Antileishmanial Activity of Amphotericin B-loaded-PLGA Nanoparticles: An Overview

 ,
, 

Abstract
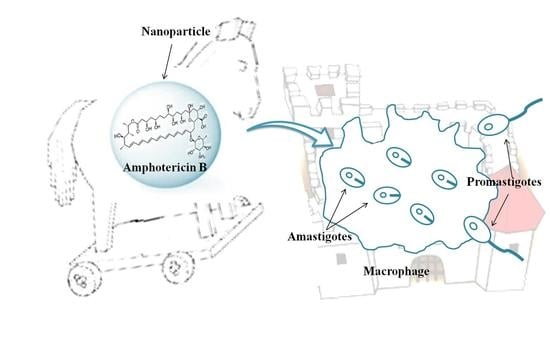
1. Introduction
1.1. AmB Pharmaceutical Formulations
1.2. Nanotechnology in the Pharmaceutical Field
2. Polymeric Nanoparticles
2.1. PLGA Nanoparticles Containing Amphotericin B
2.2. PLGA-PEG Nanoparticles Containing Amphotericin B
2.3. Selective Targeting of PLGA-Nanoparticles Containing Amphotericin B
3. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Bern, C.; Maguire, J.H.; Alvar, J. Complexities of assessing the disease burden attributable to leishmaniasis. PLoS Negl. Trop. Dis. 2008, 2. [Google Scholar] [CrossRef] [PubMed]
- Alvar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M.; WHO Leishmaniasis Control Team. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Leishmaniasis. Available online: http://apps.who.int/tdr/svc/diseases/leishmaniasis/ (accessed on 20 May 2018).
- Singh, N.; Kumar, M.; Singh, R.K. Leishmaniasis: Current status of available drugs and new potential drug targets. Asian Pac. J. Trop. Med. 2012, 5, 485–497. [Google Scholar] [CrossRef]
- Lipoldová, M.; Demant, P. Genetic susceptibility to infectious disease: Lessons from mouse models of leishmaniasis. Nat. Rev. Genet. 2006, 7, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Croft, S.L.; Sundar, S.; Fairlamb, A.H. Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 2006, 19, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Frézard, F.; Demicheli, C.; Ribeiro, R.R. Pentavalent antimonials: New perspectives for old drugs. Molecules 2009, 14, 2317–2336. [Google Scholar] [CrossRef] [PubMed]
- Guerin, P.J.; Olliaro, P.; Sundar, S.; Boelaert, M.; Croft, S.L.; Desjeux, P.; Wasunna, M.K.; Bryceson, A.D. Visceral leishmaniasis: Current status of control, diagnosis, and treatment, and a proposed research and development agenda. Lancet Infect. Dis. 2002, 2. [Google Scholar] [CrossRef]
- Maltezou, H.C. Drug resistance in visceral leishmaniasis. J. Biomed. Biotechnol. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; More, D.K.; Singh, M.K.; Singh, V.P.; Sharma, S.; Makharia, A.; Kumar, P.C.; Murray, H.W. Failure of pentavalent antimony in visceral leishmaniasis in India: Report from the center of the Indian epidemic. Clin. Infect. Dis. 2000, 31, 1104–1107. [Google Scholar] [CrossRef] [PubMed]
- Miró, G.; Cardoso, L.; Pennisi, M.G.; Oliva, G.; Baneth, G. Canine leishmaniosis-new concepts and insights on an expanding zoonosis: Part two. Trends Parasitol. 2008, 24, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Duffin, J.; Renè, P. Anti-moine; anti-biotique: The public fortunes of the secret properties of antimony potassium tartrate (tartar emetic). J. Hist. Med. Allied Sci. 1991, 46, 440–456. [Google Scholar] [CrossRef] [PubMed]
- Bern, C.; Adler-Moore, J.; Berenguer, J.; Boelaert, M.; den Boer, M.; Davidson, R.N.; Figueras, C.; Gradoni, L.; Kafetzis, D.A.; Ritmeijer, K.; et al. Liposomal amphotericin B for the treatment of visceral leishmaniasis. Clin. Infect. Dis. 2006, 43, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Laniado-Laborín, R.; Cabrales-Vargas, M.N. Amphotericin B: Side effects and toxicity. Rev. Iberoam. Micol. 2009, 26, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Kedzierski, L. Recent advances in antileishmanial drug development. Curr. Opin. Investig. Drugs. 2005, 6, 163–169. [Google Scholar] [PubMed]
- Sundar, S.; Jha, T.K.; Thakur, C.P.; Bhattacharya, S.K.; Rai, M. Oral miltefosine for the treatment of Indian visceral leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 2006, 1, S26–S33. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Murray, H.W. Availability of miltefosine for the treatment of kala-azar in India. Bull. World Health Organ. 2005, 83, 394–395. [Google Scholar] [PubMed]
- Oliva, G.; Roura, X.; Crotti, A.; Maroli, M.; Castagnaro, M.; Gradoni, L.; Lubas, G.; Paltrinieri, S.; Zatelli, A.; Zini, E. Guidelines for treatment of leishmaniasis in dogs. J. Am. Vet. Med. Assoc. 2010, 236, 1192–1198. [Google Scholar] [CrossRef] [PubMed]
- Berczi, I.; Bertók, L.; Chow, D.A.; Chow, A. Natural immunity and neuroimmune host defense. N. Y. Acad. Sci. 2000, 917, 248–257. [Google Scholar] [CrossRef]
- Gómez-Ochoa, P.; Castillo, J.A.; Gascón, M.; Zarate, J.J.; Alvarez, F.; Couto, C.G. Use of domperidone in the treatment of canine visceral leishmaniasis: A clinical trial. Vet. J. 2009, 179, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Baneth, G.; Shaw, S.E. Chemotherapy of canine leishmaniosis. Vet. Parasitol. 2002, 106, 315–324. [Google Scholar] [CrossRef]
- Paila, Y.D.; Saha, B.; Chattopadhyay, A. Amphotericin B inhibits entry of Leishmania donovani into primary macrophages. Biochem. Biophys. Res. Commun. 2010, 399, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Torrado, J.J.; Espada, R.; Ballesteros, M.P.; Torrado-Santiago, S. Amphotericin B formulations and drug targeting. J. Pharm. Sci. 2008, 97, 2405–2425. [Google Scholar] [CrossRef] [PubMed]
- Lemke, A.; Kiderlen, A.F.; Kayser, O. Amphotericin B. Appl. Microbiol. Biotechnol. 2005, 68, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Caillot, D.; Chavanet, P.; Casasnovas, O.; Solary, E.; Zanetta, G.; Buisson, M.; Wagner, O.; Cuisenier, B.; Bonnin, A.; Camerlynck, P.; et al. Clinical evaluation of a new lipid-based delivery system for intravenous administration of amphotericin B. Eur. J. Clin. Microbiol. Infect. Dis. 1992, 11, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Veerareddy, P.R.; Vobalaboina, V. Lipid-based formulations of amphotericin B. Drugs Today 2004, 40, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Askarizadeh, A.; Jaafari, M.R.; Khamesipour, A.; Badiee, A. Liposomal adjuvant development for leishmaniasis vaccines. Ther. Adv. Vaccines 2017, 5, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Simões, S.; Filipe, A.; Faneca, H.; Mano, M.; Penacho, N.; Düzgünes, N.; de Lima, M.P. Cationic liposomes for gene delivery. Expert Opin. Drug Deliv. 2005, 2, 237–254. [Google Scholar] [CrossRef] [PubMed]
- Vyas, K.S.; Rajendran, S.; Morrison, S.D.; Shakir, A.; Mardini, S.; Lemaine, V.; Nahabedian, M.Y.; Baker, S.B.; Rinker, B.D.; Vasconez, H.C. Systematic review of liposomal bupivacaine (exparel) for postoperative analgesia. Plast. Reconstr. Surg. 2016, 138, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Cosco, D.; Tsapis, N.; Nascimento, T.L.; Fresta, M.; Chapron, D.; Taverna, M.; Arpicco, S.; Fattal, E. Polysaccharide-coated liposomes by post-insertion of a hyaluronan-lipid conjugate. Colloids Surf. B Biointerfaces 2017, 158, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Paolino, D.; Cosco, D.; Gaspari, M.; Celano, M.; Wolfram, J.; Voce, P.; Puxeddu, E.; Filetti, S.; Celia, C.; Ferrari, M.; et al. Targeting the thyroid gland with thyroid-stimulating hormone (TSH)-nanoliposomes. Biomaterials 2014, 35, 7101–7109. [Google Scholar] [CrossRef] [PubMed]
- Celano, M.; Schenone, S.; Cosco, D.; Navarra, M.; Puxeddu, E.; Racanicchi, L.; Brullo, C.; Varano, E.; Alcaro, S.; Ferretti, E.; et al. Cytotoxic effects of a novel pyrazolopyrimidine derivative entrapped in liposomes in anaplastic thyroid cancer cells in vitro and in xenograft tumors in vivo. Endocr. Relat. Cancer 2008, 15, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Cosco, D.; Paolino, D.; Maiuolo, J.; Russo, D.; Fresta, M. Liposomes as multicompartmental carriers for multidrug delivery in anticancer chemotherapy. Drug Deliv. Transl. Res. 2011, 1, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Cristiano, M.C.; Cosco, D.; Celia, C.; Tudose, A.; Mare, R.; Paolino, D.; Fresta, M. Anticancer activity of all-trans retinoic acid-loaded liposomes on human thyroid carcinoma cells. Colloids Surf. B Biointerfaces 2017, 150, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Cosco, D.; Paolino, D.; Maiuolo, J.; Marzio, L.D.; Carafa, M.; Ventura, C.A.; Fresta, M. Ultradeformable liposomes as multidrug carrier of resveratrol and 5-fluorouracil for their topical delivery. Int. J. Pharm. 2015, 489, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Celia, C.; Cosco, D.; Paolino, D.; Fresta, M. Gemcitabine-loaded innovative nanocarriers vs. GEMZAR: Biodistribution, pharmacokinetic features and in vivo antitumor activity. Expert Opin. Drug Deliv. 2011, 8, 1609–1629. [Google Scholar] [CrossRef] [PubMed]
- Paolino, D.; Cosco, D.; Molinaro, R.; Celia, C.; Fresta, M. Supramolecular devices to improve the treatment of brain diseases. Drug Discov. Today 2011, 16, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Adler-Moore, J.; Proffitt, R.T. AmBisome: Liposomal formulation, structure, mechanism of action and pre-clinical experience. J. Antimicrob. Chemother. 2002, 49, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Steimbach, L.M.; Tonin, F.S.; Virtuoso, S.; Borba, H.H.; Sanches, A.C.; Wiens, A.; Fernandez-Llimós, F.; Pontarolo, R. Efficacy and safety of amphotericin B lipid-based formulations-A systematic review and meta-analysis. Mycoses 2017, 60, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Hullmann, A. Who is winning the global nanorace? Nat. Nanotechnol. 2006, 1, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, G.; Tiwari, R.; Sriwastawa, B.; Bhati, L.; Pandey, S.; Pandey, P.; Bannerjee, S.K. Drug delivery systems: An updated review. Int. J. Pharm. Investig. 2012, 2, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Van de Ven, H.; Paulussen, C.; Feijens, P.B.; Matheeussen, A.; Rombaut, P.; Kayaert, P.; Van den Mooter, G.; Weyenberg, W.; Cos, P.; Maes, L.; et al. PLGA nanoparticles and nanosuspensions with amphotericin B: Potent in vitro and in vivo alternatives to Fungizone and AmBisome. J. Control. Release 2012, 161, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Cosco, D.; Federico, C.; Maiuolo, J.; Bulotta, S.; Molinaro, R.; Paolino, D.; Tassone, P.; Fresta, M. Physicochemical features and transfection properties of chitosan/poloxamer 188/poly(D, L-lactide-co-glycolide) nanoplexes. Int. J. Nanomed. 2014, 9, 2359–2372. [Google Scholar] [CrossRef] [PubMed]
- Paolino, D.; Cosco, D.; Celano, M.; Moretti, S.; Puxeddu, E.; Russo, D.; Fresta, M. Gemcitabine-loaded biocompatible nanocapsules for the effective treatment of human cancer. Nanomedicine 2013, 8, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Cosco, D.; Di Marzio, L.; Marianecci, C.; Trapasso, E.; Paolino, D.; Celia, C.; Carafa, M.; Fresta, M. Colloidal supramolecular aggregates for therapeutic application in neuromedicine. Curr. Med. Chem. 2014, 21, 4132–4153. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.M.; Videira, M.; Gaspar, R.; Préat, V.; Florindo, H.F. Immune system targeting by biodegradable nanoparticles for cancer vaccines. J. Control. Release 2013, 168, 179–199. [Google Scholar] [CrossRef] [PubMed]
- Sarti, F.; Perera, G.; Hintzen, F.; Kotti, K.; Karageorgiou, V.; Kammona, O.; Kiparissides, C.; Bernkop-Schnürch, A. In vivo evidence of oral vaccination with PLGA nanoparticles containing the immunostimulant monophosphoryl lipid A. Biomaterials 2011, 32, 4052–4057. [Google Scholar] [CrossRef] [PubMed]
- Rietscher, R.; Schröder, M.; Janke, J.; Czaplewska, J.; Gottschaldt, M.; Scherließ, R.; Hanefeld, A.; Schubert, U.S.; Schneider, M.; Knolle, P.A.; et al. Antigen delivery via hydrophilic PEG-b-PAGE-b-PLGA nanoparticles boosts vaccination induced T cell immunity. Eur. J. Pharm. Biopharm. 2016, 102, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Abed, N.; Couvreur, P. Nanocarriers for antibiotics: A promising solution to treat intracellular bacterial infections. Int. J. Antimicrob. Agents 2014, 43, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Wu, C.; Chen, J.; Xiao, Y. Nanotechnology in the targeted drug delivery for bone diseases and bone regeneration. Int. J. Nanomed. 2013, 8, 2305–2317. [Google Scholar] [CrossRef] [PubMed]
- Grottkau, B.E.; Cai, X.; Wang, J.; Yang, X.; Lin, Y. Polymeric nanoparticles for a drug delivery system. Curr. Drug Metab. 2013, 14, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Diou, O.; Tsapis, N.; Giraudeau, C.; Valette, J.; Gueutin, C.; Bourasset, F.; Zanna, S.; Vauthier, C.; Fattal, E. Long-circulating perfluorooctyl bromide nanocapsules for tumor imaging by 19FMRI. Biomaterials 2012, 33, 5593–5602. [Google Scholar] [CrossRef] [PubMed]
- Cosco, D.; Paolino, D.; De Angelis, F.; Cilurzo, F.; Celia, C.; Di Marzio, L.; Russo, D.; Tsapis, N.; Fattal, E.; Fresta, M. Aqueous-core PEG-coated PLA nanocapsules for an efficient entrapment of water soluble anticancer drugs and a smart therapeutic response. Eur. J. Pharm. Biopharm. 2015, 89, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Svenson, S. Clinical translation of nanomedicines. Curr. Opin. Solid State Mat. Sci. 2012, 16, 287–294. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Shive, M.S.; Anderson, J.M. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 1997, 28, 5–24. [Google Scholar] [PubMed]
- Cosco, D.; Cilurzo, F.; Maiuolo, J.; Federico, C.; Di Martino, M.T.; Cristiano, M.C.; Tassone, P.; Fresta, M.; Paolino, D. Delivery of miR-34a by chitosan/PLGA nanoplexes for the anticancer treatment of multiple myeloma. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Costa Lima, S.; Rodrigues, V.; Garrido, J.; Borges, F.; Kong Thoo Lin, P.; Cordeiro da Silva, A. In vitro evaluation of bisnaphthalimidopropyl derivatives loaded into pegylated nanoparticles against Leishmania infantum protozoa. Int. J. Antimicrob. Agents 2012, 39, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Amaral, A.C.; Bocca, A.L.; Ribeiro, A.M.; Nunes, J.; Peixoto, D.L.; Simioni, A.R.; Primo, F.L.; Lacava, Z.G.; Bentes, R.; Titze-de-Almeida, R.; et al. Amphotericin B in poly(lactic-co-glycolic acid) (PLGA) and dimercaptosuccinic acid (DMSA) nanoparticles against paracoccidioidomycosis. J. Antimicrob. Chemother. 2009, 63, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Shafiq, N.; Malhotra, S. Drug-loaded PLGA nanoparticles for oral administration: Fundamental issues and challenges ahead. Crit. Rev. Ther. Drug Carr. Syst. 2012, 29, 149–182. [Google Scholar] [CrossRef]
- McClean, S.; Prosser, E.; Meehan, E.; O’Malley, D.; Clarke, N.; Ramtoola, Z.; Brayden, D. Binding and uptake of biodegradable poly-DL-lactide micro- and nanoparticles in intestinal epithelia. Eur. J. Pharm. Sci. 1998, 6, 153–163. [Google Scholar] [CrossRef]
- Mittal, G.; Sahana, D.K.; Bhardwaj, V.; Ravi Kumar, M.N. Estradiol loaded PLGA nanoparticles for oral administration: Effect of polymer molecular weight and copolymer composition on release behavior in vitro and in vivo. J. Control. Release 2007, 119, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Italia, J.L.; Yahya, M.M.; Singh, D.; Ravi Kumar, M.N. Biodegradable nanoparticles improve oral bioavailability of amphotericin B and show reduced nephrotoxicity compared to intravenous Fungizone. Pharm. Res. 2009, 26, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Sahana, D.K.; Mittal, G.; Bhardwaj, V.; Kumar, M.N. PLGA nanoparticles for oral delivery of hydrophobic drugs: influence of organic solvent on nanoparticle formation and release behavior in vitro and in vivo using estradiol as a model drug. J. Pharm. Sci. 2008, 97, 1530–1542. [Google Scholar] [CrossRef] [PubMed]
- Italia, J.L.; Sharp, A.; Carter, K.C.; Warn, P.; Kumar, M.N. Peroral amphotericin B polymer nanoparticles lead to comparable or superior in vivo antifungal activity to that of intravenous Ambisome® or Fungizone™. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, R.F.; Ribeiro, I.F.; Miranda-Vilela, A.L.; de Souza Filho, J.; Martins, O.P.; Cintra e Silva Dde, O.; Tedesco, A.C.; Lacava, Z.G.; Báo, S.N.; Sampaio, R.N. Leishmanicidal activity of amphotericin B encapsulated in PLGA–DMSA nanoparticles to treat cutaneous leishmaniasis in C57BL/6 mice. Exp. Parasitol. 2013, 135, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Nakamura, M.; Miki, H.; Ozaki, S.; Abe, M.; Matsumoto, T.; Sakamoto, W.; Yogo, T.; Ishimura, K. Magnetically responsive smart nanoparticles for cancer treatment with a combination of magnetic hyperthermia and remote-control drug release. Theranostics 2014, 4, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Al-Quadeib, B.T.; Radwan, M.A.; Siller, L.; Horrocks, B.; Wright, M.C. Stealth Amphotericin B nanoparticles for oral drug delivery: In vitro optimization. Saudi Pharm. J. 2015, 23, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Carraro, T.C.; Khalil, N.M.; Mainardes, R.M. Amphotericin B-loaded polymeric nanoparticles: formulation optimization by factorial design. Pharm. Dev. Technol. 2016, 21, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Moraes Moreira Carraro, T.C.; Altmeyer, C.; Maissar Khalil, N.; Mara Mainardes, R. Assessment of in vitro antifungal efficacy and in vivo toxicity of Amphotericin B-loaded PLGA and PLGA-PEG blend nanoparticles. J. Mycol. Med. 2017, 27, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Sahoo, G.C.; Pandey, K.; Das, V.; Das, P. Study the effects of PLGA-PEG encapsulated amphotericin B nanoparticle drug delivery system against Leishmania donovani. Drug Deliv. 2015, 22, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Owais, M.; Gupta, C.M. Targeted drug delivery to macrophages in parasitic infections. Curr. Drug Deliv. 2005, 2, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Alibakhshi, A.; Abarghooi Kahaki, F.; Ahangarzadeh, S.; Yaghoobi, H.; Yarian, F.; Arezumand, R.; Ranjbari, J.; Mokhtarzadeh, A.; de la Guardia, M. Targeted cancer therapy through antibody fragments-decorated nanomedicines. J. Control. Release 2017, 268, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Nahar, M.; Jain, N.K. Preparation, characterization and evaluation of targeting potential of amphotericin B-loaded engineered PLGA nanoparticles. Pharm. Res. 2009, 26, 2588–2598. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Sharma, S.; Khuller, G.K. Lectin-functionalized poly (lactide-co-glycolide) nanoparticles as oral/aerosolized antitubercular drug carriers for treatment of tuberculosis. J. Antimicrob. Chemother. 2004, 54, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Kassab, R.; Parrot-Lopez, H.; Fessi, H.; Menaucourt, J.; Bonaly, R.; Coulon, J. Molecular recognition by Kluyveromyces of amphotericin B-loaded, galactose-tagged, poly (lactic acid) microspheres. Bioorg. Med. Chem. 2002, 10, 1767–1775. [Google Scholar] [CrossRef]
- Shahnaz, G.; Edagwa, B.J.; McMillan, J.; Akhtar, S.; Raza, A.; Qureshi, N.A.; Yasinzai, M.; Gendelman, H.E. Development of mannose-anchored thiolated amphotericin B nanocarriers for treatment of visceral leishmaniasis. Nanomedicine 2017, 12, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic. Bioeng. Transl. Med. 2016, 1, 10–29. [Google Scholar] [CrossRef] [PubMed]
- Cosco, D.; Fattal, E.; Fresta, M.; Tsapis, N. Perfluorocarbon-loaded micro and nanosystems for medical imaging: A state of the art. J. Fluor. Chem. 2015, 171, 18–26. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Formulation | Composition | Application | Ref. |
|---|---|---|---|---|
| Fungizone® | Colloidal dispersion | AmB, sodium deoxycholate (1:2 molar ratio) | Treatment of invasive fungal infections; treatment of leishmaniasis (not as primary therapy) | [23,24,25] |
| Ambisome® | Unilamellar liposomes | Phosphatidylcholine, cholesterol, diastearoylglycerol and AmB (2:1:0.8:0.4 molar ratio) | Therapy of febrile neutropenia, aspergillosis, candidiasis, and cryptococcosis; treatment of visceral leishmaniasis (as second-line therapy) | [39] |
| Albecet® | Lipid complex | AmB, L-α dimyristoylphosphatidylcholine, L-α dimyristoylphosphatidylglycerol (1:7:3 molar ratio) | Treatment of invasive fungal infections in patients resistant or intolerant to conventional AmB therapy | [23] |
| Amphotec® | Colloidal lipid complex | AmB, cholesterol sulfate (1:1 molar ratio) | Similar antifungal efficacy of Fungizone® but less hemolytic and cytotoxic effects. | [26] |
| Composition | Preparation | Size (nm) | ZP (mV) a | Administration Route | Ref. |
|---|---|---|---|---|---|
| AmB, PLGA, PVA | Nanoprecipitation method | 86–153 | −9 | i.p. | [43] |
| AmB, PLGA, P188-P338 with Tween80 or Sodium cholate | Nanoprecipitation method | 86–153 | −31 | i.p. | [43] |
| D-AmB b, PLGA, DMSA c | Water-in-oil emulsification and solvent evaporation method | -- | - | i.p. | [61] |
| AmB, PLGA, Vitamin E-TPGS | Emulsion-diffusion evaporation method Nanoprecipitation method | 182 165 | −16 −15 | Oral | [65] |
| AMB, PLGA, Vitamin E-TPGS, DMSO, Ethanol, AmB | Emulsion-diffusion evaporation method | 113 | - | Oral | [65] |
| AmB, PLGA, Sodium cholate | Emulsion solvent evaporation method (o/w emulsification) | 154 | −46 | i.v. | [67] |
| AmB, diblock polymer PLGA–PEG with 15% PEG | Modified emulsification method | 26 to 1068 | - | Oral | [70] |
| Mannosylated | Emulsion solvent evaporation method | 157 | −26 | i.v. | [76] |
| PEG-Mannosylated | Emulsion solvent evaporation method | 178 | −34 | i.v. | [76] |
| Mannose-anchored thiolated chitosan | Emulsion solvent evaporation method | 362 | - | i.v. | [79] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palma, E.; Pasqua, A.; Gagliardi, A.; Britti, D.; Fresta, M.; Cosco, D. Antileishmanial Activity of Amphotericin B-loaded-PLGA Nanoparticles: An Overview. Materials 2018, 11, 1167. https://doi.org/10.3390/ma11071167
Palma E, Pasqua A, Gagliardi A, Britti D, Fresta M, Cosco D. Antileishmanial Activity of Amphotericin B-loaded-PLGA Nanoparticles: An Overview. Materials. 2018; 11(7):1167. https://doi.org/10.3390/ma11071167
Chicago/Turabian StylePalma, Ernesto, Antonella Pasqua, Agnese Gagliardi, Domenico Britti, Massimo Fresta, and Donato Cosco. 2018. "Antileishmanial Activity of Amphotericin B-loaded-PLGA Nanoparticles: An Overview" Materials 11, no. 7: 1167. https://doi.org/10.3390/ma11071167
APA StylePalma, E., Pasqua, A., Gagliardi, A., Britti, D., Fresta, M., & Cosco, D. (2018). Antileishmanial Activity of Amphotericin B-loaded-PLGA Nanoparticles: An Overview. Materials, 11(7), 1167. https://doi.org/10.3390/ma11071167