Modification of a Shockley-Type Surface State on Pt(111) upon Deposition of Gold Thin Layers

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Calculation Details
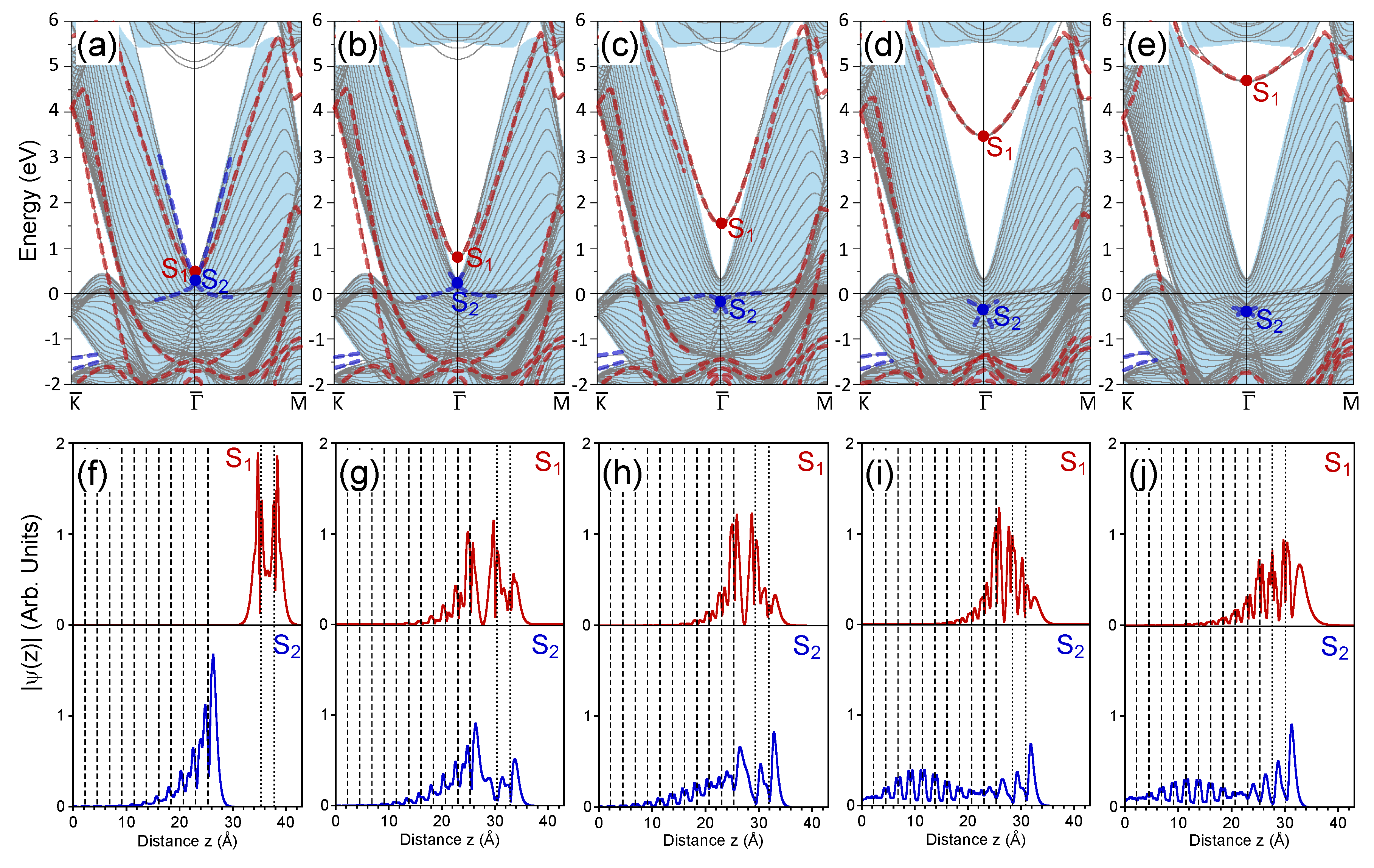
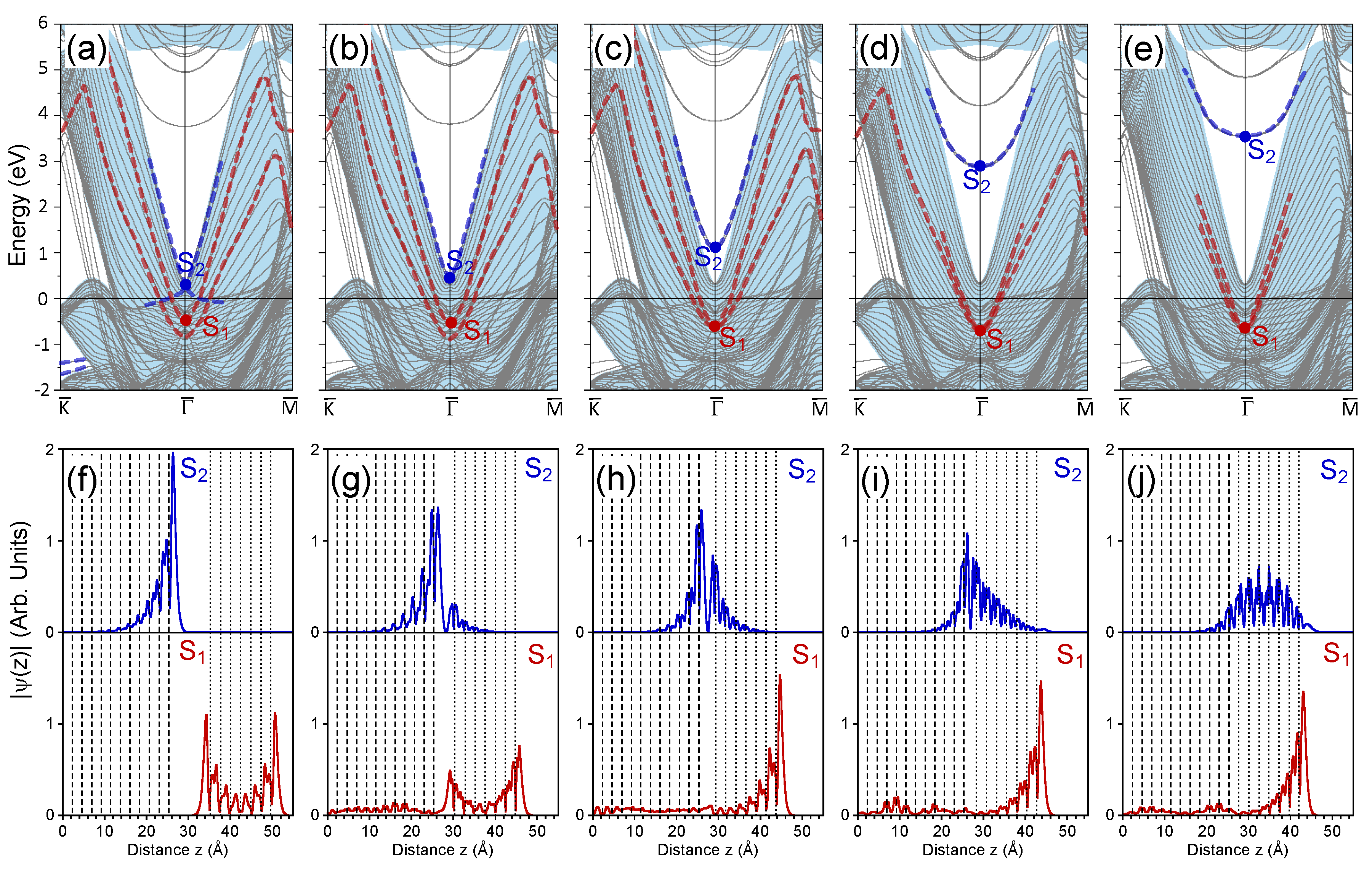
3. Calculation Results and Discussion
3.1. 1 ML Au/Pt(111)
3.2. 2 ML Au/Pt(111)
3.3. 3 ML Au/Pt(111)
3.4. 7 ML Au/Pt(111)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| STM | Scanning tunneling microscopy |
| SBZ | Surface Brillouin zone |
| ML | Monolayer |
| VASP | Vienna Ab-initio Simulation Package |
| GGA | Generalized gradient approximation |
| LDA | Local density approximation |
References
- Zangwill, A. Physics at Surfaces; Cambridge University Press: Cambridge, UK, 1988; ISBN 978-0521347525. [Google Scholar]
- Davison, S.G.; Stȩślicka, M. Basic Theory of Surface States; Oxford University Press: Oxford, UK, 1992; ISBN 9780198518969. [Google Scholar]
- Lüth, H. Surface and Interfaces of Solids; Springer: Berlin, Germany, 2010; ISBN 978-3-642-13591-0. [Google Scholar]
- Echenique, P.M.; Pendry, J.B. Existence of detection of Rydberg states at surfaces. J. Phys. C 1978, 11, 2065–2075. [Google Scholar] [CrossRef]
- Höfer, U.; Shumay, I.L.; Reuss, C.; Thomann, U.; Wallauer, W.; Fauster, T. Time-resolved coherent photoelectron spectroscopy of quantized electronic states on metal surfaces. Science 1997, 277, 1480–1482. [Google Scholar] [CrossRef]
- Echenique, P.M.; Berndt, R.; Chulkov, E.V.; Fauster, T.; Goldmann, A.; Höfer, U. Decay of electronic excitations at metal surfaces. Surf. Sci. Rep. 2004, 52, 219–318. [Google Scholar] [CrossRef]
- Feibelman, P.J.; Hamann, D.R. Modification of transition-metal electronic structures by P, S, Cl, an Li adatoms. Surf. Sci. 1985, 149, 48–66. [Google Scholar] [CrossRef]
- Bartynski, R.A.; Jepsen, E.; Gustafsson, T.; Plummer, E.W. Angle-resolved. photoemission investigation of the electronic structure of Be surface states. Phys. Rev. B 1985, 32, 1921–1926. [Google Scholar] [CrossRef]
- Chulkov, E.V.; Silkin, V.M.; Shirykalov, E.N. Surface electronic structure of Be(0001) and Mg(0001). Surf. Sci. 1987, 188, 287–300. [Google Scholar] [CrossRef]
- Kevan, S.; Gaylord, R. High-resolution photoemission study of the electronic structure of the noble-metal (111) surfaces. Phys. Rev. B 1987, 36, 5809–5818. [Google Scholar] [CrossRef]
- Lashell, S.; McDougall, B.; Jensen, E. Spin splitting of an Au(111) surface state band observed with angle resolved photoelectron spectroscopy. Phys. Rev. Lett. 1996, 77, 3419–3422. [Google Scholar] [CrossRef] [PubMed]
- Reinert, F.; Nicolay, G.; Schmidt, S.; Ehm, D.; Hüfner, S. Direct measurements of the L-gap surface states on the (111) face of noble metals by photoelectron spectroscopy. Phys. Rev. B 2001, 63, 115415. [Google Scholar] [CrossRef]
- Reinert, F. Spin-orbit interaction in the photoemission spectra of noble metal surface states. Phys. J. Condens. Matter 2003, 15, S693–S705. [Google Scholar] [CrossRef]
- Hoesch, M.; Muntwiler, M.; Petrov, V.N.; Hengsberger, M.; Patthey, L.; Shi, M.; Falub, M.; Greber, T.; Osterwalder, J. Spin structure of the Shockley surface state on Au(111). Phys. Rev. B 2004, 69, 241401. [Google Scholar] [CrossRef]
- Yan, B.; Stadtmüller, B.; Haag, N.; Jakobs, S.; Seidel, J.; Jungkenn, D.; Mathias, S.; Cinchetti, M.; Aeschlimann, M.; Felser, C. Topological states on the gold surface. Nat. Commun. 2015, 6, 10167. [Google Scholar] [CrossRef] [PubMed]
- Hulbert, S.L.; Johnson, P.D.; Weinert, M. High-resolution inverse-photoemission study of the Pd(111) surface. Phys. Rev. B 1986, 34, 3670–3673. [Google Scholar] [CrossRef]
- Di, W.; Smith, K.E.; Kevan, S.D. Angle-resolved photoemission study of the clean and hydrogen-covered Pt(111) surface. Phys. Rev. B 1992, 45, 3652–3658. [Google Scholar] [CrossRef]
- Donath, M.; Passek, F.; Dose, V. Surface state contribution to the magnetic mooment of Ni(111). Phys. Rev. Lett. 1993, 70, 2802–2805. [Google Scholar] [CrossRef] [PubMed]
- Passek, F.; Donath, M. Magnetic surface state becomes nonmagnetic by oxygen adsorption. Phys. Rev. Lett. 1993, 71, 2122–2125. [Google Scholar] [CrossRef]
- Dong, W.; Kresse, G.; Furthmüller, J.; Hafner, J. Chemisorption of H on Pd(111): An ab initio approach with ultrasoft pseudopotentials. Phys. Rev. B 1996, 54, 2157–2166. [Google Scholar] [CrossRef]
- Heinrichsmeier, M.; Fleszar, A.; Hanke, W.; Eguiluz, A.G. Nonlocal density-functional calculations of the surface electronic structure of metals: Application to aluminum and palladium. Phys. Rev. B 1998, 57, 14974–14982. [Google Scholar] [CrossRef]
- Schäfer, A.; Shumay, I.L.; Wiets, M.; Weinelt, M.; Fauster, T.; Chulkov, E.V.; Silkin, V.M.; Echenique, P.M. Lifetimes of unoccupied surface states on Pd(111). Phys. Rev. B 2000, 61, 13159–13163. [Google Scholar] [CrossRef]
- Link, S.; Durr, H.A.; Bihlmayer, G.; Blügel, S.; Eberhardt, W.; Chulkov, E.V.; Silkin, V.M.; Echenique, P.M. Femtosecond electron dynamics of image-potential states on clean and oxygen-covered Pt(111). Phys. Rev. B 2001, 63, 115420. [Google Scholar] [CrossRef]
- Wiebe, J.; Meier, F.; Hashimoto, K.; Bihlmayer, G.; Blügel, S.; Ferriani, P.; Heinze, S.; Wiesendanger, R. Unoccupied surface state on Pt(111) revealed by scanning tunneling spectroscopy. Phys. Rev. B 2005, 72, 193406. [Google Scholar] [CrossRef]
- Sklyadneva, I.Y.; Heid, R.; Silkin, V.M.; Melzer, A.; Bohnen, K.P.; Echenique, P.M.; Fauster, T.; Chulkov, E.V. Unusually weak electron-phonon coupling in the Shockley surface state on Pd(111). Phys. Rev. B 2009, 80, 045429. [Google Scholar] [CrossRef]
- Memmel, N. Monitoring and modifying properties of metal surfaces by electronic surface states. Surf. Sci. Rep. 1998, 32, 91–163. [Google Scholar] [CrossRef]
- Lindgren, S.Å.; Walldén, L. Discrete valence-electron states in thin metal overlayers on a metal. Phys. Rev. Lett. 1987, 59, 3003–3006. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, S.Å.; Walldén, L. Discrete valence electron states for Na overlayers on Cu(111). Phys. Rev. B 1988, 38, 3060–3067. [Google Scholar] [CrossRef]
- Chulkov, E.V.; Silkin, V.M. Electronic structure of the Al(001) surface with adsorbed Na half monolayer. Surf. Sci. 1989, 215, 385–393. [Google Scholar] [CrossRef]
- Díez-Muiño, R.; Sánchez-Portal, D.; Silkin, V.M.; Chulkov, E.V.; Echenique, P.M. Time-dependent electron phenomena at surafces. Proc. Natl. Acad. Sci. USA 2011, 108, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Stampfl, C.; Scheffler, M. Theory of alkali-metal adsorption on close-packed metal surfaces. Surf. Rev. Lett. 1995, 2, 317–343. [Google Scholar] [CrossRef]
- Diehl, R.; McGrath, R. Structural studies of alkali metal adsorption and coadsorption on metal surfaces. Surf. Sci. Rep. 1996, 23, 43–171. [Google Scholar] [CrossRef]
- Carlsson, J.M.; Hellsing, B. First-principles investigation of the quantum-well system Na on Cu(111). Phys. Rev. B 2000, 61, 13973–13982. [Google Scholar] [CrossRef]
- Kirchmann, P.S.; Wolf, M.; Dil, J.H.; Horn, K.; Bovensiepen, U. Quantum size effects in Pb/Si(111) investigated by laser-induced photoemission. Phys. Rev. B 2007, 76, 075406. [Google Scholar] [CrossRef]
- Algdal, J.; Balasubramanian, I.; Breitholtz, M.; Chis, V.; Hellsing, B.; Lindgren, S.A.; Wallden, L. Sodium and potassium monolayers on Be(0001) investigated by photoemission and electronic structure calcualtions. Phys. Rev. B 2008, 78, 085102. [Google Scholar] [CrossRef]
- Caravati, S.; Trioni, M.I. Structural and electronic properties of Na/Cu(111) at different coverages by first principles. Eur. Phys. J. B 2010, 75, 101–106. [Google Scholar] [CrossRef]
- Kirchmann, P.S.; Rettig, L.; Zubizarreta, X.; Silkin, V.M.; Chulkov, E.V.; Bovensiepen, U. Quasiparticle lifetimes in metallic quantum-well nanostructures. Nat. Phys. 2010, 6, 782–785. [Google Scholar] [CrossRef]
- Rybkin, A.G.; Shikin, A.M.; Marchenko, D.; Varykhalov, A.; Rader, O. Spin-dependent avoided-crossing effect on quantum-well states in Al/W(110). Phys. Rev. B 2012, 85, 045425. [Google Scholar] [CrossRef]
- Chiang, T.C. Photoemission studies of quantum well states in thin films. Surf. Sci. Rep. 2000, 39, 181–235. [Google Scholar] [CrossRef]
- Fischer, R.; Schuppler, S.; Fischer, N.; Fauster, T.; Steinmann, W. Image states and local work functin for Ag/Pd(111). Phys. Rev. Lett. 1993, 70, 654–657. [Google Scholar] [CrossRef]
- Fischer, R.; Fauster, T.; Steinmann, W. 3-dimensional localization of electrons on Ag islands. Phys. Rev. B 1993, 48, 15496–15499. [Google Scholar] [CrossRef]
- Lazić, P.; Crljen, Ž.; Brako, R. Localization and hybridization of the electronic states in thin films of Ag on V(100). Phys. Rev. B 2005, 71, 155402. [Google Scholar] [CrossRef]
- Moras, P.; Bihlmayer, G.; Vescovo, E.; Sheverdyaeva, P.M.; Papagno, M.; Ferrari, L.; Carbone, C. Spin-polarized confined states in Ag films on Fe (110). J. Phys. Coondens. Matter 2017, 29, 495806. [Google Scholar] [CrossRef]
- Chen, C.T.; Smith, N.V. Unoccupied surface states on clean and oxygen-covered Cu(110) and Cu(111). Phys. Rev. B 1989, 40, 7487–7490. [Google Scholar] [CrossRef]
- Bertel, E. Unoccupied electronic states in adsorbate systems. Appl. Phys. A 1991, 53, 356–368. [Google Scholar] [CrossRef]
- Lenac, Z.; Sunjic, M.; Conrad, H.; Kordesch, M.E. Image-potential states on clean and hydrogen-covered Pd surfaces—Analysis of a one-dimensional model. Phys. Rev. B 1987, 36, 9500–9506. [Google Scholar] [CrossRef]
- Rangelov, G.; Memmel, N.; Bertel, E.; Dose, V. The bonding of hydrogen on nickel studied by inverse photoemission. Surf. Sci. 1990, 236, 250–258. [Google Scholar] [CrossRef]
- Sachtler, J.W.A.; Van Hove, M.A.; Biberian, J.P.; Somorjai, G.A. Enhanced reactivity of ordered monolayers of gold on Pt(100) and platinum on Au(100) single-crystal surfaces. Phys. Rev. Lett. 1980, 45, 1601–1603. [Google Scholar] [CrossRef]
- Sachtler, J.W.A.; Somorjai, G.A. Influence of ensemble size on CO chemisorption and catalytic n-hexane conversation by Au-Pt(111) bimetallic single-crystal surfaces. J. Catal. 1983, 81, 77–94. [Google Scholar] [CrossRef]
- Yeates, R.C.; Somorjai, G.A. Surface-structure sensitivity of alloy catalysis: Catalytic conversion of n-hexane over Au-Pt(111) and Au-Pt(100) alloy crystal surfaces. J. Catal. 1987, 103, 208–212. [Google Scholar] [CrossRef]
- Eyrich, M.; Diemant, T.; Hartmann, H.; Bansmann, J.; Behm, R.J. Interaction of CO with structurally well-defined monolayer PtAu/Pt(111) surface alloys. J. Phys. Chem. C 2012, 116, 11154–11165. [Google Scholar] [CrossRef]
- Krupski, K.; Moors, M.; Jozwik, P.; Kobiela, T.; Krupski, A. Structure determination of Au on Pt(111) surface: LEED, STM and DFT study. Materials 2015, 8, 2935–2952. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Available online: http://www.vasp.at (accessed on 17 December 2018).
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Koelling, D.D.; Harmon, B.N. A technique for relativistic spin-polarised calculations. J. Phys. C 1977, 10, 3107. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillonin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Wolfschmidt, H.; Baier, C.; Gsell, S.; Fischer, M.; Schreck, M.; Stimming, U. STM, SECPM, AFM and electrochemistry on single crystalline surfaces. Materials 2010, 3, 4196–4213. [Google Scholar] [CrossRef] [PubMed]
- Kesmodel, L.L.; Somorjai, G.A. Structure determination of the platinum (111) crystal face by low-energy-electron diffraction. Phys. Rev. B 1975, 11, 630–637. [Google Scholar] [CrossRef]
- Adams, D.L.; Nielsen, H.B.; van Hove, M.A. Quantitative analysis of low-energy-electron-diffraction: Application to Pt(111). Phys. Rev. B 1979, 20, 4789–4806. [Google Scholar] [CrossRef]
- Feder, R.; Pleyer, H.; Bauer, P.; Mueller, N. Spin polarization in low-energy electron diffraction: Surface analysis of Pt(111). Surf. Sci. 1981, 109, 419–434. [Google Scholar] [CrossRef]
- Hayek, K.; Glassl, H.; Gutmann, A.; Leonhard, H.; Prutton, M.; Tear, S.P.; Weltoncook, M.R. A LEED analysis of the structure of Pt(111)( × )R30o-S. Surf. Sci. 1985, 152, 419–425. [Google Scholar] [CrossRef]
- Ogletree, D.F.; van Hove, M.A.; Somorjai, G.A. LEED intensity analysis of the structure of clean Pt(111) and of CO adsorbed on Pt(111) in the c(4 × 2) arrangement. Surf. Sci. 1986, 173, 351–365. [Google Scholar] [CrossRef]
- Materer, N.; Starke, U.; Barbieri, A.; Doll, R.; Heinz, K.; van Hove, M.A.; Somorjai, G.A. Reliability of detailed LEED structural analysis: Pt(111) and Pt(111-p(2 × 2)-O. Surf. Sci. 1995, 325, 207–222. [Google Scholar] [CrossRef]
- Waseda, Y.; Hirata, K.; Ohtani, M. High-temperature thermal expansion of platinum, tantalum, molybdenum, and tungsten measured by X-ray diffraction. High Temp. High Press. 1975, 7, 221–226. [Google Scholar]
- Felici, R.; Pedio, M.; Borgatti, F.; Iannotta, S.; Capozi, M.; Ciullo, G.; Stierle, A. X-ray diffraction characterization of Pt(111) surface nanopattering induced by C60 adsorption. Nat. Mater. 2005, 4, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Feibelman, P.J. First-principles calculations of stress induced by gas adsorption on Pt(111). Phys. Rev. B 1997, 56, 2175–2182. [Google Scholar] [CrossRef]
- Da Silva, J.L.F.; Stampfl, C.; Scheffler, M. Converged properties of clean metal surfaces by all electron first-principles calculations. Surf. Sci. 2006, 600, 703–715. [Google Scholar] [CrossRef]
- Singh-Miller, N.E.; Marzari, N. Surface energies, work functions, and surface relaxations of low-index metallic surfaces from first principles. Phys. Rev. B 2009, 80, 235407. [Google Scholar] [CrossRef]
- Herrera-Suárez, H.J.; Rubio-Ponce, A.; Olguín, D. Electronic band structure of the Pt(111) surface: An ab initio and tight-binding Study—I. Comp. Mater. Sci. 2012, 56, 141–146. [Google Scholar] [CrossRef]
- Dal Corso, A. Clean Ir(111) and Pt(111) electronic surface states: A first-principle fully relativistic. investigation. Surf. Sci. 2015, 637, 106–115. [Google Scholar] [CrossRef]
- Silkin, I.V.; Koroteev, Y.M.; Echenique, P.M.; Chulkov, E.V. Formation of surface and quantum-well states in ultra thin Pt films on the Au(111) surface. Materials 2017, 10, 1409. [Google Scholar] [CrossRef]
- Bychkov, Y.A.; Rashba, E.I. Properties of a 2D electron-gas with lifted spectral degeneracy. JETP Lett. 1984, 39, 78–81. [Google Scholar]
- Ramstad, A.; Raaen, S.; Barrett, N. Electronic structure of the La–Pt(111) surface alloy. Surf. Sci. 2000, 448, 179–186. [Google Scholar] [CrossRef]
- Kobiela, T.; Moors, M.; Linhart, W.; Cebula, I.; Krupski, A.; Becker, C.; Wandelt, K. Characterization of bimetallic Au-Pt(111) surfaces. Thin Solid Films 2010, 518, 3650–3657. [Google Scholar] [CrossRef]
- Frantzeskakis, E.; Pons, S.; Crepaldi, A.; Brune, H.; Kern, K.; Grioni, M. Ag-coverage-dependent symmetry of the electronic states of the Pt(111)-Ag-Bi interface: The ARPES view of a structural transition. Phys. Rev. B 2011, 84, 245443. [Google Scholar] [CrossRef]
- Silkin, V.M.; Zhao, J.; Guinea, F.; Chulkov, E.V.; Echenique, P.M.; Petek, H. Image potential states in graphene. Phys. Rev. B 2009, 80, 121408. [Google Scholar] [CrossRef]
- Chulkov, E.V.; Silkin, V.M.; Echenique, P.M. Image potential states on metal surfaces:binding energies and wave functions. Surf. Sci. 1999, 437, 330. [Google Scholar] [CrossRef]
- Sarría, I.; Osma, J.; Chulkov, E.V.; Pitarke, J.M.; Echenique, P.M. Self-energy of image states on copper surfaces. Phys. Rev. B 1999, 60, 11795–11803. [Google Scholar] [CrossRef]
- Mazzarello, R.; Dal Corso, A.; Tosatti, E. Spin-orbit modifications and splittings of deep surface states on clean Au(111). Surf. Sci. 2008, 602, 893–905. [Google Scholar] [CrossRef]
- Sheverdyaeva, P.M.; Requist, R.; Moras, P.; Mahatha, S.K.; Papagno, M.; Ferrari, L.; Tosatti, E.; Carbone, C. Energy-momentum mapping of d-derived Au(111) states in a thin film. Phys. Rev. B 2016, 93, 035113. [Google Scholar] [CrossRef]
- Chulkov, E.V.; Silkin, V.M.; Machado, M. Quasiparticle dynamics on metal surfaces. Surf. Sci. 2001, 482, 693–701. [Google Scholar] [CrossRef]
- Varykhalov, A.; Marchenko, D.; Scholz, M.R.; Rienks, E.D.L.; Kim, T.K.; Bihlmayer, G.; Sánchez-Barriga, J.; Rader, O. Ir(111) surface state with giant Rashba splitting persists under graphene in air. Phys. Rev. Lett. 2012, 108, 066804. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silkin, I.V.; Koroteev, Y.M.; Silkin, V.M.; Chulkov, E.V. Modification of a Shockley-Type Surface State on Pt(111) upon Deposition of Gold Thin Layers. Materials 2018, 11, 2569. https://doi.org/10.3390/ma11122569
Silkin IV, Koroteev YM, Silkin VM, Chulkov EV. Modification of a Shockley-Type Surface State on Pt(111) upon Deposition of Gold Thin Layers. Materials. 2018; 11(12):2569. https://doi.org/10.3390/ma11122569
Chicago/Turabian StyleSilkin, Igor V., Yury M. Koroteev, Vyacheslav M. Silkin, and Evgueni V. Chulkov. 2018. "Modification of a Shockley-Type Surface State on Pt(111) upon Deposition of Gold Thin Layers" Materials 11, no. 12: 2569. https://doi.org/10.3390/ma11122569
APA StyleSilkin, I. V., Koroteev, Y. M., Silkin, V. M., & Chulkov, E. V. (2018). Modification of a Shockley-Type Surface State on Pt(111) upon Deposition of Gold Thin Layers. Materials, 11(12), 2569. https://doi.org/10.3390/ma11122569