Lithium Attachment to C60 and Nitrogen- and Boron-Doped C60: A Mechanistic Study




Abstract
1. Introduction
2. Methods
3. Results
3.1. Bandstructure of Li–C60/C59N/C59B Complexes
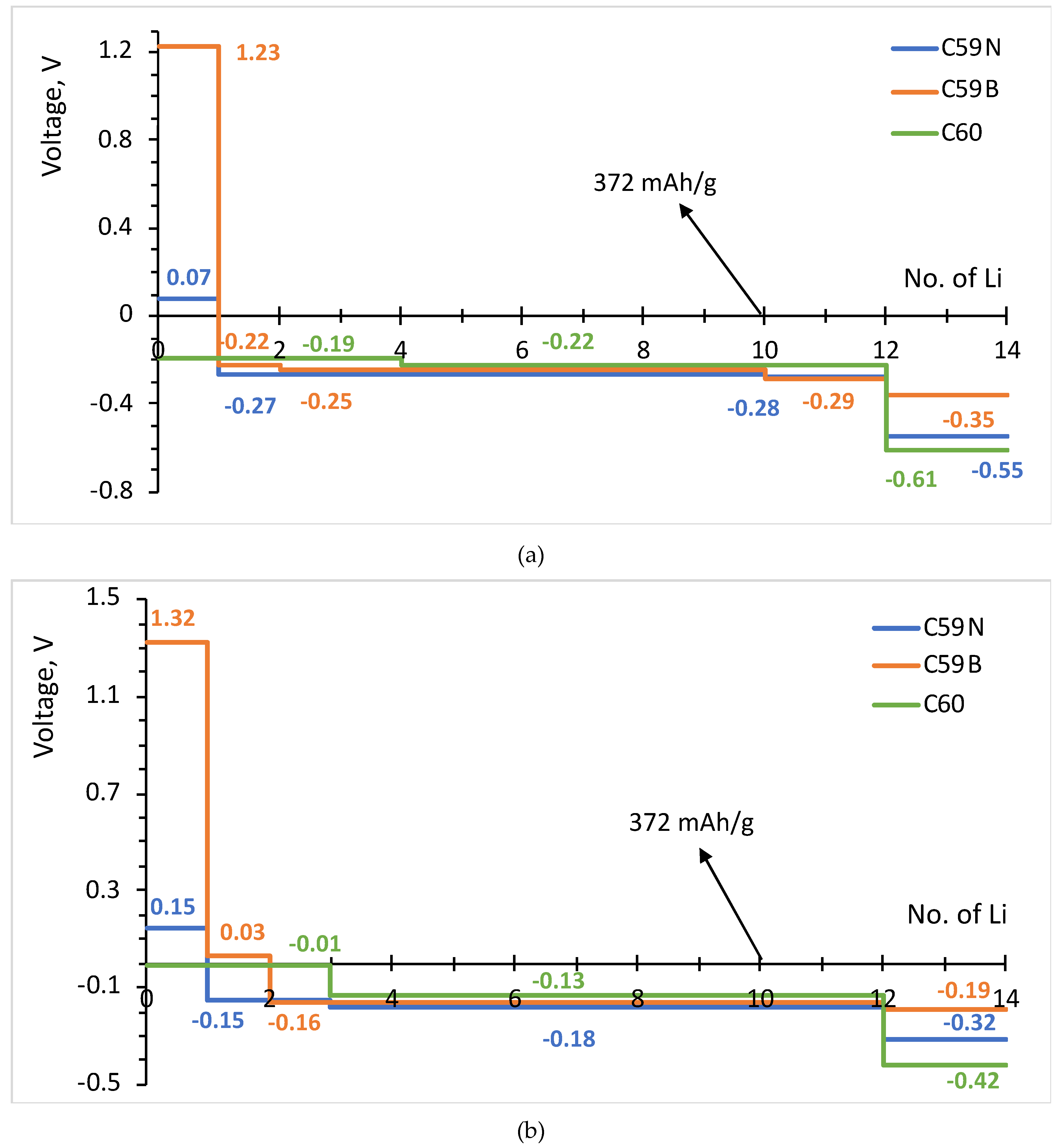
3.2. Formation Energies and Voltage–Capacity Curves
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kawabe, S.; Kawai, T.; Sugimoto, R.; Yagasaki, E.; Yoshino, K. Electrochemical properties of fullerene derivative polymers as electrode materials. Jpn. J. Appl. Phys. 1997, 36, L1055–L1058. [Google Scholar] [CrossRef]
- Chabre, Y.; Djurado, D.; Armand, M.; Romanow, W.R.; Coustel, N.; McCauley, J.P., Jr.; Fischer, J.E.; Smith, A.B., III. Electrochemical intercalation of lithium into solid C60. J. Am. Chem. Soc. 1992, 114, 764–766. [Google Scholar] [CrossRef]
- Lemont, S.; Ghanbaja, J.; Billaud, D. Electrochemical intercalation of sodium ions into fullerene. Mater. Res. Bull. 1994, 29, 465–472. [Google Scholar] [CrossRef]
- Yildirim, T.; Zhou, O.; Fischer, J.E.; Bykovetz, N.; Strongin, R.A.; Cichy, M.A.; Smith III, A.B.; Lin, C.L.; Jelinek, R. Intercalation of sodium heteroclusters into the C60 lattice. Nature 1992, 360, 568–571. [Google Scholar] [CrossRef]
- Scaravonati, S.; Magnani, G.; Gaboardi, M.; Allodi, G.; Ricco, M.; Pontiroli, D. Electrochemical intercalation of fullerene and hydrofullerene with sodium. Carbon 2018, 130, 11–18. [Google Scholar] [CrossRef]
- Jehoulet, C.; Bard, A.J.; Wudl, F. Electrochemical reduction and oxidation of C60 films. J. Am. Chem. Soc. 1991, 113, 5456–5457. [Google Scholar] [CrossRef]
- Jehoulet, C.; Obeng, Y.S.; Kim, Y.T.; Zhou, F.; Bard, A.J. Electrochemistry and Langmuir trough studies of fullerene C60 and C70 films. J. Am. Chem. Soc. 1992, 114, 4237–4247. [Google Scholar] [CrossRef]
- Xie, Q.; Perez-Cordero, E.; Echegoyen, L. Electrochemical detection of C606- and C706-: Enhanced stability of fullerides in solution. J. Am. Chem. Soc. 1992, 114, 3978–3980. [Google Scholar] [CrossRef]
- Tycko, R.; Dabbagh, G.; Rosseinsky, M.J.; Murphy, D.W.; Fleming, R.M.; Ramirez, A.P.; Tully, J.C. 13C NMR spectroscopy of KxC60: Phase separation, molecular dynamics, and metallic properties. Science 1991, 253, 884–886. [Google Scholar] [CrossRef]
- Chen, Y.; Manzhos, S. Lithium and sodium storage on tetracyanoethylene (TCNE) and TCNE-(doped)-graphene complexes: a computational study. Mater. Chem. Phys. 2015, 156, 180–187. [Google Scholar] [CrossRef]
- Madian, M.; Eychmüller, A.; Giebeler, L. Current advances in TiO2-based nanostructure electrodes for high performance lithium ion batteries. Batteries 2018, 4, 7. [Google Scholar] [CrossRef]
- Saravanan, K.; Ananthanarayanan, K.; Balaya, P. Mesoporous TiO2 with high packing density for superior lithium storage. Energy Environ. Sci. 2010, 3, 939–948. [Google Scholar] [CrossRef]
- Wang, F.F.; Wang, C.; Liu, R.Q.; Tian, D.; Li, N. Experimental study on the preparation of Ag nanoparticle doped fullerenol for lithium ion battery application. J. Phys. Chem. C 2012, 116, 10461–10467. [Google Scholar] [CrossRef]
- Sood, P.; Kim, K.C.; Jang, S.S. Electrochemical and electronic properties of nitrogen doped fullerene and its derivatives for lithium-ion battery applications. J. Energy Chem. 2018, 27, 528–534. [Google Scholar] [CrossRef]
- Lueder, J.; Legrain, F.; Chen, Y.; Manzhos, S. Doping of active electrode materials for electrochemical batteries: an electronic structure perspective. MRS Commun. 2017, 7, 523–540. [Google Scholar] [CrossRef]
- Legrain, F.; Manzhos, S. A first-principles comparative study of lithium, sodium, and magnesium storage in pure and gallium-doped germanium: competition between interstitial and substitutional sites. J. Chem. Phys. 2017, 146, 034706. [Google Scholar] [CrossRef] [PubMed]
- Legrain, F.; Manzhos, S. Aluminum doping improves the energetics of lithium, sodium, and magnesium storage in silicon: a first-principles study. J. Power Sources 2015, 274, 65–70. [Google Scholar] [CrossRef]
- Legrain, F.; Malyi, O.I.; Manzhos, S. Comparative computational study of the energetics of Li, Na, and Mg storage in amorphous and crystalline silicon. Comput. Mater. Sci. 2014, 94, 214–217. [Google Scholar] [CrossRef]
- Legrain, F.; Malyi, O.I.; Manzhos, S. Comparative computational study of the diffusion of Li, Na, and Mg in silicon including the effect of vibrations. Solid State Ionics 2013, 253, 157–163. [Google Scholar] [CrossRef]
- Legrain, F.; Malyi, O.I.; Tan, T.L.; Manzhos, S. Computational study of Mg insertion into amorphous silicon: advantageous energetics over crystalline silicon for Mg storage. MRS Proc. 2013, 1540, mrss13-1540-e03-06. [Google Scholar] [CrossRef]
- Tan, T.L.; Malyi, O.I.; Legrain, F.; Manzhos, S. Role of inter-dopant interactions on the diffusion of Li and Na atoms in bulk Si anodes. MRS Proc. 2013, 1541, mrss13-1541-f06-13. [Google Scholar] [CrossRef]
- Malyi, O.I.; Tan, T.L.; Manzhos, S. A computational study of the insertion of Li, Na, and Mg atoms into Si(111) nanosheets. Nano Energy 2013, 2, 1149–1157. [Google Scholar] [CrossRef]
- Malyi, O.I.; Tan, T.L.; Manzhos, S. A comparative computational study of structures, diffusion, and dopant interactions between Li and Na insertion into Si. Appl. Phys. Express 2013, 6, 027301. [Google Scholar] [CrossRef]
- Malyi, O.I.; Tan, T.L.; Manzhos, S. In search of high performance anode materials for Mg batteries: computational studies of Mg in Ge, Si, and Sn. J. Power Sources 2013, 233, 341–345. [Google Scholar] [CrossRef]
- Kulish, V.; Malyi, O.I.; Ng, M.F.; Chen, Z.; Manzhos, S.; Wu, P. Controlling Na diffusion by rational design of Si-based layered architectures. Phys. Chem. Chem. Phys. 2014, 16, 4260–4267. [Google Scholar] [CrossRef] [PubMed]
- Kulish, V.; Koch, D.; Manzhos, S. Insertion of mono- vs. bi- vs. trivalent atoms in prospective active electrode materials for electrochemical batteries: an ab initio perspective. Energies 2017, 10, 2061. [Google Scholar] [CrossRef]
- Legrain, F.; Malyi, O.I.; Manzhos, S. Insertion energetics of lithium, sodium, and magnesium in crystalline and amorphous titanium dioxide: a comparative first-principles study. J. Power Sources 2015, 278, 197–202. [Google Scholar] [CrossRef]
- Koch, D.; Kulish, V.; Manzhos, S. A first-principles study of the potassium insertion in crystalline vanadium oxide phases as possible potassium-ion battery cathode materials. MRS Commun. 2017, 7, 819–825. [Google Scholar] [CrossRef]
- Kulish, V.; Manzhos, S. Comparison of Li, Na, Mg and Al-ion insertion in vanadium pentoxides and vanadium dioxides. RSC Adv. 2017, 7, 18643. [Google Scholar] [CrossRef]
- Koch, D.; Manzhos, S. A comparative first-principles study of lithium, sodium and magnesium insertion energetics in brookite titanium dioxide. MRS Adv. 2019, 4, 837–842. [Google Scholar] [CrossRef]
- Koch, D.; Manzhos, S. First-principles study of the calcium insertion in layered and non-layered phases of vanadia. MRS Adv. 2018, 3, 3507–3512. [Google Scholar] [CrossRef]
- Manzhos, S. Organic electrode materials for lithium and post-lithium batteries: an ab initio perspective on design. Curr. Opin. Green Sustain. Chem. 2019, 17, 8–14. [Google Scholar] [CrossRef]
- Lueder, J.; Cheow, M.H.; Manzhos, S. Understanding doping strategies in the design of organic electrode materials for Li and Na ion batteries: an electronic structure perspective. Phys. Chem. Chem. Phys. 2017, 19, 13195–13209. [Google Scholar] [CrossRef] [PubMed]
- Sk, M.A.; Manzhos, S. Sodium interaction with disodium terephthalate molecule: an ab initio study. MRS Adv. 2016, 1, 3579–3584. [Google Scholar] [CrossRef]
- Sk, M.A.; Manzhos, S. Exploring the sodium storage mechanism in disodium terephthalate as anode for organic battery using density-functional theory calculations. J. Power Sources 2016, 324, 572–581. [Google Scholar] [CrossRef]
- Padhy, H.; Chen, Y.; Lüder, J.; Gajella, S.R.; Manzhos, S.; Balaya, P. Charge and discharge processes and sodium storage in disodium pyridine-2,5-dicarboxylate anode - insights from experiments and theory. Adv. Energy Mater. 2018, 8, 1701572. [Google Scholar] [CrossRef]
- Chen, Y.; Lueder, J.; Manzhos, S. Disodium pyridine dicarboxylate vs disodium terephthalate as anode materials for organic Na ion batteries: effect of molecular structure on voltage from the molecular modeling perspective. MRS Adv. 2017, 2, 3231–3235. [Google Scholar] [CrossRef][Green Version]
- Chen, Y.; Manzhos, S. Comparative computational study of lithium and sodium insertion in van der Waals and covalent tetracyanoethylene (TCNE) -based crystals as promising materials for organic lithium and sodium ion batteries. Phys. Chem. Chem. Phys. 2016, 18, 8874–8880. [Google Scholar] [CrossRef] [PubMed]
- Moutanabbir, O.; Isheim, D.; Blumtritt, H.; Senz, S.; Pippel, E.; Seidman, D.N. Colossal injection of catalyst atoms into silicon nanowires. Nature 2013, 496, 78–82. [Google Scholar] [CrossRef]
- Urban, A.; Seo, D.H.; Ceder, G. Computational understanding of Li-ion batteries. npj Comput. Mater. 2016, 2, 16002. [Google Scholar] [CrossRef]
- Chen, Y.; Manzhos, S. Voltage and capacity control of polyaniline based organic cathodes: An ab initio study. J. Power Sources 2016, 336, 126–131. [Google Scholar] [CrossRef]
- Chen, Y.; Manzhos, S. A computational study of lithium interaction with tetracyanoethylene (TCNE) and tetracyaniquinodimethane (TCNQ) molecules. Phys. Chem. Chem. Phys. 2016, 18, 1470–1477. [Google Scholar] [CrossRef] [PubMed]
- Sk, M.A.; Chen, Y.; Manzhos, S. Orbital order switching in molecular calculations using GGA functionals: qualitative errors in materials modeling for electrochemical power sources and how to fix them. Chem. Phys. Lett. 2016, 659, 270–276. [Google Scholar] [CrossRef]
- Chen, Y.; Lueder, J.; Ng, M.F.; Sullivan, M.; Manzhos, S. Polyaniline and CN-functionalized polyaniline as organic cathodes for lithium and sodium ion batteries: a combined molecular dynamics and Density Functional Tight Binding Study in solid state. Phys. Chem. Chem. Phys. 2018, 20, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision D.3; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Sanville, E.; Kenny, S.D.; Smith, R.; Henkelman, G. An improved grid-based algorithm for Bader charge allocation. J. Comp. Chem. 2007, 28, 899–908. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Tenderholt, A.L.; Langner, K.M. CCLIB: a library for package-independent computational chemistry algorithms. J. Comp. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to Solid State Physics, 8th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar]
- Kaxiras, E. Atomic and Electronic Structure of Solids; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Otero, G.; Biddau, G.; Sanchez-Sanchez, C.; Caillard, R.; Lopez, M.F.; Rogero, C.; Palomares, F.J.; Cabello, N.; Basanta, M.A.; Ortega, J.; et al. Fullerenes from aromatic precursors by surface-catalysed cyclodehydrogenation. Nature 2018, 454, 865–869. [Google Scholar] [CrossRef]
- Arabnejad, S.; Pal, A.; Yamashita, K.; Manzhos, S. Effect of nuclear motion on charge transport in fullerenes: a combined Density Functional Tight Binding – Density Functional Theory investigation. Front. Energy Res. 2019, 7, 3. [Google Scholar] [CrossRef]
- Pal, A.; Lai, K.W.; Chia, Y.J.; Jeon, I.; Matsuo, Y.; Manzhos, S. Comparative Density Functional Theory – Density Functional Tight Binding Study of fullerene derivatives: effects due to fullerene size, addends, and crystallinity on band structure, charge transport and optical properties. Phys. Chem. Chem. Phys. 2017, 19, 28330–28343. [Google Scholar] [CrossRef] [PubMed]
- Konarev, D.V.; Lyubovskaya, R.N.; Khasanov, S.S.; Otsuka, A.; Saito, G. Formation and properties of (C60−)2 dimers of fullerenes bonded by one and two σ-bonds in ionic complexes. Mol. Cryst. Liq. Cryst. 2007, 468, 227–579. [Google Scholar] [CrossRef]
- Hummelen, J.C.; Knight, B.; Pavlovich, J.; González, R.; Wudl, F. Isolation of the heterofullerene C59N as its dimer (C59N)2. Science 1995, 269, 1554–1556. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bader Charge, |e| | B3LYP | PBE | ||||
|---|---|---|---|---|---|---|
| C60 | C59N | C59B | C60 | C59N | C59B | |
| 1 Li | 0.91 | 0.90 | 0.90 | 0.90 | 0.90 | 0.90 |
| 2 Li | 0.90 | 0.90 | 0.90 | 0.90 | 0.90 | 0.89 |
| 3 Li | 0.90 | 0.89 | 0.89 | 0.90 | 0.90 | 0.89 |
| ... | ... | ... | ... | ... | ... | ... |
| 12 Li | 0.86 | 0.85/0.34 | 0.86 | 0.86 | 0.84/0.45 | 0.86 |
| 14 Li | 0.84/0.59 | 0.85/0.74 | 0.84/0.62 | 0.85/0.74 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Cho, C.-R.; Manzhos, S. Lithium Attachment to C60 and Nitrogen- and Boron-Doped C60: A Mechanistic Study. Materials 2019, 12, 2136. https://doi.org/10.3390/ma12132136
Chen Y, Cho C-R, Manzhos S. Lithium Attachment to C60 and Nitrogen- and Boron-Doped C60: A Mechanistic Study. Materials. 2019; 12(13):2136. https://doi.org/10.3390/ma12132136
Chicago/Turabian StyleChen, Yingqian, Chae-Ryong Cho, and Sergei Manzhos. 2019. "Lithium Attachment to C60 and Nitrogen- and Boron-Doped C60: A Mechanistic Study" Materials 12, no. 13: 2136. https://doi.org/10.3390/ma12132136
APA StyleChen, Y., Cho, C.-R., & Manzhos, S. (2019). Lithium Attachment to C60 and Nitrogen- and Boron-Doped C60: A Mechanistic Study. Materials, 12(13), 2136. https://doi.org/10.3390/ma12132136