Phenotypic Variation during Biofilm Formation: Implications for Anti-Biofilm Therapeutic Design



Abstract
1. Introduction
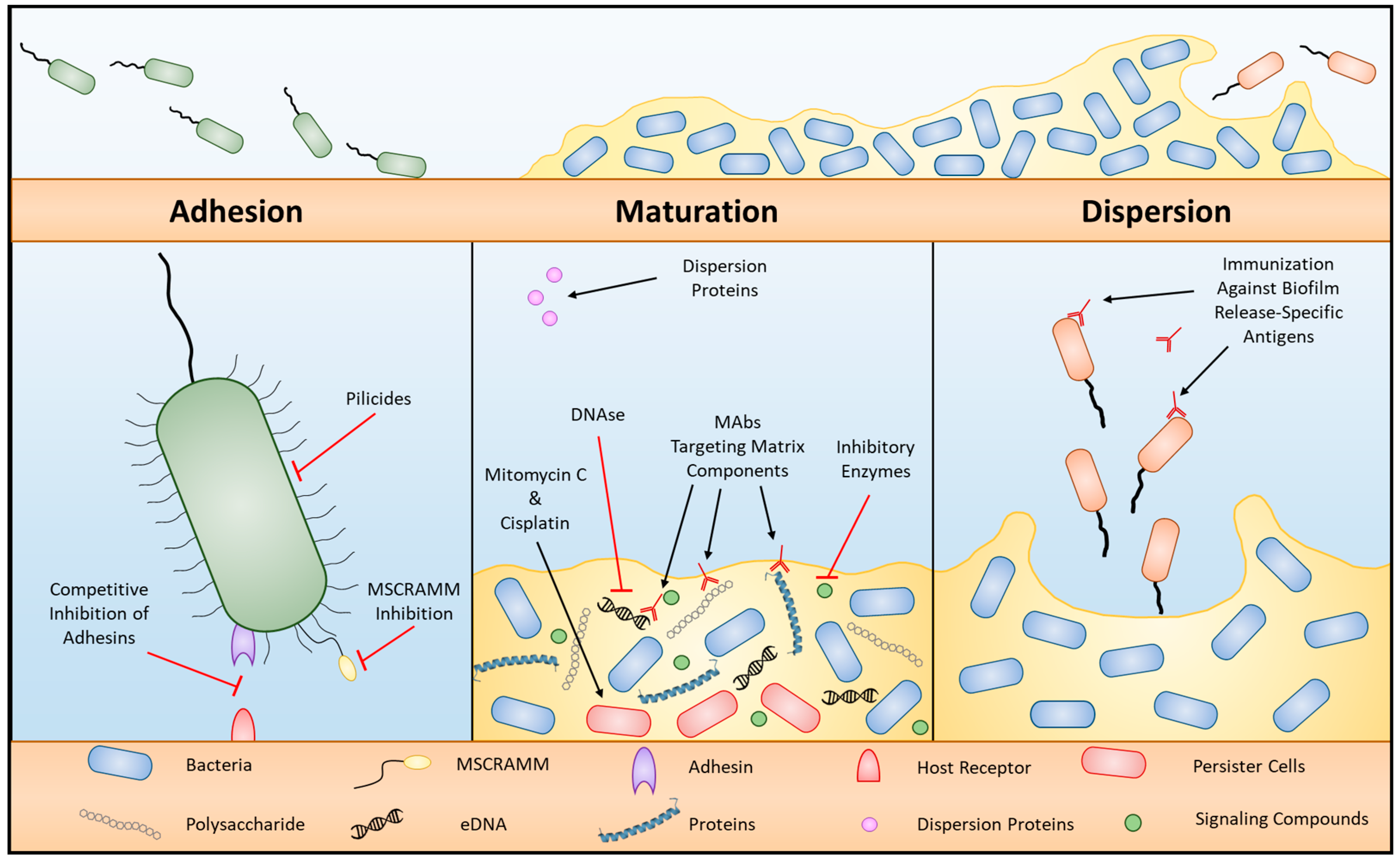
2. Biofilm Development Overview
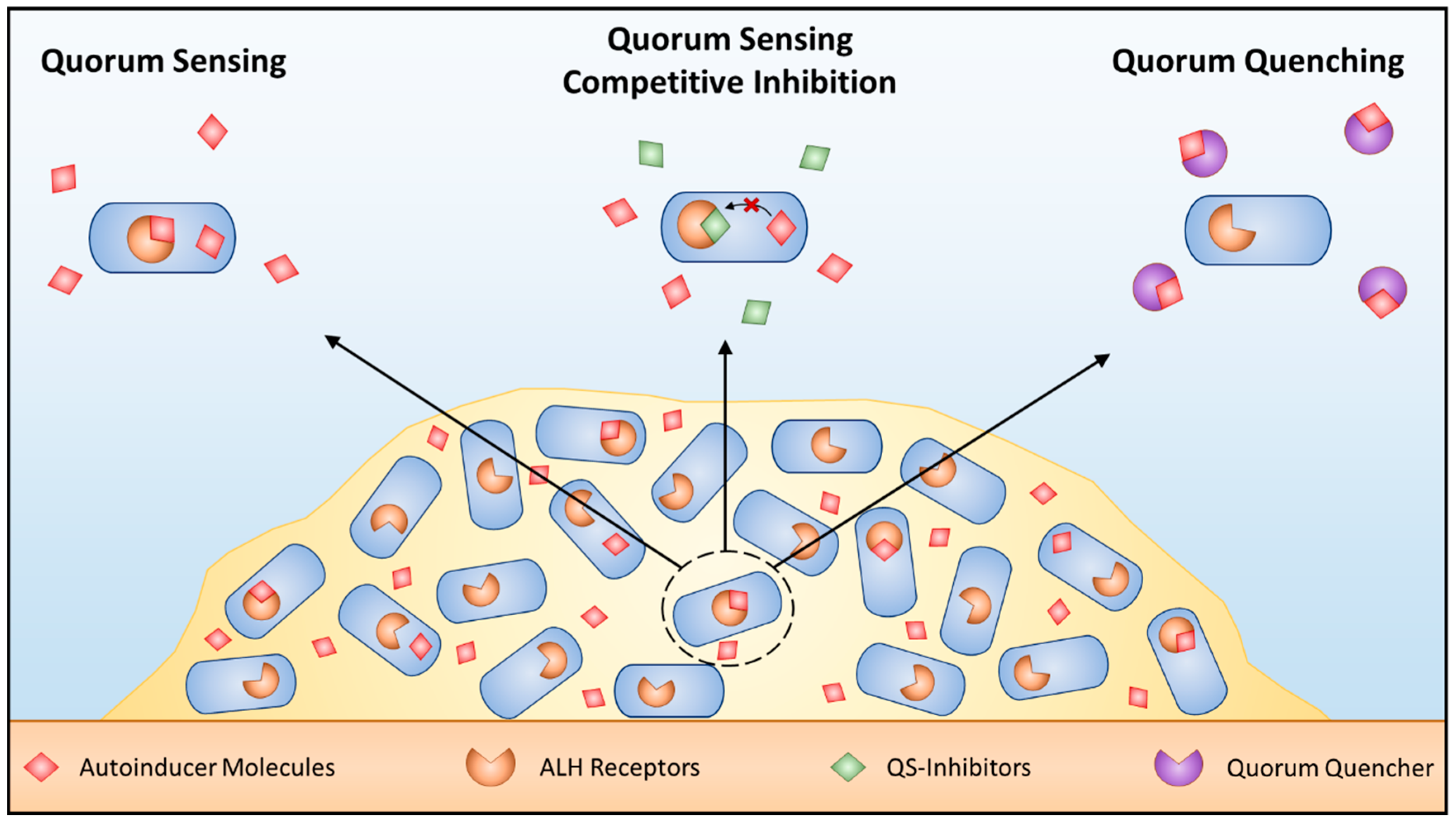
Quorum Sensing During Biofilm Formation
3. Bacterial Adhesion
Anti-Adhesion Therapies
4. Biofilm Maturation
4.1. Extracellular Matrix Producers
4.2. Persister Cells
4.3. Anti-Biofilm Strategies
5. Dispersion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- National Institutes of Health. Available online: https://grants.nih.gov/grants/guide/pa-files/PA-07-288.html (accessed on 3 October 2017).
- Ribet, D.; Cossart, P. How bacterial pathogens colonize their hosts and invade deeper tissues. Microbes Infect. 2015, 17, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.; Marks, L.R.; Pettigrew, M.M.; Hakansson, A.P. Streptococcus pneumoniae biofilm formation and dispersion during colonization and disease. Front. Cell. Infect. Microbiol. 2014, 4, 194. [Google Scholar] [CrossRef] [PubMed]
- Pettigrew, M.M.; Marks, L.R.; Kong, Y.; Gent, J.F.; Roche-Hakansson, H.; Hakansson, A.P. Dynamic changes in the Streptococcus pneumoniae transcriptome during transition from biofilm formation to invasive disease upon influenza a virus infection. Infect. Immun. 2014, 82, 4607–4619. [Google Scholar] [CrossRef] [PubMed]
- Lebeaux, D.; Ghigo, J.-M.; Beloin, C. Biofilm-related infections: Bridging the gap between clinical management and fundamental aspects of recalcitrance toward antibiotics. Microbiol. Mol. Biol. Rev. 2014, 78, 510–543. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 2007, 5, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.S.; Costerton, J.W. Antibiotic resistance of bacteria in biofilms. Lancet 2001, 358, 135–138. [Google Scholar] [CrossRef]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial biofilms: A common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Hoiby, N.; Ciofu, O.; Bjarnsholt, T. Pseudomonas aeruginosa biofilms in cystic fibrosis. Future Microbiol. 2010, 5, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Rafiei, M.; Kiani, F.; Sayehmiri, F.; Sayehmiri, K.; Sheikhi, A.; Zamanian Azodi, M. Study of Porphyromonas gingivalis in periodontal diseases: A systematic review and meta-analysis. Med. J. Islam. Repub. Iran. 2017, 31, 62. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, V.D.; Bijie, H.; Maki, D.G.; Mehta, Y.; Apisarnthanarak, A.; Medeiros, E.A.; Leblebicioglu, H.; Fisher, D.; Álvarez-Moreno, C.; Khader, I.A. International nosocomial infection control consortium (INICC) report, data summary of 36 countries, for 2004–2009. Am. J. Infect. Control 2012, 40, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Lentino, J.R. Prosthetic joint infections: Bane of orthopedists, challenge for infectious disease specialists. Clin. Infect. Dis. 2003, 36, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Balsells, E.; Guillot, L.; Nair, H.; Kyaw, M.H. Serotype distribution of Streptococcus pneumoniae causing invasive disease in children in the post-PCV era: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0177113. [Google Scholar] [CrossRef] [PubMed]
- Hanage, W.P. Serotype replacement in invasive pneumococcal disease: Where do we go from here? J. Infect. Dis. 2007, 196, 1282–1284. [Google Scholar] [CrossRef] [PubMed]
- Boles, B.R.; Thoendel, M.; Singh, P.K. Self-generated diversity produces “insurance effects” in biofilm communities. Proc. Natl. Acad. Sci. USA 2004, 101, 16630–16635. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.S.; Franklin, M.J. Physiological heterogeneity in biofilms. Nat. Rev. Microbiol. 2008, 6, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Elias, S.; Banin, E. Multi-species biofilms: Living with friendly neighbors. FEMS Microbiol. Rev. 2012, 36, 990–1004. [Google Scholar] [CrossRef] [PubMed]
- Marks, L.R.; Davidson, B.A.; Knight, P.R.; Hakansson, A.P. Interkingdom signaling induces Streptococcus pneumoniae biofilm dispersion and transition from asymptomatic colonization to disease. mBio 2013, 4, e00438-13. [Google Scholar] [CrossRef] [PubMed]
- Cécile, R.; Laurent, G.; Jean, G. Biofilm-detached cells, a transition from a sessile to a planktonic phenotype: A comparative study of adhesion and physiological characteristics in Pseudomonas aeruginosa. FEMS Microbiol. Lett. 2008, 290, 135–142. [Google Scholar]
- Greene, S.E.; Pinkner, J.S.; Chorell, E.; Dodson, K.W.; Shaffer, C.L.; Conover, M.S.; Livny, J.; Hadjifrangiskou, M.; Almqvist, F.; Hultgren, S.J. Pilicide ec240 disrupts virulence circuits in uropathogenic Escherichia coli. mBio 2014, 5, e02038-14. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Papavlassopoulos, H.; Chandrasekaran, V.; Grabosch, C.; Beiroth, F.; Lindhorst, T.K.; Röhl, C. Inhibition of bacterial adhesion to live human cells: Activity and cytotoxicity of synthetic mannosides. FEBS Lett. 2012, 586, 1459–1465. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Loimaranta, V.; Joosten, J.A.; Khan, A.S.; Hacker, J.; Pieters, R.J.; Finne, J. Inhibition of p-fimbriated Escherichia coli adhesion by multivalent galabiose derivatives studied by a live-bacteria application of surface plasmon resonance. J. Antimicrob. Chemother. 2007, 60, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Mora, M.; Bensi, G.; Capo, S.; Falugi, F.; Zingaretti, C.; Manetti, A.G.O.; Maggi, T.; Taddei, A.R.; Grandi, G.; Telford, J.L. Group A Streptococcus produce pilus-like structures containing protective antigens and Lancefield T antigens. Proc. Natl. Acad. Sci. USA 2005, 102, 15641–15646. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Hu, P.; Zhou, S.Y.; Li, Q.; Chen, W.M. Morin inhibits sortase a and subsequent biofilm formation in Streptococcus mutans. Curr. Microbiol. 2014, 68, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Suree, N.; Yi, S.W.; Thieu, W.; Marohn, M.; Damoiseaux, R.; Chan, A.; Jung, M.E.; Clubb, R.T. Discovery and structure–activity relationship analysis of Staphylococcus aureus sortase a inhibitors. Bioorg. Med. Chem. 2009, 17, 7174–7185. [Google Scholar] [CrossRef] [PubMed]
- Guendouze, A.; Plener, L.; Bzdrenga, J.; Jacquet, P.; Rémy, B.; Elias, M.; Lavigne, J.-P.; Daudé, D.; Chabrière, E. Effect of quorum quenching lactonase in clinical isolates of Pseudomonas aeruginosa and comparison with quorum sensing inhibitors. Front. Microbiol. 2017, 8, 227. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Tian, X.-Q.; Wei, J.-W.; Ding, L.-L.; Qian, W.; Liu, Z.; Wang, F.-F. BsmR degrades c-di-GMP to modulate biofilm formation of nosocomial pathogen Stenotrophomonas maltophilia. Sci. Rep. 2017, 7, 4665. [Google Scholar] [CrossRef] [PubMed]
- Zuberi, A.; Misba, L.; Khan, A.U. CRISPR interference (crispri) inhibition of luxs gene expression in E. coli: An approach to inhibit biofilm. Front. Cell. Infect. Microbiol. 2017, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Kim, J.; Hur, J.K.; Lee, S.-S. CRISPR-based genome editing of clinically important Escherichia coli SE15 isolated from indwelling urinary catheters of patients. J. Med. Microbiol. 2017, 66, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.B. Therapeutic potential of biofilm-dispersing enzymes. IJAO 2009, 32, 545–554. [Google Scholar] [CrossRef]
- Shak, S.; Capon, D.J.; Hellmiss, R.; Marsters, S.A.; Baker, C.L. Recombinant human DNAse I reduces the viscosity of cystic fibrosis sputum. Proc. Natl. Acad. Sci. USA 1990, 87, 9188–9192. [Google Scholar] [CrossRef] [PubMed]
- Kelly-Quintos, C.; Cavacini, L.A.; Posner, M.R.; Goldmann, D.; Pier, G.B. Characterization of the opsonic and protective activity against Staphylococcus aureus of fully human monoclonal antibodies specific for the bacterial surface polysaccharide poly-n-acetylglucosamine. Infect. Immun. 2006, 74, 2742–2750. [Google Scholar] [CrossRef] [PubMed]
- Kwan, B.W.; Chowdhury, N.; Wood, T.K. Combatting bacterial infections by killing persister cells with mitomycin c. Environ. Microbiol. 2015, 17, 4406–4414. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, N.; Wood, T.L.; Martinez-Vazquez, M.; Garcia-Contreras, R.; Wood, T.K. DNA-crosslinker cisplatin eradicates bacterial persister cells. Biotechnol. Bioeng. 2016, 113, 1984–1992. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.N.H.; Morozov, A.; Planzos, P.; Zelaya, H.M. The fatty acid signaling molecule cis-2-decenoic acid increases metabolic activity and reverts persister cells to an antimicrobial-susceptible state. Appl. Environ. Microbiol. 2014, 80, 6976–6991. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hill, A.; Beitelshees, M.; Sha, S.; Lovell, J.F.; Davidson, B.A.; Knight, P.R.; Hakansson, A.P.; Pfeifer, B.A.; Jones, C.H. Directed vaccination against pneumococcal disease. Proc. Natl. Acad. Sci. USA 2016, 113, 6898–6903. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.H.; Zhang, G.; Nayerhoda, R.; Beitelshees, M.; Hill, A.; Rostami, P.; Li, Y.; Davidson, B.A.; Knight, P.; Pfeifer, B.A. Comprehensive vaccine design for commensal disease progression. Sci. Adv. 2017, 3, e1701797. [Google Scholar] [CrossRef] [PubMed]
- Roilides, E.; Simitsopoulou, M.; Katragkou, A.; Walsh, T.J. How biofilms evade host defenses. Microbiol. Spectr. 2015, 3, 3. [Google Scholar]
- Thurlow, L.R.; Hanke, M.L.; Fritz, T.; Angle, A.; Aldrich, A.; Williams, S.H.; Engebretsen, I.L.; Bayles, K.W.; Horswill, A.R.; Kielian, T. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J. Immunol. 2011, 186, 6585–6596. [Google Scholar] [CrossRef] [PubMed]
- Medini, D.; Serruto, D.; Parkhill, J.; Relman, D.A.; Donati, C.; Moxon, R.; Falkow, S.; Rappuoli, R. Microbiology in the post-genomic era. Nat. Rev. Microbiol. 2008, 6, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Landini, P.; Antoniani, D.; Burgess, J.G.; Nijland, R. Molecular mechanisms of compounds affecting bacterial biofilm formation and dispersal. Appl. Microbiol. Biotechnol. 2010, 86, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Garrett, T.R.; Bhakoo, M.; Zhang, Z. Bacterial adhesion and biofilms on surfaces. Prog. Nat. Sci. Mater. 2008, 18, 1049–1056. [Google Scholar] [CrossRef]
- O’Toole, G.; Kaplan, H.B.; Kolter, R. Biofilm formation as microbial development. Annu. Rev. Microbiol. 2000, 54, 49. [Google Scholar] [CrossRef] [PubMed]
- Hermansson, M. The DLVO theory in microbial adhesion. Colloids Surf. B Biointerfaces 1999, 14, 105–119. [Google Scholar] [CrossRef]
- Abbot, E.L.; Smith, W.D.; Siou, G.P.; Chiriboga, C.; Smith, R.J.; Wilson, J.A.; Hirst, B.H.; Kehoe, M.A. Pili mediate specific adhesion of Streptococcus pyogenes to human tonsil and skin. Cell. Microbiol. 2007, 9, 1822–1833. [Google Scholar] [CrossRef] [PubMed]
- Maisey, H.C.; Hensler, M.; Nizet, V.; Doran, K.S. Group B Streptococcal pilus proteins contribute to adherence to and invasion of brain microvascular endothelial cells. J. Bacteriol. 2007, 189, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Manetti, A.G.; Zingaretti, C.; Falugi, F.; Capo, S.; Bombaci, M.; Bagnoli, F.; Gambellini, G.; Bensi, G.; Mora, M.; Edwards, A.M.; et al. Streptococcus pyogenes pili promote pharyngeal cell adhesion and biofilm formation. Mol. Microbiol. 2007, 64, 968–983. [Google Scholar] [CrossRef] [PubMed]
- Flemming, H.C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.B. Biofilm dispersal: Mechanisms, clinical implications, and potential therapeutic uses. J. Dent. Res. 2010, 89, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Cvitkovitch, D.G.; Li, Y.-H.; Ellen, R.P. Quorum sensing and biofilm formation in streptococcal infections. J. Clin. Investig. 2003, 112, 1626–1632. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, A.; Wood, T.K. Bacterial quorum sensing: Signals, circuits, and implications for biofilms and disease. Annu. Rev. Biomed. Eng. 2008, 10, 145–167. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Tian, X. Quorum sensing and bacterial social interactions in biofilms. Sensors (Basel) 2012, 12, 2519–2538. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Schauder, S.; Potier, N.; Van Dorsselaer, A.; Pelczer, I.; Bassler, B.L.; Hughson, F.M. Structural identification of a bacterial quorum-sensing signal containing boron. Nature 2002, 415, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Li, H.; Vuong, C.; Vadyvaloo, V.; Wang, J.; Yao, Y.; Otto, M.; Gao, Q. Role of the luxS quorum-sensing system in biofilm formation and virulence of Staphylococcus epidermidis. Infect. Immun. 2006, 74, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Vidal, J.E.; Ludewick, H.P.; Kunkel, R.M.; Zahner, D.; Klugman, K.P. The luxS-dependent quorum-sensing system regulates early biofilm formation by Streptococcus pneumoniae strain d39. Infect. Immun. 2011, 79, 4050–4060. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Ou, Y.; Yang, L.; Zhu, Y.; Tolker-Nielsen, T.; Molin, S.; Qu, D. Role of autolysin-mediated DNA release in biofilm formation of Staphylococcus epidermidis. Microbiology 2007, 153, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Cotter, P.A.; Stibitz, S. C-di-GMP-mediated regulation of virulence and biofilm formation. Curr. Opin. Microbiol. 2007, 10, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Simm, R.; Morr, M.; Kader, A.; Nimtz, M.; Romling, U. GGDEF and EAL domains inversely regulate cyclic di-gmp levels and transition from sessility to motility. Mol. Microbiol. 2004, 53, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Ha, D.G.; O’Toole, G.A. C-di-GMP and its effects on biofilm formation and dispersion: A Pseudomonas aeruginosa review. Microbiol. Spectr. 2015, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Tischler, A.D.; Camilli, A. Cyclic diguanylate (c-di-GMP) regulates Vibrio cholerae biofilm formation. Mol. Microbiol. 2004, 53, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, D.; Waters, C.M. A tangled web: Regulatory connections between quorum sensing and cyclic di-GMP. J. Bacteriol. 2012, 194, 4485–4493. [Google Scholar] [CrossRef] [PubMed]
- Hengge, R. Principles of c-di-GMP signalling in bacteria. Nat. Rev. Microbiol. 2009, 7, 263. [Google Scholar] [CrossRef] [PubMed]
- Valentini, M.; Filloux, A. Biofilms and cyclic di-GMP (c-di-GMP) signaling: Lessons from Pseudomonas aeruginosa and other bacteria. J. Biol. Chem. 2016, 291, 12547–12555. [Google Scholar] [CrossRef] [PubMed]
- Hickman, J.W.; Tifrea, D.F.; Harwood, C.S. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc. Natl. Acad. Sci. USA 2005, 102, 14422–14427. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Heine, S.; Entian, M.; Sauer, K.; Frankenberg-Dinkel, N. No-induced biofilm dispersion in Pseudomonas aeruginosa is mediated by an MHYT domain-coupled phosphodiesterase. J. Bacteriol. 2013, 195, 3531–3542. [Google Scholar] [CrossRef] [PubMed]
- Yoda, I.; Koseki, H.; Tomita, M.; Shida, T.; Horiuchi, H.; Sakoda, H.; Osaki, M. Effect of surface roughness of biomaterials on Staphylococcus epidermidis adhesion. BMC Microbiol. 2014, 14, 234. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Sharma, P.K.; Busscher, H.J.; van der Mei, H.C.; Krom, B.P. Role of extracellular DNA in initial bacterial adhesion and surface aggregation. Appl. Environ. Microbiol. 2010, 76, 3405–3408. [Google Scholar] [CrossRef] [PubMed]
- Sjollema, J.; van der Mei, H.C.; Hall, C.L.; Peterson, B.W.; de Vries, J.; Song, L.; Jong, E.D.d.; Busscher, H.J.; Swartjes, J.J.T.M. Detachment and successive re-attachment of multiple, reversibly-binding tethers result in irreversible bacterial adhesion to surfaces. Sci. Rep. 2017, 7, 4369. [Google Scholar] [CrossRef] [PubMed]
- Sauer, M.M.; Jakob, R.P.; Eras, J.; Baday, S.; Eriş, D.; Navarra, G.; Bernèche, S.; Ernst, B.; Maier, T.; Glockshuber, R. Catch-bond mechanism of the bacterial adhesin FimH. Nat. Commun. 2016, 7, 10738. [Google Scholar] [CrossRef] [PubMed]
- Kline, K.A.; Fälker, S.; Dahlberg, S.; Normark, S.; Henriques-Normark, B. Bacterial adhesins in host-microbe interactions. Cell. Host Microbe 2009, 5, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Proft, T.; Baker, E.N. Pili in Gram-negative and Gram-positive bacteria—Structure, assembly and their role in disease. Cell. Mol. Life Sci. 2009, 66, 613–635. [Google Scholar] [CrossRef] [PubMed]
- Patti, J.M.; Allen, B.L.; McGavin, M.J.; Hook, M. MSCRAMM-mediated adherence of microorganisms to host tissues. Annu. Rev. Microbiol. 1994, 48, 585–617. [Google Scholar] [CrossRef] [PubMed]
- Walsh, E.J.; Miajlovic, H.; Gorkun, O.V.; Foster, T.J. Identification of the Staphylococcus aureus MSCRAMM clumping factor b (ClfB) binding site in the alphac-domain of human fibrinogen. Microbiology 2008, 154, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Jensch, I.; Gamez, G.; Rothe, M.; Ebert, S.; Fulde, M.; Somplatzki, D.; Bergmann, S.; Petruschka, L.; Rohde, M.; Nau, R.; et al. PavB is a surface-exposed adhesin of Streptococcus pneumoniae contributing to nasopharyngeal colonization and airways infections. Mol. Microbiol. 2010, 77, 22–43. [Google Scholar] [CrossRef] [PubMed]
- Nobbs, A.H.; Lamont, R.J.; Jenkinson, H.F. Streptococcus adherence and colonization. Microbiol. Mol. Biol. Rev. 2009, 73, 407–450. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J.; Geoghegan, J.A.; Ganesh, V.K.; Höök, M. Adhesion, invasion and evasion: The many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 2014, 12, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Hair, P.S.; Ward, M.D.; Semmes, O.J.; Foster, T.J.; Cunnion, K.M. Staphylococcus aureus clumping factor A binds to complement regulator factor I and increases factor I cleavage of C3b. J. Infect. Dis. 2008, 198, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Hair, P.S.; Echague, C.G.; Sholl, A.M.; Watkins, J.A.; Geoghegan, J.A.; Foster, T.J.; Cunnion, K.M. Clumping factor A interaction with complement factor I increases C3b cleavage on the bacterial surface of Staphylococcus aureus and decreases complement-mediated phagocytosis. Infect. Immun. 2010, 78, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Sharp, J.A.; Echague, C.G.; Hair, P.S.; Ward, M.D.; Nyalwidhe, J.O.; Geoghegan, J.A.; Foster, T.J.; Cunnion, K.M. Staphylococcus aureus surface protein SdrE binds complement regulator factor H as an immune evasion tactic. PLoS ONE 2012, 7, e38407. [Google Scholar] [CrossRef] [PubMed]
- Ghasemian, A.; Najar Peerayeh, S.; Bakhshi, B.; Mirzaee, M. The microbial surface components recognizing adhesive matrix molecules (MSCRAMMs) genes among clinical isolates of Staphylococcus aureus from hospitalized children. Iran. J. Pathol. 2015, 10, 258–264. [Google Scholar] [PubMed]
- McCarthy, A.J.; Lindsay, J.A. Genetic variation in Staphylococcus aureus surface and immune evasion genes is lineage associated: Implications for vaccine design and host-pathogen interactions. BMC Microbiol. 2010, 10, 173. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Ishiwa, A. The role of carbohydrates in infection strategies of enteric pathogens. Trop. Med. Health 2015, 43, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Pieters, R.J. Intervention with bacterial adhesion by multivalent carbohydrates. Med. Res. Rev. 2007, 27, 796–816. [Google Scholar] [CrossRef] [PubMed]
- Cozens, D.; Read, R.C. Anti-adhesion methods as novel therapeutics for bacterial infections. Expert Rev. Anti-Infect. Ther. 2012, 10, 1457–1468. [Google Scholar] [CrossRef] [PubMed]
- Svensson, A.; Larsson, A.; Emtenäs, H.; Hedenström, M.; Fex, T.; Hultgren, S.J.; Pinkner, J.S.; Almqvist, F.; Kihlberg, J. Design and evaluation of pilicides: Potential novel antibacterial agents directed against uropathogenic Escherichia coli. ChemBioChem 2001, 2, 915–918. [Google Scholar] [CrossRef]
- Pinkner, J.S.; Remaut, H.; Buelens, F.; Miller, E.; Aberg, V.; Pemberton, N.; Hedenstrom, M.; Larsson, A.; Seed, P.; Waksman, G.; et al. Rationally designed small compounds inhibit pilus biogenesis in uropathogenic bacteria. Proc. Natl. Acad. Sci. USA 2006, 103, 17897–17902. [Google Scholar] [CrossRef] [PubMed]
- Chorell, E.; Pinkner, J.S.; Phan, G.; Edvinsson, S.; Buelens, F.; Remaut, H.; Waksman, G.; Hultgren, S.J.; Almqvist, F. Design and synthesis of C-2 substituted thiazolo and dihydrothiazolo ring-fused 2-pyridones: Pilicides with increased antivirulence activity. J. Med. Chem. 2010, 53, 5690–5695. [Google Scholar] [CrossRef] [PubMed]
- Margarit, I.; Rinaudo, C.D.; Galeotti, C.L.; Maione, D.; Ghezzo, C.; Buttazzoni, E.; Rosini, R.; Runci, Y.; Mora, M.; Buccato, S.; et al. Preventing bacterial infections with pilus-based vaccines: The Group B Streptococcus paradigm. J. Infect. Dis. 2009, 199, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Jiang, L.; Song, Q.; Yang, J.; Chen, Z.; Guo, Z.; Zhou, D.; Du, Z.; Song, Y.; Wang, J.; et al. Protein microarray for profiling antibody responses to Yersinia pestis live vaccine. Infect. Immun. 2005, 73, 3734–3739. [Google Scholar] [CrossRef] [PubMed]
- Williamson, E.D.; Flick-Smith, H.C.; LeButt, C.; Rowland, C.A.; Jones, S.M.; Waters, E.L.; Gwyther, R.J.; Miller, J.; Packer, P.J.; Irving, M. Human immune response to a plague vaccine comprising recombinant F1 and V antigens. Infect. Immun. 2005, 73, 3598–3608. [Google Scholar] [CrossRef] [PubMed]
- Strindelius, L.; Filler, M.; Sjoholm, I. Mucosal immunization with purified flagellin from salmonella induces systemic and mucosal immune responses in C3H/HeJ mice. Vaccine 2004, 22, 3797–3808. [Google Scholar] [CrossRef] [PubMed]
- Tchesnokova, V.; Aprikian, P.; Kisiela, D.; Gowey, S.; Korotkova, N.; Thomas, W.; Sokurenko, E. Type 1 fimbrial adhesin FimH elicits an immune response that enhances cell adhesion of Escherichia coli. Infect. Immun. 2011, 79, 3895–3904. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Cai, S.; Gu, G.; Guo, Z.; Long, Z. Recent progress in the development of sortase A inhibitors as novel anti-bacterial virulence agents. RSC Adv. 2015, 5, 49880–49889. [Google Scholar] [CrossRef]
- Cascioferro, S.; Cusimano, M.G.; Schillaci, D. Antiadhesion agents against gram-positive pathogens. Future Microbiol. 2014, 9, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Ofek, I.; Hasty, D.L.; Sharon, N. Anti-adhesion therapy of bacterial diseases: Prospects and problems. FEMS Immunol. Med. Microbiol. 2003, 38, 181–191. [Google Scholar] [CrossRef]
- Krachler, A.M.; Orth, K. Targeting the bacteria–host interface: Strategies in anti-adhesion therapy. Virulence 2013, 4, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Sauer, K.; Camper, A.K.; Ehrlich, G.D.; Costerton, J.W.; Davies, D.G. Pseudomonas aeruginosa displays multiple phenotypes during development as a biofilm. J. Bacteriol. 2002, 184, 1140–1154. [Google Scholar] [CrossRef] [PubMed]
- Vlamakis, H.; Aguilar, C.; Losick, R.; Kolter, R. Control of cell fate by the formation of an architecturally complex bacterial community. Genes Dev. 2008, 22, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Van Gestel, J.; Vlamakis, H.; Kolter, R. Division of labor in biofilms: The ecology of cell differentiation. Microbiol. Spectr. 2015, 3, MB-0002. [Google Scholar] [CrossRef] [PubMed]
- Branda, S.S.; Vik, A.; Friedman, L.; Kolter, R. Biofilms: The matrix revisited. Trends Microbiol. 2005, 13, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, D.J.; Wyckoff, T.J.; Starkey, M.; Keyser, R.; Azadi, P.; O’Toole, G.A.; Parsek, M.R. Alginate is not a significant component of the extracellular polysaccharide matrix of PA14 and PAO1 Pseudomonas aeruginosa biofilms. Proc. Natl. Acad. Sci. USA 2003, 100, 7907–7912. [Google Scholar] [CrossRef] [PubMed]
- Ghafoor, A.; Hay, I.D.; Rehm, B.H. Role of exopolysaccharides in Pseudomonas aeruginosa biofilm formation and architecture. Appl. Environ. Microbiol. 2011, 77, 5238–5246. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.D.; Starkey, M.; Kremer, S.; Parsek, M.R.; Wozniak, D.J. Identification of Psl, a locus encoding a potential exopolysaccharide that is essential for pseudomonas aeruginosa PAO1 biofilm formation. J. Bacteriol. 2004, 186, 4466–4475. [Google Scholar] [CrossRef] [PubMed]
- Owlia, P.; Nosrati, R.; Alaghehbandan, R.; Lari, A.R. Antimicrobial susceptibility differences among mucoid and non-mucoid Pseudomonas aeruginosa isolates. GMS Hyg. Infect. Control 2014, 9, 2. [Google Scholar]
- Cabral, D.A.; Loh, B.A.; Speert, D.P. Mucoid Pseudomonas aeruginosa resists nonopsonic phagocytosis by human neutrophils and macrophages. Pediatr. Res. 1987, 22, 429. [Google Scholar] [CrossRef] [PubMed]
- Cywes-Bentley, C.; Skurnik, D.; Zaidi, T.; Roux, D.; DeOliveira, R.B.; Garrett, W.S.; Lu, X.; O’Malley, J.; Kinzel, K.; Zaidi, T.; et al. Antibody to a conserved antigenic target is protective against diverse prokaryotic and eukaryotic pathogens. Proc. Natl. Acad. Sci. USA 2013, 110, E2209–E2218. [Google Scholar] [CrossRef] [PubMed]
- Arciola, C.R.; Campoccia, D.; Ravaioli, S.; Montanaro, L. Polysaccharide intercellular adhesin in biofilm: Structural and regulatory aspects. Front. Cell. Infect. Microbiol. 2015, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Whitchurch, C.B.; Tolker-Nielsen, T.; Ragas, P.C.; Mattick, J.S. Extracellular DNA required for bacterial biofilm formation. Science 2002, 295, 1487. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, X.; Liu, H.; Zhang, L.; Guo, Y.; Yu, S.; Wozniak, D.J.; Ma, L.Z. The exopolysaccharide Psl-eDNA interaction enables the formation of a biofilm skeleton in Pseudomonas aeruginosa. Environ. Microbiol. Rep. 2015, 7, 330–340. [Google Scholar] [CrossRef] [PubMed]
- De Aldecoa, A.L.I.; Zafra, O.; González-Pastor, J.E. Mechanisms and regulation of extracellular DNA release and its biological roles in microbial communities. Front. Microbiol. 2017, 8, 1390. [Google Scholar] [CrossRef] [PubMed]
- Rice, K.C.; Mann, E.E.; Endres, J.L.; Weiss, E.C.; Cassat, J.E.; Smeltzer, M.S.; Bayles, K.W. The cidA murein hydrolase regulator contributes to DNA release and biofilm development in Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2007, 104, 8113–8118. [Google Scholar] [CrossRef] [PubMed]
- Sharma-Kuinkel, B.K.; Mann, E.E.; Ahn, J.-S.; Kuechenmeister, L.J.; Dunman, P.M.; Bayles, K.W. The Staphylococcus aureus LytSR two-component regulatory system affects biofilm formation. J. Bacteriol. 2009, 191, 4767–4775. [Google Scholar] [CrossRef] [PubMed]
- Jurcisek, J.A.; Brockman, K.L.; Novotny, L.A.; Goodman, S.D.; Bakaletz, L.O. Nontypeable Haemophilus influenzae; releases DNA and DNABII proteins via a T4SS-like complex and come of the type IV pilus machinery. Proc. Natl. Acad. Sci. USA 2017, 114, E6632–E6641. [Google Scholar] [CrossRef] [PubMed]
- Toyofuku, M.; Roschitzki, B.; Riedel, K.; Eberl, L. Identification of proteins associated with the Pseudomonas aeruginosa biofilm extracellular matrix. J. Proteome Res. 2012, 11, 4906–4915. [Google Scholar] [CrossRef] [PubMed]
- Borlee, B.R.; Goldman, A.D.; Murakami, K.; Samudrala, R.; Wozniak, D.J.; Parsek, M.R. Pseudomonas aeruginosa uses a cyclic-di-GMP-regulated adhesin to reinforce the biofilm extracellular matrix. Mol. Microbiol. 2010, 75, 827–842. [Google Scholar] [CrossRef] [PubMed]
- Dillon, S.C.; Dorman, C.J. Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nat. Rev. Microbiol. 2010, 8, 185. [Google Scholar] [CrossRef] [PubMed]
- Archer, N.K.; Mazaitis, M.J.; Costerton, J.W.; Leid, J.G.; Powers, M.E.; Shirtliff, M.E. Staphylococcus aureus biofilms: Properties, regulation, and roles in human disease. Virulence 2011, 2, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.K.; Knabel, S.J.; Kwan, B.W. Bacterial persister cell formation and dormancy. Appl. Environ. Microbiol. 2013, 79, 7116–7121. [Google Scholar] [CrossRef] [PubMed]
- Brackman, G.; Coenye, T. Quorum sensing inhibitors as anti-biofilm agents. Curr. Pharm. Des. 2015, 21, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Solano, C.; Echeverz, M.; Lasa, I. Biofilm dispersion and quorum sensing. Curr. Opin. Microbiol. 2014, 18, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Boles, B.R.; Horswill, A.R. Agr-mediated dispersal of Staphylococcus aureus biofilms. PLoS Pathog. 2008, 4, e1000052. [Google Scholar] [CrossRef] [PubMed]
- LaSarre, B.; Federle, M.J. Exploiting quorum sensing to confuse bacterial pathogens. Microbiol. Mol. Biol. Rev. 2013, 77, 73–111. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.-H.; Gusti, A.R.; Zhang, Q.; Xu, J.-L.; Zhang, L.-H. Identification of quorum-quenching N-acyl homoserine lactonases from Bacillus species. Appl. Environ. Microbiol. 2002, 68, 1754–1759. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Xu, J.L.; Hu, J.; Wang, L.H.; Ong, S.L.; Leadbetter, J.R.; Zhang, L.H. Acyl-homoserine lactone acylase from Ralstonia strain XJ12B represents a novel and potent class of quorum-quenching enzymes. Mol. Microbiol. 2003, 47, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.-H.; Wang, L.-H.; Xu, J.-L.; Zhang, H.-B.; Zhang, X.-F.; Zhang, L.-H. Quenching quorum-sensing-dependent bacterial infection by an n-acyl homoserine lactonase. Nature 2001, 411, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Parsek, M.R. Controlling the connections of cells to the biofilm matrix. J. Bacteriol. 2016, 198, 12–14. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Romao, S.; Memmi, G.; Oggioni, M.R.; Trombe, M.-C. LuxS impacts on LytA-dependent autolysis and on competence in Streptococcus pneumoniae. Microbiology 2006, 152, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Verez-Bencomo, V.; Fernández-Santana, V.; Hardy, E.; Toledo, M.E.; Rodríguez, M.C.; Heynngnezz, L.; Rodriguez, A.; Baly, A.; Herrera, L.; Izquierdo, M.; et al. A synthetic conjugate polysaccharide vaccine against Haemophilus influenzae type b. Science 2004, 305, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, M.E.; Andrews, N.; Kaczmarski, E.B.; Miller, E. Efficacy of meningococcal serogroup C conjugate vaccine in teenagers and toddlers in England. Lancet 2001, 357, 195. [Google Scholar] [CrossRef]
- Jefferson, K. What drives bacteria to produce a biofilm? FEMS Microbiol. Lett. 2004, 236, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.; Kohn, S.; Hwang, S.H.; Hassett, D.J.; Sauer, K. Bdla, a chemotaxis regulator essential for biofilm dispersion in Pseudomonas aeruginosa. J. Bacteriol. 2006, 188, 7335–7343. [Google Scholar] [CrossRef] [PubMed]
- Hall-Stoodley, L.; Nistico, L.; Sambanthamoorthy, K.; Dice, B.; Nguyen, D.; Mershon, W.J.; Johnson, C.; Hu, F.Z.; Stoodley, P.; Ehrlich, G.D.; et al. Characterization of biofilm matrix, degradation by DNAse treatment and evidence of capsule downregulation in Streptococcus pneumoniae clinical isolates. BMC Microbiol. 2008, 8, 173. [Google Scholar] [CrossRef] [PubMed]
- Tetz, G.V.; Artemenko, N.K.; Tetz, V.V. Effect of DNAse and antibiotics on biofilm characteristics. Antimicrob. Agents Chemother. 2009, 53, 1204–1209. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Accavitti, M.A.; Bryers, J.D. Inhibition of biofilm formation by monoclonal antibodies against Staphylococcus epidermidis RP62A accumulation-associated protein. Clin. Diagn. Lab. Immunol. 2005, 12, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Novotny, L.A.; Jurcisek, J.A.; Goodman, S.D.; Bakaletz, L.O. Monoclonal antibodies against DNA-binding tips of DNABII proteins disrupt biofilms in vitro and induce bacterial clearance in vivo. EBioMedicine 2016, 10, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Ray, V.A.; Hill, P.J.; Stover, K.C.; Roy, S.; Sen, C.K.; Yu, L.; Wozniak, D.J.; DiGiandomenico, A. Anti-Psl targeting of Pseudomonas aeruginosa biofilms for neutrophil-mediated disruption. Sci. Rep. 2017, 7, 16065. [Google Scholar] [CrossRef] [PubMed]
- Skurnik, D.; Davis, J.M.R.; Benedetti, D.; Moravec, K.L.; Cywes-Bentley, C.; Roux, D.; Traficante, D.C.; Walsh, R.L.; Maira-Litràn, T.; Cassidy, S.K.; et al. Targeting pan-resistant bacteria with antibodies to a broadly conserved surface polysaccharide expressed during infection. J. Infect. Dis. 2012, 205, 1709–1718. [Google Scholar] [CrossRef] [PubMed]
- Skurnik, D.; Cywes-Bentley, C.; Pier, G.B. The exceptionally broad-based potential of active and passive vaccination targeting the conserved microbial surface polysaccharide PNAG. Expert Rev. Vaccines 2016, 15, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Wood Thomas, K. Strategies for combating persister cell and biofilm infections. Microb. Biotechnol. 2017, 10, 1054–1056. [Google Scholar] [CrossRef] [PubMed]
- Chua, S.L.; Liu, Y.; Yam, J.K.H.; Chen, Y.; Vejborg, R.M.; Tan, B.G.C.; Kjelleberg, S.; Tolker-Nielsen, T.; Givskov, M.; Yang, L. Dispersed cells represent a distinct stage in the transition from bacterial biofilm to planktonic lifestyles. Nat. Commun. 2014, 5, 4462. [Google Scholar] [CrossRef] [PubMed]
- Uppuluri, P.; Lopez-Ribot, J.L. Go forth and colonize: Dispersal from clinically important microbial biofilms. PLoS Pathog. 2016, 12, e1005397. [Google Scholar] [CrossRef] [PubMed]
- Sauer, K.; Cullen, M.C.; Rickard, A.H.; Zeef, L.A.H.; Davies, D.G.; Gilbert, P. Characterization of nutrient-induced dispersion in Pseudomonas aeruginosa PAO1 biofilm. J. Bacteriol. 2004, 186, 7312–7326. [Google Scholar] [CrossRef] [PubMed]
- Kragh, K.N.; Hutchison, J.B.; Melaugh, G.; Rodesney, C.; Roberts, A.E.L.; Irie, Y.; Jensen, P.Ø.; Diggle, S.P.; Allen, R.J.; Gordon, V.; et al. Role of multicellular aggregates in biofilm formation. mBio 2016, 7, e00237-16. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Reddy, D.N. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Pathogen | Disease | Colonization Site | Incidence Rate A | Fatality Rate A |
|---|---|---|---|---|
| Streptococcus pneumoniae | Pneumonia | Nasopharynx | 9.5 A | 1.14 A |
| Staphylococcus aureus (MRSA) | Skin infection | Nasopharynx, Skin | 22.72 A | 2.88 A |
| Group A Streptococcus | Strep throat | Pharynx | 5.8 A | 0.58 A |
| Haemophilus influenzae | Bacteremia | Nasopharynx | 1.99 A | 0.29 A |
| Neisseria meningitidis | Meningitis | Nasopharynx | 0.12 A | 0.01 A |
| Legionellosis | Atypical pneumonia | Lungs | 1.42 A | 0.1 A |
| Moraxella catarrhalis | Otitis media | Nasopharynx | N/A | 0 A |
| Group B Streptococcus | Septicemia | Gastrointestinal tract | 9.6 A | 0.53 A |
| Porphyromonas gingivalis | Periodontal disease | Oral Cavity | 9.24 B | - |
| Escherichia coli Pseudomonas aeruginosa Klebsiella pneumoniae | Catheter- Associated Urinary Tract Infection (CAUTI) | Bladder Catheter | 3.3 C | 17.3 C |
| Ventilator-Associated Pneumonia (VAP) | Ventilator | 3.3 C | 15.2 C | |
| Escherichia coli Staphylococcus aureus Pseudomonas aeruginosa | Prosthetic Joint Infections (PJI) | Prosthetic Joints (e.g., hip, knee) | 1.52.5 D | 2.5 D |
| Target | Bacteria | Anti-Microbial Strategy | Reference |
|---|---|---|---|
| Anti-Adhesion Phenotype Strategies | |||
| Type I Pili | Escherichia coli | Pilicide ec240 | [20] |
| SAMan | [21] | ||
| P-fimbrate | Escherichia coli | Synthetic galabinose | [22] |
| Spy0128 and Spy0130 | Group A Streptococcus | Vaccination | [23] |
| StrA | Streptococcus mutans | Morin | [24] |
| StrA | Staphylococcus aureus | pyrazolethione and pyridazinone | [25] |
| Anti-Biofilm Phenotype Strategies | |||
| AHL Molecules | Pseudomonas aeruginosa | SsoPox-W263I | [26] |
| c-di-GMP | Stenotrophomonas maltophilia | BsmR | [27] |
| LuxS | Streptococcus pneumoniae | CRISPR | [28,29] |
| PIA | Staphylococcus | dispersin B | [30] |
| eDNA | Pseudomonas aeruginosa | DNAse I (Pulmozyme®) | [31] |
| PNAG | S. aureus | Monoclonal Antibody | [32] |
| Persister Cells | Escherichia coli | Mitomycin C | [33] |
| Persister Cells | Pseudomonas aeruginosa | Cisplatin | [34] |
| Persister Cells | Pseudomonas aeruginosa Escherichia coli | cis-2-Decenoic Acid | [35] |
| Anti-Dispersed Bacteria Phenotype Strategies | |||
| GlpO | Streptococcus pneumoniae | Vaccine with GlpO Antigen | [36,37] |
| PncO | Streptococcus pneumoniae | Vaccine with PncO Antigen | [36,37] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beitelshees, M.; Hill, A.; Jones, C.H.; Pfeifer, B.A. Phenotypic Variation during Biofilm Formation: Implications for Anti-Biofilm Therapeutic Design. Materials 2018, 11, 1086. https://doi.org/10.3390/ma11071086
Beitelshees M, Hill A, Jones CH, Pfeifer BA. Phenotypic Variation during Biofilm Formation: Implications for Anti-Biofilm Therapeutic Design. Materials. 2018; 11(7):1086. https://doi.org/10.3390/ma11071086
Chicago/Turabian StyleBeitelshees, Marie, Andrew Hill, Charles H. Jones, and Blaine A. Pfeifer. 2018. "Phenotypic Variation during Biofilm Formation: Implications for Anti-Biofilm Therapeutic Design" Materials 11, no. 7: 1086. https://doi.org/10.3390/ma11071086
APA StyleBeitelshees, M., Hill, A., Jones, C. H., & Pfeifer, B. A. (2018). Phenotypic Variation during Biofilm Formation: Implications for Anti-Biofilm Therapeutic Design. Materials, 11(7), 1086. https://doi.org/10.3390/ma11071086