Defect Chemistry, Sodium Diffusion and Doping Behaviour in NaFeO2 Polymorphs as Cathode Materials for Na-Ion Batteries: A Computational Study




Abstract

1. Introduction
2. Computational Methods
3. Results and Discussion
3.1. NaFeO2 Crystal Structures
3.2. Intrinsic Defect Processes
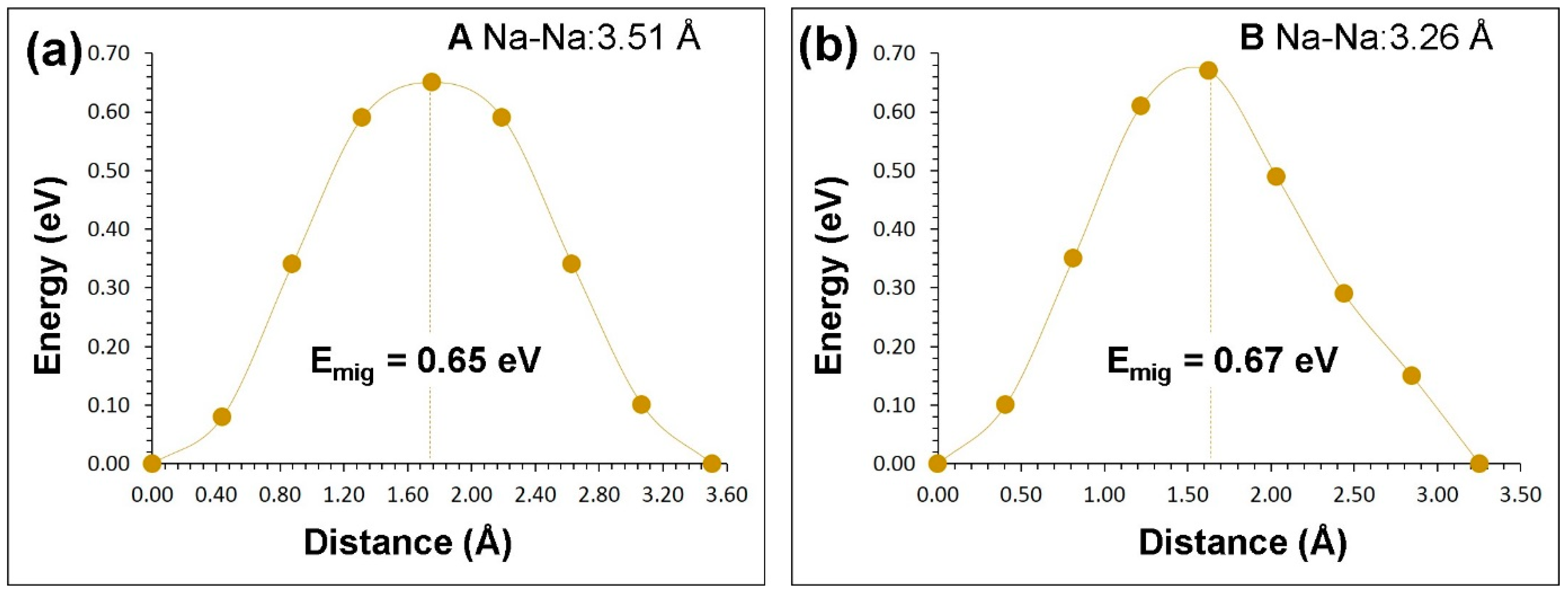
3.3. Sodium-Ion Diffusion
3.4. Dopant Substitution
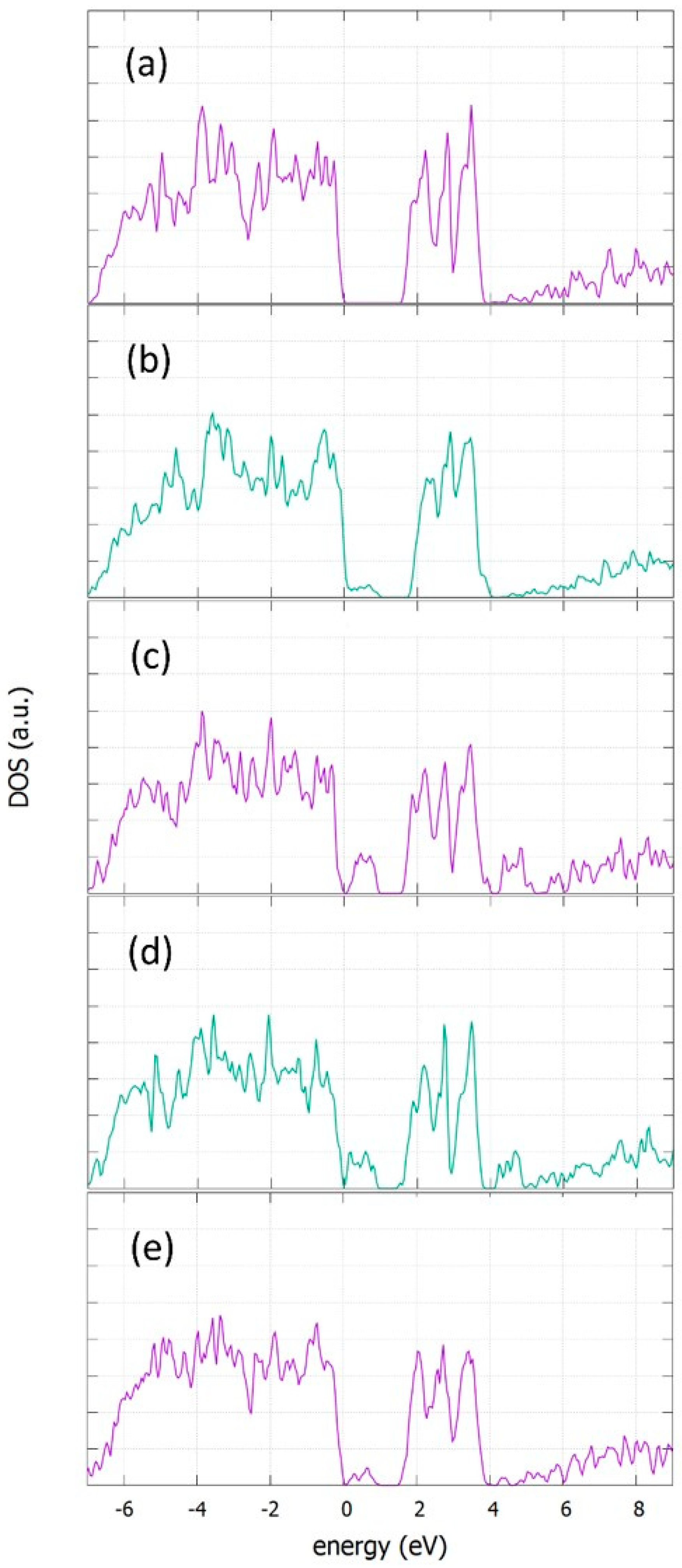
3.5. Density of States
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Armand, M.; Tarascon, J.M. Building better batteries. Nature 2008, 451, 652. [Google Scholar] [CrossRef] [PubMed]
- Padhi, A.K.; Nanjundaswamy, K.S.; Goodenough, J.B. Phospho-olivines as Positive-Electrode Materials for Rechargeable Lithium Batteries. J. Electrochem. Soc. 1997, 144, 1188–1194. [Google Scholar] [CrossRef]
- Nytén, A.; Abouimrane, A.; Armand, M.; Gustafsson, T.; Thomas, J.O. Electrochemical performance of Li2FeSiO4 as a new Li-battery cathode material. Electrochem. Commun. 2005, 7, 156–160. [Google Scholar] [CrossRef]
- Masquelier, C.; Croguennec, L. Polyanionic (Phosphates, Silicates, Sulfates) Frameworks as Electrode Materials for Rechargeable Li (or Na) Batteries. Chem. Rev. 2013, 113, 6552–6591. [Google Scholar] [CrossRef] [PubMed]
- Afyon, S.; Wörle, M.; Nesper, R. A Lithium-Rich Compound Li7Mn(BO3)3 Containing Mn2+ in Tetrahedral Coordination: A Cathode Candidate for Lithium-Ion Batteries. Angew. Chem. Int. Ed. 2013, 52, 12541–12544. [Google Scholar] [CrossRef]
- Simon, P.; Gogotsi, Y.; Dunn, B. Where Do Batteries End and Supercapacitors Begin? Science 2014, 343, 1210–1211. [Google Scholar] [CrossRef] [PubMed]
- Jeżowski, P.; Fic, K.; Crosnier, O.; Brousse, T.; Béguin, F. Lithium rhenium(vii) oxide as a novel material for graphite pre-lithiation in high performance lithium-ion capacitors. J. Mater. Chem. A 2016, 4, 12609–12615. [Google Scholar] [CrossRef]
- Jeżowski, P.; Fic, K.; Crosnier, O.; Brousse, T.; Béguin, F. Use of sacrificial lithium nickel oxide for loading graphitic anode in Li-ion capacitors. Electrochim. Acta 2016, 206, 440–445. [Google Scholar] [CrossRef]
- Jeżowski, P.; Crosnier, O.; Deunf, E.; Poizot, P.; Béguin, F.; Brousse, T. Safe and recyclable lithium-ion capacitors using sacrificial organic lithium salt. Nat. Mater. 2017, 17, 167. [Google Scholar] [CrossRef]
- Jagadale, A.; Zhou, X.; Xiong, R.; Dubal, D.P.; Xu, J.; Yang, S. Lithium ion capacitors (LICs): Development of the materials. Energy Storage Mater. 2019, 19, 314–329. [Google Scholar] [CrossRef]
- Ellis, B.L.; Nazar, L.F. Sodium and sodium-ion energy storage batteries. Curr. Opin. Solid State Mater. Sci. 2012, 16, 168–177. [Google Scholar] [CrossRef]
- Yabuuchi, N.; Kubota, K.; Dahbi, M.; Komaba, S. Research Development on Sodium-Ion Batteries. Chem. Rev. 2014, 114, 11636–11682. [Google Scholar] [CrossRef] [PubMed]
- Palomares, V.; Casas-Cabanas, M.; Castillo-Martinez, E.; Han, M.H.; Rojo, T. Update on Na-based battery materials. A growing research path. Energy Environ. Sci. 2013, 6, 2312–2337. [Google Scholar] [CrossRef]
- Oh, S.-M.; Myung, S.-T.; Hassoun, J.; Scrosati, B.; Sun, Y.-K. Reversible NaFePO4 electrode for sodium secondary batteries. Electrochem. Commun. 2012, 22, 149–152. [Google Scholar] [CrossRef]
- Fang, Y.; Liu, Q.; Xiao, L.; Ai, X.; Yang, H.; Cao, Y. High-Performance Olivine NaFePO4 Microsphere Cathode Synthesized by Aqueous Electrochemical Displacement Method for Sodium Ion Batteries. ACS Appl. Mater. Interfaces 2015, 7, 17977–17984. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Song, X.; Du, Y.; Peng, C.; Lin, M.; Xi, S.; Tian, B.; Zheng, J.; Wu, Y.; Pan, F.; et al. High-performance NaFePO4 formed by aqueous ion-exchange and its mechanism for advanced sodium ion batteries. J. Mater. Chem. A 2016, 4, 4882–4892. [Google Scholar] [CrossRef]
- Kawabe, Y.; Yabuuchi, N.; Kajiyama, M.; Fukuhara, N.; Inamasu, T.; Okuyama, R.; Nakai, I.; Komaba, S. Synthesis and electrode performance of carbon coated Na2FePO4F for rechargeable Na batteries. Electrochem. Commun. 2011, 13, 1225–1228. [Google Scholar] [CrossRef]
- Kosova, N.V.; Podugolnikov, V.R.; Devyatkina, E.T.; Slobodyuk, A.B. Structure and electrochemistry of NaFePO4 and Na2FePO4F cathode materials prepared via mechanochemical route. Mater. Res. Bull. 2014, 60, 849–857. [Google Scholar] [CrossRef]
- Jian, Z.; Zhao, L.; Pan, H.; Hu, Y.-S.; Li, H.; Chen, W.; Chen, L. Carbon coated Na3V2(PO4)3 as novel electrode material for sodium ion batteries. Electrochem. Commun. 2012, 14, 86–89. [Google Scholar] [CrossRef]
- Jiang, X.; Yang, L.; Ding, B.; Qu, B.; Ji, G.; Lee, J.Y. Extending the cycle life of Na3V2(PO4)3 cathodes in sodium-ion batteries through interdigitated carbon scaffolding. J. Mater. Chem. 2016, 4, 14669–14674. [Google Scholar] [CrossRef]
- Kovrugin, V.M.; David, R.; Chotard, J.-N.; Recham, N.; Masquelier, C. A High Voltage Cathode Material for Sodium Batteries: Na3V(PO4)2. Inorg. Chem. 2018, 57, 8760–8768. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yoon, G.; Kim, H.; Park, Y.-U.; Kang, K. Na3V(PO4)2: A New Layered-Type Cathode Material with High Water Stability and Power Capability for Na-Ion Batteries. Chem. Mater. 2018, 30, 3683–3689. [Google Scholar] [CrossRef]
- Nose, M.; Nakayama, H.; Nobuhara, K.; Yamaguchi, H.; Nakanishi, S.; Iba, H. Na4Co3(PO4)2P2O7: A novel storage material for sodium-ion batteries. J. Power Sources 2013, 234, 175–179. [Google Scholar] [CrossRef]
- Treacher, J.C.; Wood, S.M.; Islam, M.S.; Kendrick, E. Na2CoSiO4 as a cathode material for sodium-ion batteries: Structure, electrochemistry and diffusion pathways. Phys. Chem. Chem. Phys. 2016, 18, 32744–32752. [Google Scholar] [CrossRef] [PubMed]
- Vassilaras, P.; Ma, X.; Li, X.; Ceder, G. Electrochemical Properties of Monoclinic NaNiO2. J. Electrochem. Soc. 2013, 160, A207–A211. [Google Scholar] [CrossRef]
- Kikkawa, S.; Miyazaki, S.; Koizumi, M. Deintercalated NaCoO2 and LiCoO2. J. Solid State Chem. 1986, 62, 35–39. [Google Scholar] [CrossRef]
- Ma, X.; Chen, H.; Ceder, G. Electrochemical Properties of Monoclinic NaMnO2. J. Electrochem. Soc. 2011, 158, A1307–A1312. [Google Scholar] [CrossRef]
- Komaba, S.; Takei, C.; Nakayama, T.; Ogata, A.; Yabuuchi, N. Electrochemical intercalation activity of layered NaCrO2 vs. LiCrO2. Electrochem. Commun. 2010, 12, 355–358. [Google Scholar] [CrossRef]
- Didier, C.; Guignard, M.; Denage, C.; Szajwaj, O.; Ito, S.; Saadoune, I.; Darriet, J.; Delmas, C. Electrochemical Na-Deintercalation from NaVO2. Electrochem. Solid-State Lett. 2011, 14, A75–A78. [Google Scholar] [CrossRef]
- Wu, D.; Li, X.; Xu, B.; Twu, N.; Liu, L.; Ceder, G. NaTiO2: A layered anode material for sodium-ion batteries. Energy Environ. Sci. 2015, 8, 195–202. [Google Scholar] [CrossRef]
- Maazaz, A.; Delmas, C.; Hagenmuller, P. A study of the NaxTiO2 system by electrochemical deintercalation. J. Inclusion Phenom. 1983, 1, 45–51. [Google Scholar] [CrossRef]
- Takeda, Y.; Nakahara, K.; Nishijima, M.; Imanishi, N.; Yamamoto, O.; Takano, M.; Kanno, R. Sodium deintercalation from sodium iron oxide. Mater. Res. Bull. 1994, 29, 659–666. [Google Scholar] [CrossRef]
- Guo, S.; Yi, J.; Sun, Y.; Zhou, H. Recent advances in titanium-based electrode materials for stationary sodium-ion batteries. Energy Environ. Sci. 2016, 9, 2978–3006. [Google Scholar] [CrossRef]
- Pan, H.; Hu, Y.-S.; Chen, L. Room-temperature stationary sodium-ion batteries for large-scale electric energy storage. Energy Environ. Sci. 2013, 6, 2338–2360. [Google Scholar] [CrossRef]
- Fang, Y.; Chen, Z.; Xiao, L.; Ai, X.; Cao, Y.; Yang, H. Recent Progress in Iron-Based Electrode Materials for Grid-Scale Sodium-Ion Batteries. Small 2018, 14, 1703116. [Google Scholar] [CrossRef]
- Kikkawa, S.; Miyazaki, S.; Koizumi, M. Sodium deintercalation from α-NaFeO2. Mater. Res. Bull. 1985, 20, 373–377. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, L.; Dimov, N.; Okada, S.; Nishida, T. Electrochemical and Thermal Properties of α-NaFeO2 Cathode for Na-Ion Batteries. J. Electrochem. Soc. 2013, 160, A3077–A3081. [Google Scholar] [CrossRef]
- Yabuuchi, N.; Yoshida, H.; Komaba, S. Crystal Structures and Electrode Performance of Alpha-NaFeO2 for Rechargeable Sodium Batteries. Electrochemistry 2012, 80, 716–719. [Google Scholar] [CrossRef]
- Ohzuku, T.; Ueda, A. Why transition metal (di) oxides are the most attractive materials for batteries. Solid State Ionics 1994, 69, 201–211. [Google Scholar] [CrossRef]
- Okada, S.; Yamaki, J. Iron-Based Rare-Metal-Free Cathodes. In Lithium Ion Rechargeable Battries; Ozawa, K., Ed.; Wiley-VCH: Weinheim, Germany, 2009; p. 57. [Google Scholar]
- Grey, I.E.; Hill, R.J.; Hewat, A.W. A neutron powder diffraction study of the β to γ phase transformation in NaFeO2. Z. Kristallog. 1990, 193, 51–69. [Google Scholar] [CrossRef]
- Singh, S.; Tovstolytkin, A.; Lotey, G.S. Magnetic properties of superparamagnetic β-NaFeO2 nanoparticles. J. Magn. Magn. Mater. 2018, 458, 62–65. [Google Scholar] [CrossRef]
- Viret, M.; Rubi, D.; Colson, D.; Lebeugle, D.; Forget, A.; Bonville, P.; Dhalenne, G.; Saint-Martin, R.; André, G.; Ott, F. β-NaFeO2, a new room-temperature multiferroic material. Mater. Res. Bull. 2012, 47, 2294–2298. [Google Scholar] [CrossRef]
- Watanabe, H.; Fukase, M. Weak Ferromagnetism in β-NaFeO2. J. Phys. Soc. Japan 1961, 16, 1181–1184. [Google Scholar] [CrossRef]
- Kuganathan, N.; Islam, M.S. Li2MnSiO4 Lithium Battery Material: Atomic-Scale Study of Defects, Lithium Mobility, and Trivalent Dopants. Chem. Mater. 2009, 21, 5196–5202. [Google Scholar] [CrossRef]
- Armstrong, A.R.; Kuganathan, N.; Islam, M.S.; Bruce, P.G. Structure and Lithium Transport Pathways in Li2FeSiO4 Cathodes for Lithium Batteries. JACS 2011, 133, 13031–13035. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.A.J.; Kuganathan, N.; Islam, M.S. Defect chemistry and lithium-ion migration in polymorphs of the cathode material Li2MnSiO4. J. Mater. Chem. A 2013, 1, 4207–4214. [Google Scholar] [CrossRef]
- Kuganathan, N.; Iyngaran, P.; Chroneos, A. Lithium diffusion in Li5FeO4. Sci. Rep. 2018, 8, 5832. [Google Scholar] [CrossRef]
- Kordatos, A.; Kuganathan, N.; Kelaidis, N.; Iyngaran, P.; Chroneos, A. Defects and lithium migration in Li2CuO2. Sci. Rep. 2018, 8, 6754. [Google Scholar] [CrossRef]
- Kuganathan, N.; Ganeshalingam, S.; Chroneos, A. Defects, Dopants and Lithium Mobility in Li9V3(P2O7)3 (PO4)2. Sci. Rep. 2018, 8, 8140. [Google Scholar] [CrossRef]
- Kuganathan, N.; Kordatos, A.; Chroneos, A. Li2SnO3 as a Cathode Material for Lithium-ion Batteries: Defects, Lithium Ion Diffusion and Dopants. Sci. Rep. 2018, 8, 12621. [Google Scholar] [CrossRef]
- Kuganathan, N.; Chroneos, A. Defects, Dopants and Sodium Mobility in Na2MnSiO4. Sci. Rep. 2018, 8, 14669. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Chroneos, A. Defects and dopant properties of Li3V2(PO4)3. Sci. Rep. 2019, 9, 333. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Kordatos, A.; Chroneos, A. Defect Chemistry and Li-ion Diffusion in Li2RuO3. Sci. Rep. 2019, 9, 550. [Google Scholar] [CrossRef]
- Kuganathan, N.; Kordatos, A.; Fitzpatrick, M.E.; Vovk, R.V.; Chroneos, A. Defect process and lithium diffusion in Li2TiO3. Solid State Ionics 2018, 327, 93–98. [Google Scholar] [CrossRef]
- Kuganathan, N.; Kordatos, A.; Anurakavan, S.; Iyngaran, P.; Chroneos, A. Li3SbO4 lithium-ion battery material: Defects, lithium ion diffusion and tetravalent dopants. Mater. Chem. Phys. 2019, 225, 34–41. [Google Scholar] [CrossRef]
- Kuganathan, N.; Iyngaran, P.; Vovk, R.; Chroneos, A. Defects, dopants and Mg diffusion in MgTiO3. Sci. Rep. 2019, 9, 4394. [Google Scholar] [CrossRef]
- Kuganathan, N.; Kordatos, A.; Kelaidis, N.; Chroneos, A. Defects, Lithium Mobility and Tetravalent Dopants in the Li3NbO4 Cathode Material. Sci. Rep. 2019, 9, 2192. [Google Scholar] [CrossRef]
- Kuganathan, N.; Tsoukalas, L.H.; Chroneos, A. Defects, dopants and Li-ion diffusion in Li2SiO3. Solid State Ionics 2019, 335, 61–66. [Google Scholar] [CrossRef]
- Kuganathan, N.; Sgourou, E.N.; Panayiotatos, Y.; Chroneos, A. Defect Process, Dopant Behaviour and Li Ion Mobility in the Li2MnO3 Cathode Material. Energies 2019, 12, 1329. [Google Scholar] [CrossRef]
- Kuganathan, N.; Chroneos, A. Defect Chemistry and Na-Ion Diffusion in Na3Fe2(PO4)3 Cathode Material. Materials 2019, 12, 1348. [Google Scholar] [CrossRef]
- Kuganathan, N.; Chroneos, A. Na3V(PO4)2 cathode material for Na ion batteries: Defects, dopants and Na diffusion. Solid State Ionics 2019, 336, 75–79. [Google Scholar] [CrossRef]
- Gale, J.D.; Rohl, A.L. The General Utility Lattice Program (GULP). Mol. Simulat. 2003, 29, 291–341. [Google Scholar] [CrossRef]
- Gale, J.D. GULP: A computer program for the symmetry-adapted simulation of solids. J. Chem. Soc. Faraday Trans. 1997, 93, 629–637. [Google Scholar] [CrossRef]
- Mott, N.F.; Littleton, M.J. Conduction in polar crystals. I. Electrolytic conduction in solid salts. Trans. Faraday Soc. 1938, 34, 485–499. [Google Scholar] [CrossRef]
- Grimes, R.W.; Busker, G.; McCoy, M.A.; Chroneos, A.; Kilner, J.A.; Chen, S.-P. The Effect of Ion Size on Solution Mechanism and Defect Cluster Geometry. Ber. Bunsengest. Phys. Chem. 1997, 101, 1204–1210. [Google Scholar] [CrossRef]
- Seymour, I.D.; Chroneos, A.; Kilner, J.A.; Grimes, R.W. Defect processes in orthorhombic LnBaCo2O5.5 double perovskites. Phys. Chem. Chem. Phys. 2011, 13, 15305–15310. [Google Scholar] [CrossRef]
- Jay, E.E.; Rushton, M.J.D.; Chroneos, A.; Grimes, R.W.; Kilner, J.A. Genetics of superionic conductivity in lithium lanthanum titanates. Phys. Chem. Chem. Phys. 2015, 17, 178–183. [Google Scholar] [CrossRef]
- Segall, M.D.; Philip, J.D.L.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717. [Google Scholar] [CrossRef]
- Clark Stewart, J.; Segall Matthew, D.; Pickard Chris, J.; Hasnip Phil, J.; Probert Matt, I.J.; Refson, K.; Payne Mike, C. First principles methods using CASTEP. In Zeitschrift für Kristallographie Crystalline Materials; The University of York: York, UK, 2005; Volume 220, p. 567. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Kubota, K.; Asari, T.; Yoshida, H.; Yaabuuchi, N.; Shiiba, H.; Nakayama, M.; Komaba, S. Understanding the Structural Evolution and Redox Mechanism of a NaFeO2–NaCoO2 Solid Solution for Sodium-Ion Batteries. Adv. Funct. Mater. 2016, 26, 6047–6059. [Google Scholar] [CrossRef]
- Perdew, J.P. Density functional theory and the band gap problem. Int. J. Quantum Chem. 1985, 28, 497–523. [Google Scholar] [CrossRef]
- Kröger, F.A.; Vink, H.J. Relations between the Concentrations of Imperfections in Crystalline Solids. In Solid State Physics; Seitz, F., Turnbull, D., Eds.; Academic Press: Eindhoven, The Netherlands, 1956; Volume 3, pp. 307–435. [Google Scholar]
- Nytén, A.; Kamali, S.; Häggström, L.; Gustafsson, T.; Thomas, J.O. The lithium extraction/insertion mechanism in Li2FeSiO4. J. Mater. Chem. 2006, 16, 2266–2272. [Google Scholar] [CrossRef]
- Liu, H.; Choe, M.-J.; Enrique, R.A.; Orvañanos, B.; Zhou, L.; Liu, T.; Thornton, K.; Grey, C.P. Effects of Antisite Defects on Li Diffusion in LiFePO4 Revealed by Li Isotope Exchange. J. Phys. Chem. C 2017, 121, 12025–12036. [Google Scholar] [CrossRef]
- Kempaiah Devaraju, M.; Duc Truong, Q.; Hyodo, H.; Sasaki, Y.; Honma, I. Synthesis, characterization and observation of antisite defects in LiNiPO4 nanomaterials. Sci. Rep. 2015, 5, 11041. [Google Scholar] [CrossRef] [PubMed]
- Politaev, V.V.; Petrenko, A.A.; Nalbandyan, V.B.; Medvedev, B.S.; Shvetsova, E.S. Crystal structure, phase relations and electrochemical properties of monoclinic Li2MnSiO4. J. Solid State Chem. 2007, 180, 1045–1050. [Google Scholar] [CrossRef]
- Guo, S.; Sun, Y.; Liu, P.; Yi, J.; He, P.; Zhang, X.; Zhu, Y.; Senga, R.; Suenaga, K.; Chen, M.; et al. Cation-mixing stabilized layered oxide cathodes for sodium-ion batteries. Sci. Bull. 2018, 63, 376–384. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hexagonal (m)32 | |||
| Parameter | Calc | Expt | |∆|(%) |
| a = b (Å) | 3.0687 | 3.0221 | 1.54 |
| c (Å) | 16.0917 | 16.0817 | 0.06 |
| α = β (°) | 90.00 | 90.00 | 0.00 |
| γ (°) | 120.00 | 120.00 | 0.00 |
| Orthorhombic (Pn21a)41 | |||
| a (Å) | 5.7911 | 5.6823 | 1.92 |
| b (Å) | 5.3862 | 5.4258 | 0.73 |
| c (Å) | 7.1186 | 7.2351 | 1.61 |
| α = β = γ(°) | 90.00 | 90.00 | 0.00 |
| Migration Path | Na–Na Separation (Å) | Activation Energy (eV) |
|---|---|---|
| P | 3.07 | 0.64 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuganathan, N.; Kelaidis, N.; Chroneos, A. Defect Chemistry, Sodium Diffusion and Doping Behaviour in NaFeO2 Polymorphs as Cathode Materials for Na-Ion Batteries: A Computational Study. Materials 2019, 12, 3243. https://doi.org/10.3390/ma12193243
Kuganathan N, Kelaidis N, Chroneos A. Defect Chemistry, Sodium Diffusion and Doping Behaviour in NaFeO2 Polymorphs as Cathode Materials for Na-Ion Batteries: A Computational Study. Materials. 2019; 12(19):3243. https://doi.org/10.3390/ma12193243
Chicago/Turabian StyleKuganathan, Navaratnarajah, Nikolaos Kelaidis, and Alexander Chroneos. 2019. "Defect Chemistry, Sodium Diffusion and Doping Behaviour in NaFeO2 Polymorphs as Cathode Materials for Na-Ion Batteries: A Computational Study" Materials 12, no. 19: 3243. https://doi.org/10.3390/ma12193243
APA StyleKuganathan, N., Kelaidis, N., & Chroneos, A. (2019). Defect Chemistry, Sodium Diffusion and Doping Behaviour in NaFeO2 Polymorphs as Cathode Materials for Na-Ion Batteries: A Computational Study. Materials, 12(19), 3243. https://doi.org/10.3390/ma12193243