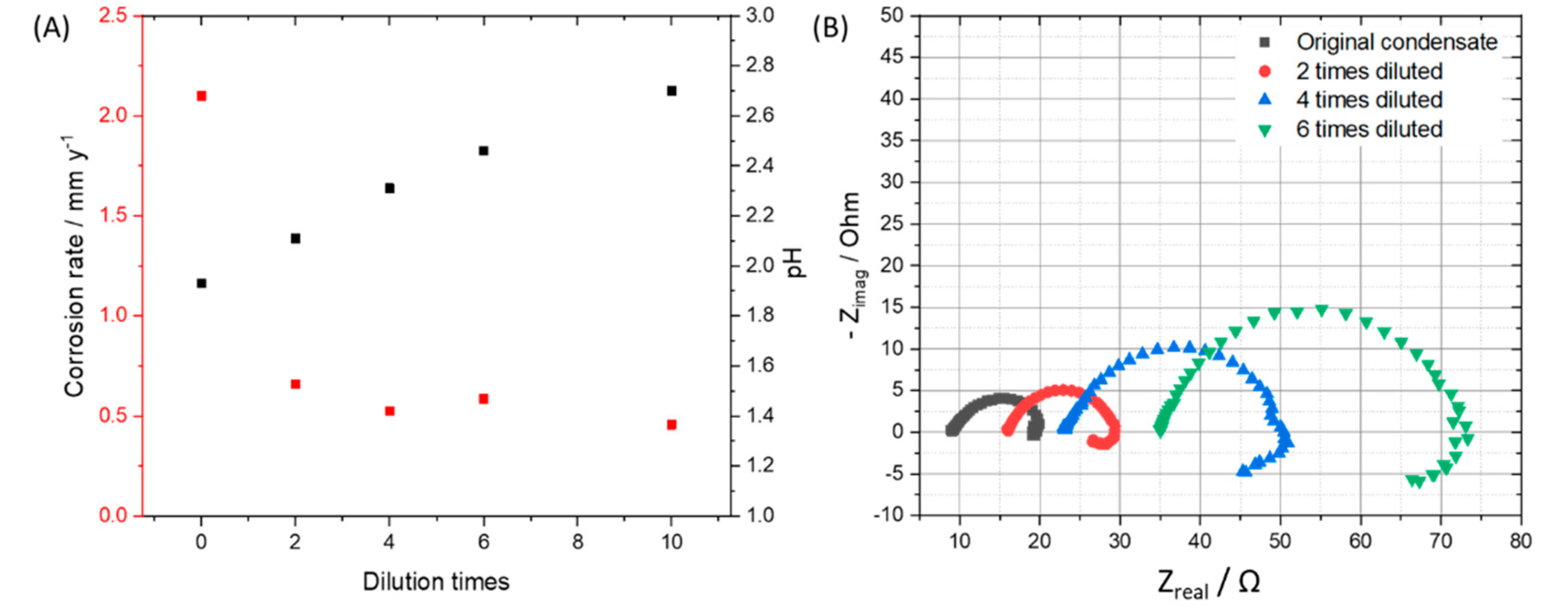
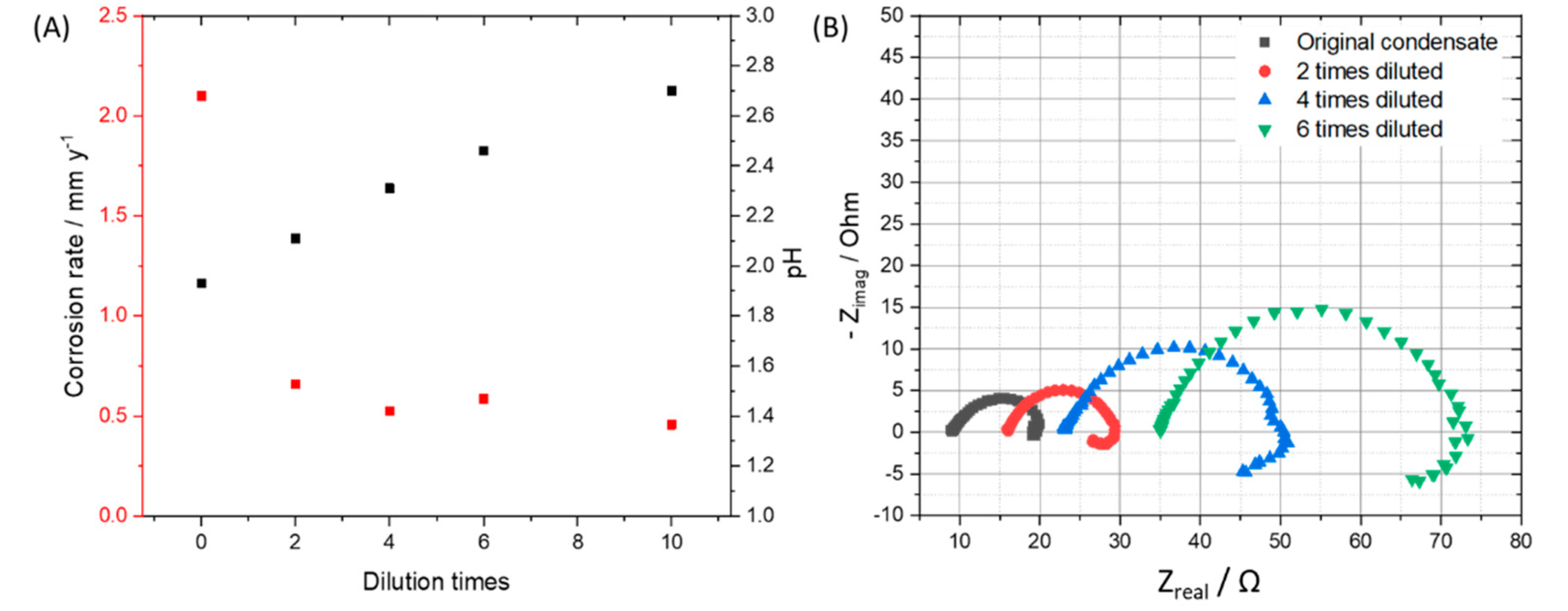
3.2. Effects of Condensate Concentration
3.3. Effects of Each Acidic Component in Condensate on the Corrosion Behavior
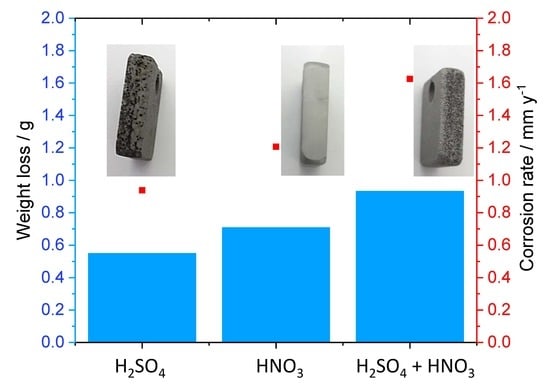
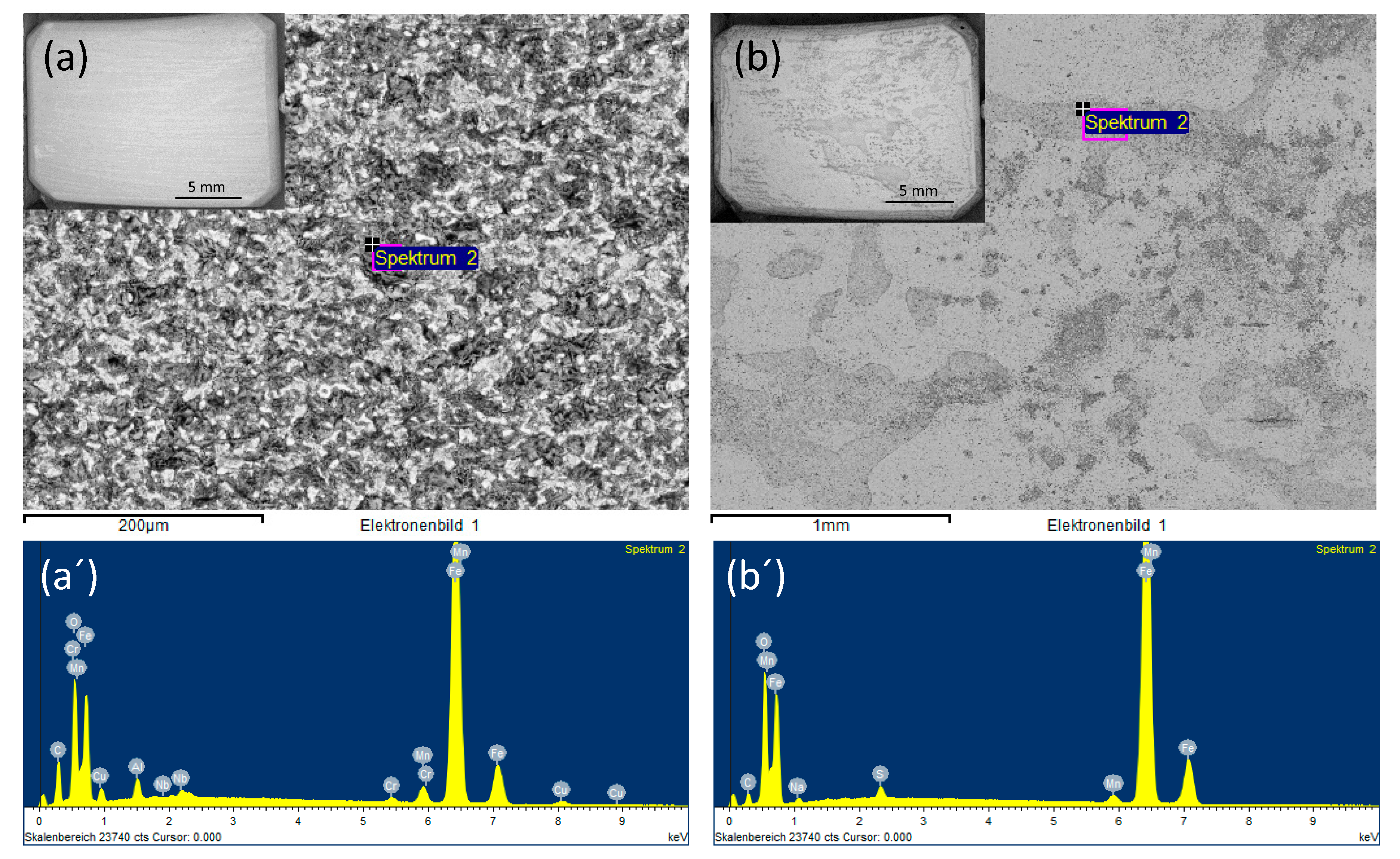
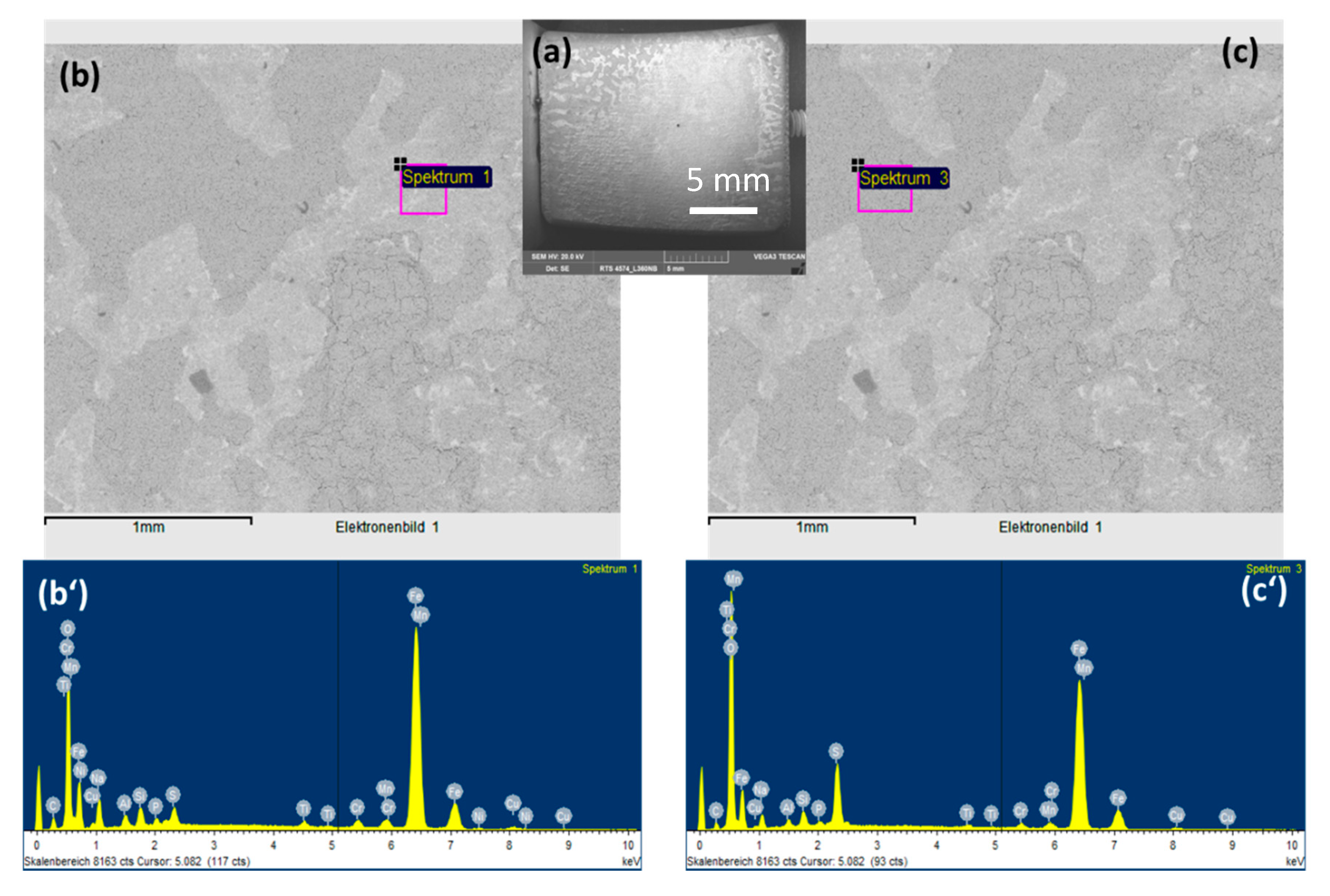
3.3.1. Role of Each Component on the Surface Morphology and Corrosion Rate
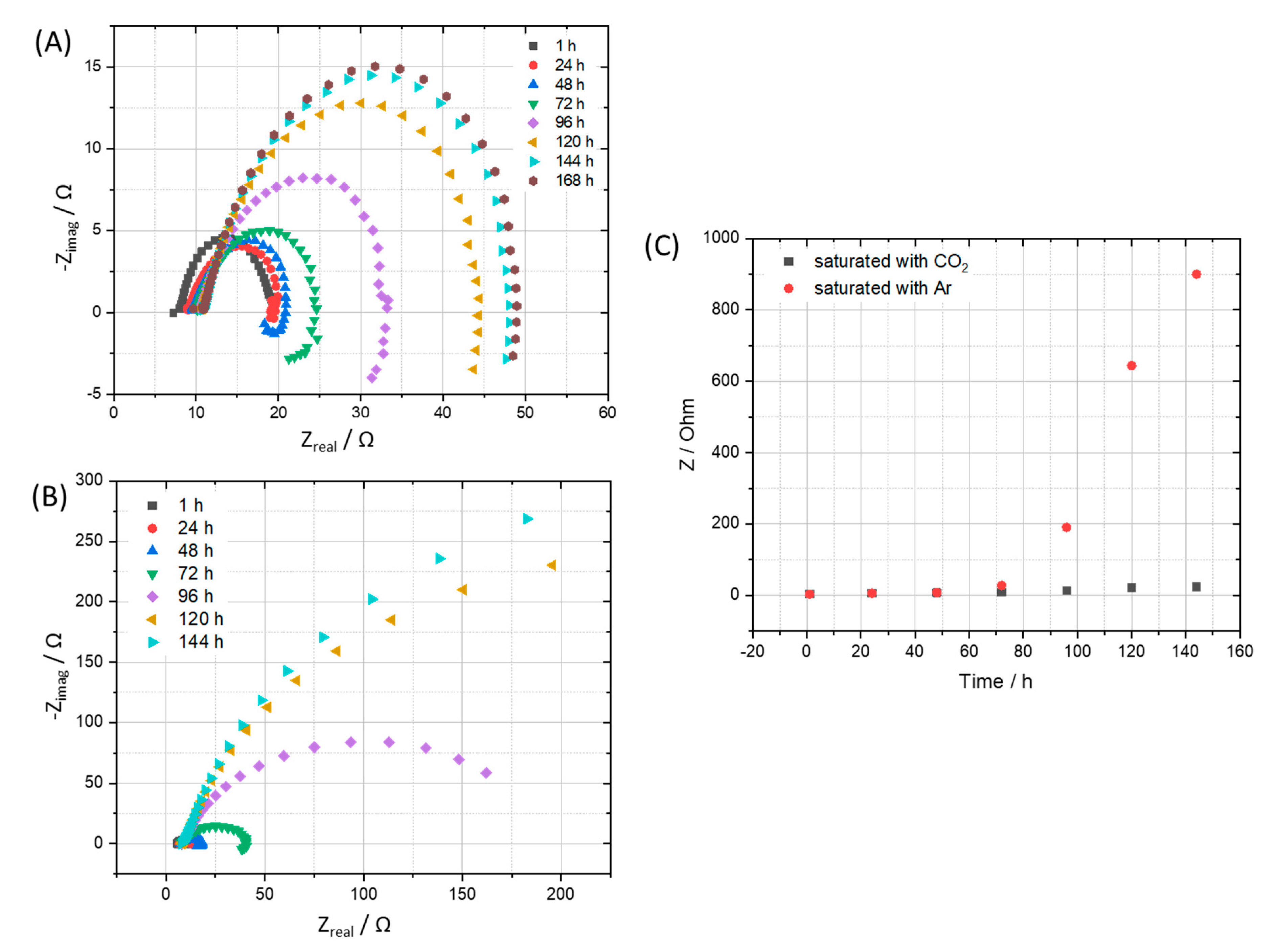
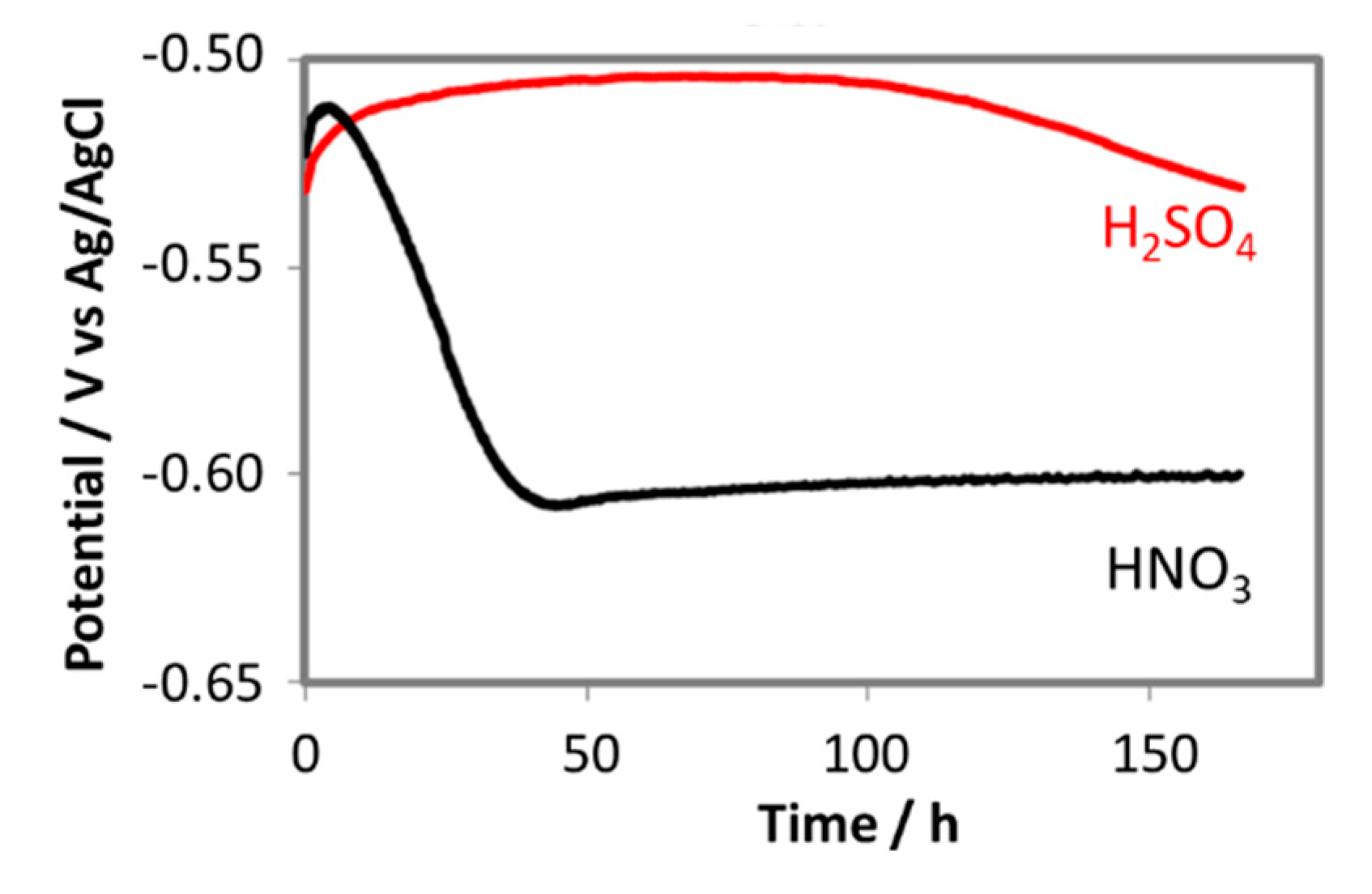
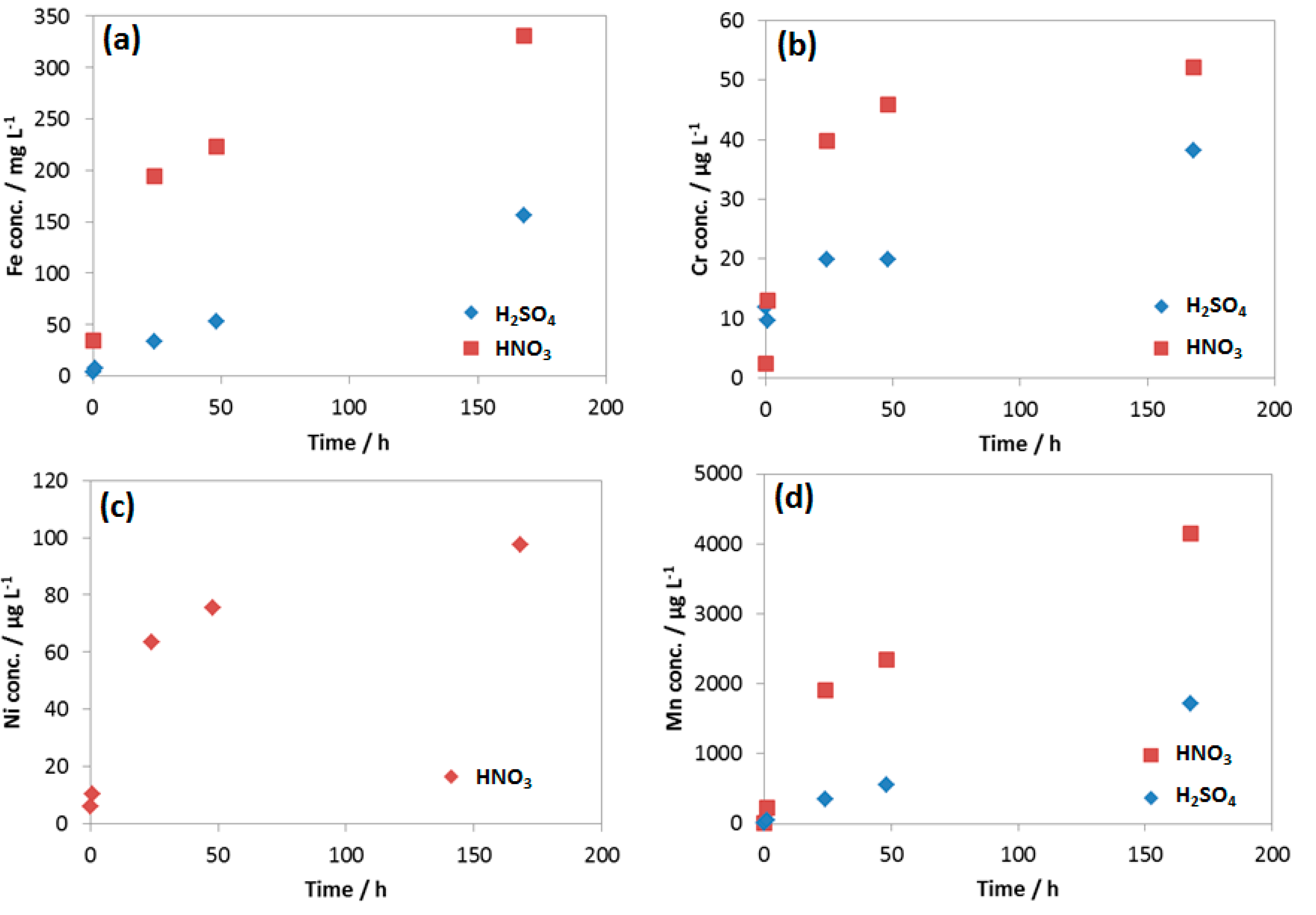
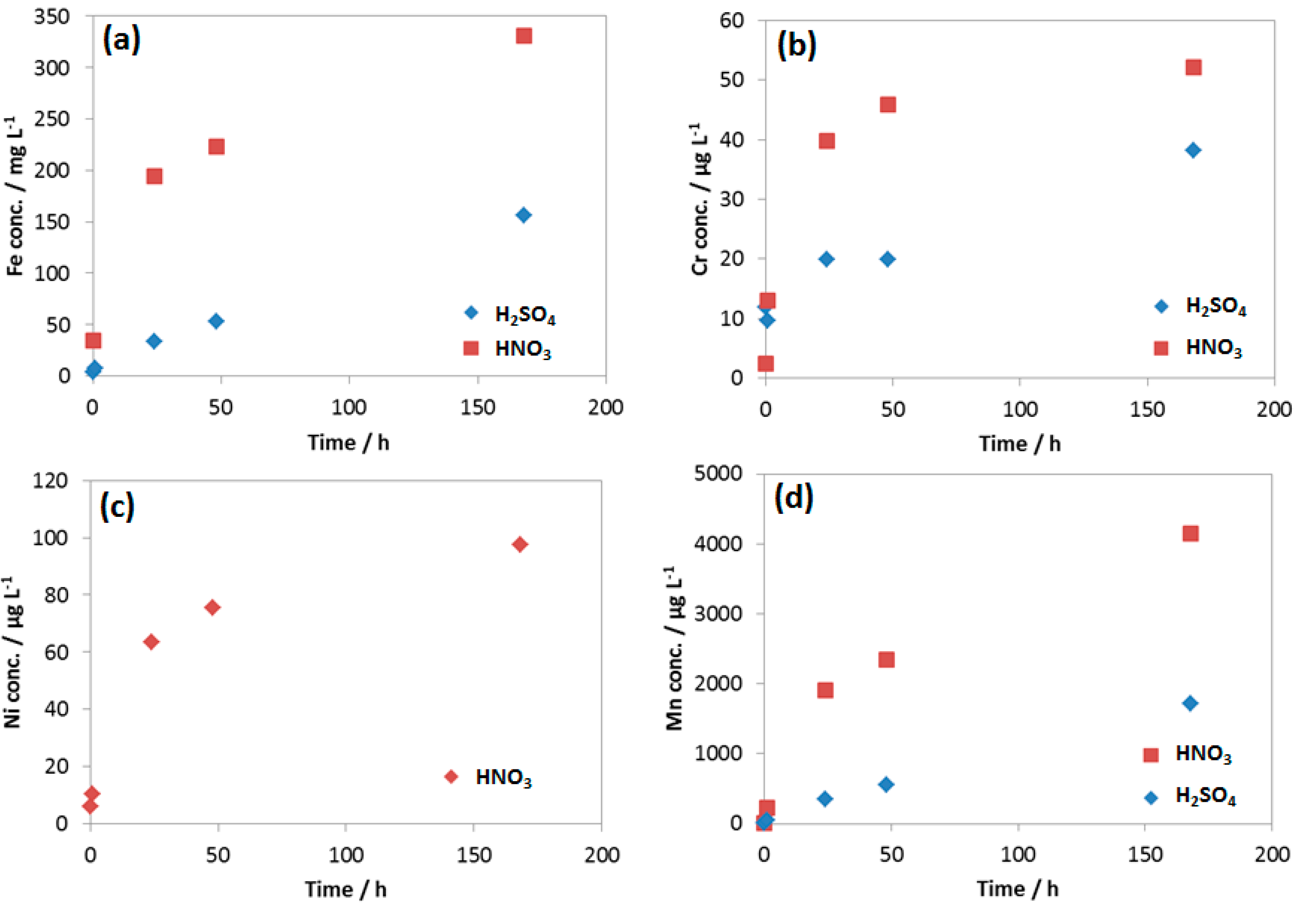
3.3.2. Kinetic of Corrosion Process
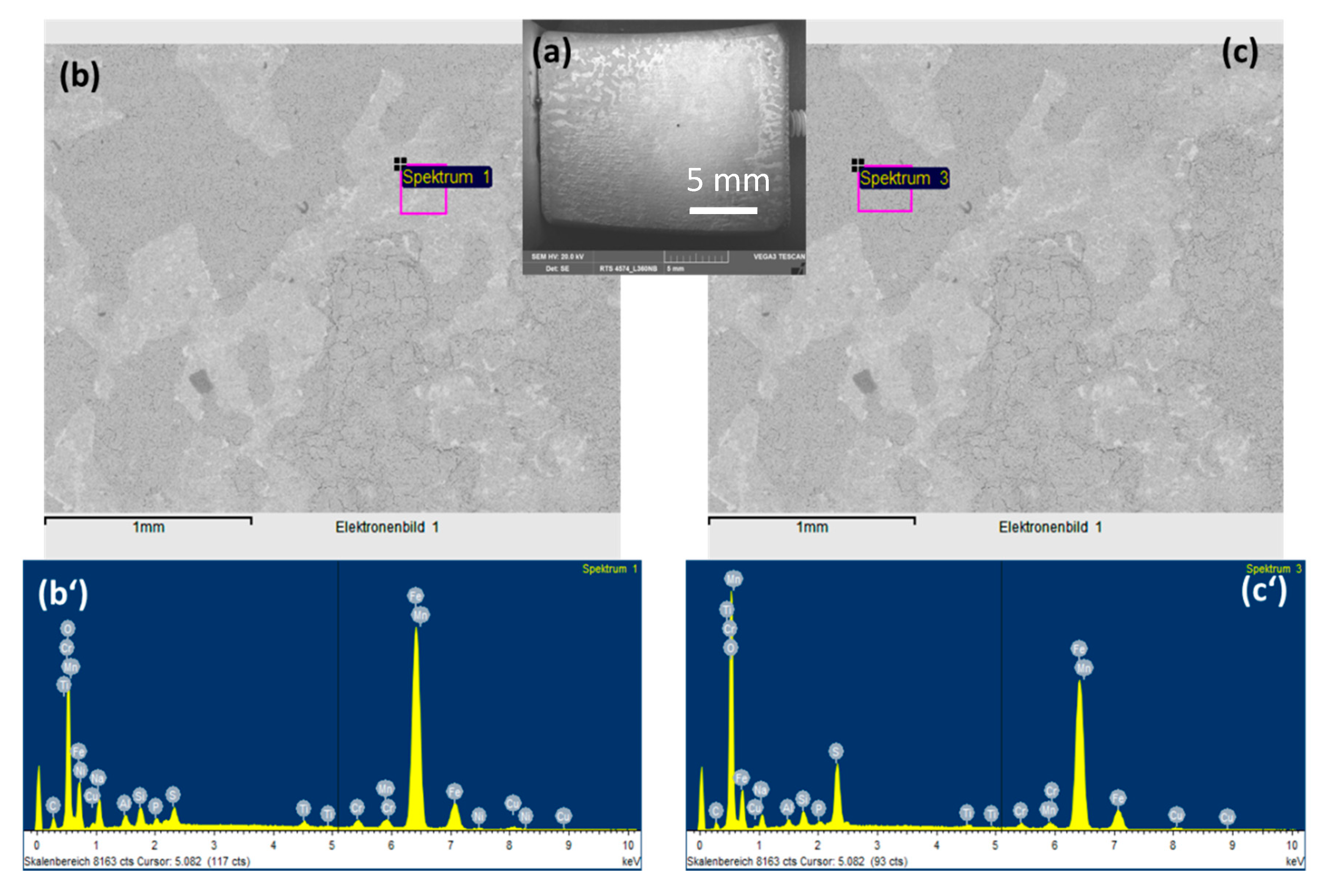
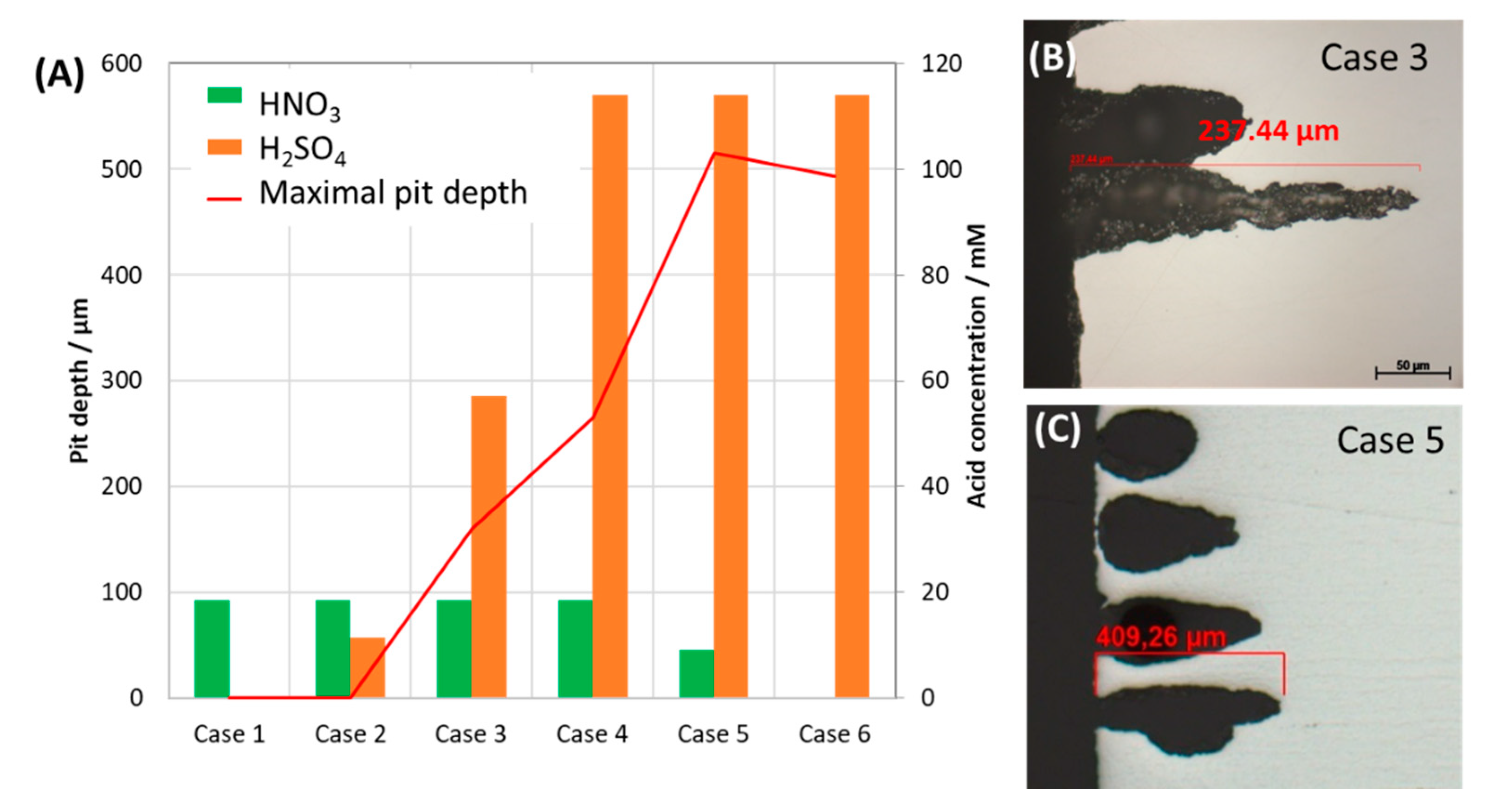
3.4. Effects of Flow Rate, Microstructure of Steel and Renewal of Condensate on Pitting Behavior
3.4.1. Effects of CO2 Flow Rate and Microstructure of Steel
3.4.2. Effects of Condensate Renewal
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Met-Office. Global Climate in Context as the World Approaches 1 °C above Pre-Industrial for the First Time. 2016. Available online: http://www.metoffice.gov.uk/research/news/2015/global-average-temperature-2015 (accessed on 1 November 2018).
- Stocker, T.F.; Qin, D.; Plattner, G.-K.; Tignor, M.; Allen, S.K.; Boschung, J.; Nauels, A.; Xia, Y.; Bex, V.; Midgley, P.M. IPCC, Summary for Policymakers. In Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovermmental Panel on Climate Change; Cambridge University Press: New York, NY, USA, 2013. [Google Scholar]
- International Energy Agency. CO2 Emissions from Fuel Combustion-Highlights. 2016. Available online: https://emis.vito.be/sites/emis.vito.be/files/articles/3331/2016/CO2EmissionsfromFuelCombustion_Highlights_2016.pdf (accessed on 1 November 2018).
- Aminu, M.D.; Nabavi, S.A.; Rochelle, C.A.; Manovic, V. A review of developments in carbon dioxide storage. Appl. Energy 2017, 208, 1389–1419. [Google Scholar] [CrossRef]
- European Climate Foundation. Roadmap 2015. A Practical Guide to a Prosperous, Low-Carbon Europe; European Climate Foundation: Brussels, Belgium, 2010. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, Y.; Zhang, W.; Wang, J.; Soltanian, M.R.; Olabi, A.G. Effectiveness of amino acid salt solutions in capturing CO2: A review. Renew. Sustain. Energy Rev. 2018, 98, 179–188. [Google Scholar] [CrossRef]
- Yan, J.; Zhang, Z. Carbon Capture, Utilization and Storage (CCUS). Appl. Energy 2019, 235, 1289–1299. [Google Scholar] [CrossRef]
- Czernichowski-Lauriol, I.; Berenblyum, R.; Bigi, S.; Car, M.; Gastine, M.; Persoglia, S.; Poulsen, N.; Schmidt-Hattenberger, C.; Stead, R.; Vincent, C.J.; et al. CO2GeoNet actions in Europe for advancing CCUS through global cooperation. Energy Procedia 2018, 154, 73–79. [Google Scholar] [CrossRef]
- Koytsoumpa, E.I.; Bergins, C.; Kakaras, E. The CO2 economy: Review of CO2 capture and reuse technologies. J. Supercrit. Fluids 2018, 132, 3–16. [Google Scholar] [CrossRef]
- Spitz, T.; Avagyan, V.; Ascui, F.; Bruce, A.R.W.; Chalmers, H.; Lucquiaud, M. On the variability of CO2 feed flows into CCS transportation and storage networks. Int. J. Greenh. Gas Control 2018, 74, 296–311. [Google Scholar] [CrossRef]
- Morland, B.H.; Dugstad, A.; Tjelta, M.; Svenningsen, G. Formation of strong acids in dense phase CO2. In Proceedings of the CORROSION 2018, NACE International, Phoenix, AZ, USA, 15–19 April 2018. Paper No. 11429. [Google Scholar]
- Hua, Y.; Barker, R.; Neville, A. The influence of SO2 on the tolerable water content to avoid pipeline corrosion during the transportation of supercritical CO2. Int. J. Greenh. Gas Control 2015, 37, 412–423. [Google Scholar] [CrossRef]
- Xiang, Y.; Wang, Z.; Xu, M. A mechanistic model for pipeline steel corrosion in supercritical CO2–SO2–O2–H2O environments. J. Supercrit. Fluids 2013, 82, 1–12. [Google Scholar] [CrossRef]
- Dugstad, A.; Halseid, M.; Morland, B. Corrosion and bulk phase reactions in CO2 transport pipelines with impurities: Review of recent published studies. Energy Proc. 2014, 63, 2547–2556. [Google Scholar] [CrossRef]
- Ruhl, A.S.; Kranzmann, A. Corrosion behavior of various steels in a continuous flow of carbon dioxide containing impurities. Int. J. Greenh. Gas Control 2012, 9, 85–90. [Google Scholar] [CrossRef]
- Yevtushenko, O.; Bettge, D.; Baessler, R.; Bohraus, S. Corrosion of CO2 transport and injection pipeline steels due to the condensation effects caused by SO2 and NO2 impurities. Mater. Corros. 2015, 66, 334–341. [Google Scholar] [CrossRef]
- Haupt, S.; Strehblow, H.-H. A combined surface analytical and electrochemical study of the formation of passive layers on FeCr alloys in 0.5 M H2SO4. Corros. Sci. 1995, 37, 43–54. [Google Scholar] [CrossRef]
- Olsson, C.-O.A.; Landolt, D. Passive films on stainless steels—Chemistry, structure and growth. Electrochim. Acta 2003, 48, 1093–1104. [Google Scholar] [CrossRef]
- Choi, Y.-S.; Nesic, S.; Young, D. Effect of Impurities on the Corrosion Behavior of CO2 Transmission Pipeline Steel in Supercritical CO2-Water Environments. Environ. Sci. Technol. 2010, 44, 9233–9238. [Google Scholar] [CrossRef] [PubMed]
- Dugstad, A. Fundamental Aspects of CO2 Metal loss corrosion part I: Mechanism. In Proceedings of the CORROSION 2006, NACE International, San Diego, CA, USA, 12–16 March 2006. Paper No. 06112. [Google Scholar]
- Kahyarian, A.; Brown, B.; Nesic, S. Electrochemistry of CO2 corrosion of mild steel: Effect of CO2 on iron dissolution reaction. Corros. Sci. 2017, 129, 146–151. [Google Scholar] [CrossRef]
- Dugstad, A. Mechanism of Protective Film Formation During CO2 Corrosion of Carbon Steel. In Proceedings of the CORROSION 1998, NACE International, Houston, TX, USA, 22–27 March 1998. Paper No. 31. [Google Scholar]
- Le, Q.H.; Baessler, R.; Knauer, S.; Jaeger, P.; Kratzig, A.; Bettge, D.; Kranzmann, A. Droplet Corrosion of CO2 Transport Pipeline Steels in Simulated Oxyfuel Flue Gas. Corrosion 2018, 74, 1406–1420. [Google Scholar] [CrossRef]
- Paschke, B.; Kather, A. Corrosion of Pipeline and Compressor Materials Due to Impurities in Separated CO2 from Fossil-Fuelled Power Plants. Energy Procedia 2012, 23, 207–215. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C | Si | Mn | Cr | Cu | Ni | Al | P | S | Mo | N |
|---|---|---|---|---|---|---|---|---|---|---|
| 0.126 | 0.094 | 1.303 | 0.031 | 0.029 | 0.029 | 0.036 | 0.0142 | 0.006 | 0.003 | 0.008 |
| Materials | Carbon Steel L360NB (comparable to X52) and L485MB (X70) |
|---|---|
| Purging gas and flow rate | Pure CO2 (compared to pure Ar), 50–60 mL/min |
| Testing temperature, pressure | 278 K, <1 bar |
| Aqueous phase | 500 mL synthetic condensate made of varied concentration of H2SO4 and HNO3 pH was adjusted by adding NaOH to 2.13. Prior to immersing of metal coupons, the solution was purged with Ar for 30 min and then saturated with CO2 (pH ≈ 1.9). |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quynh Hoa, L.; Baessler, R.; Bettge, D. On the Corrosion Mechanism of CO2 Transport Pipeline Steel Caused by Condensate: Synergistic Effects of NO2 and SO2. Materials 2019, 12, 364. https://doi.org/10.3390/ma12030364
Quynh Hoa L, Baessler R, Bettge D. On the Corrosion Mechanism of CO2 Transport Pipeline Steel Caused by Condensate: Synergistic Effects of NO2 and SO2. Materials. 2019; 12(3):364. https://doi.org/10.3390/ma12030364
Chicago/Turabian StyleQuynh Hoa, Le, Ralph Baessler, and Dirk Bettge. 2019. "On the Corrosion Mechanism of CO2 Transport Pipeline Steel Caused by Condensate: Synergistic Effects of NO2 and SO2" Materials 12, no. 3: 364. https://doi.org/10.3390/ma12030364
APA StyleQuynh Hoa, L., Baessler, R., & Bettge, D. (2019). On the Corrosion Mechanism of CO2 Transport Pipeline Steel Caused by Condensate: Synergistic Effects of NO2 and SO2. Materials, 12(3), 364. https://doi.org/10.3390/ma12030364