1. Introduction
The understanding of friction origins and the search for new effective lubricants have been the focus of tribology studies [
1]. Layered bulk materials, such as graphite, hexagonal boron nitride (
h-BN), and molybdenum disulfide (MoS
2), exhibit excellent lubricating properties and have been used to reduce friction and wear in mechanical systems for a long time [
2]. The two-dimensional (2D) strong covalent bonding in plane and the weak van der Waals (vdW) interaction between adjacent layers can effectively explain the lubricating properties of these traditional solid lubricants [
2,
3]. Surprisingly, these multilayer or even single-layer bulk materials possess excellent frictional characteristics due to their high load carrying capacity and passivating effect, which can serve as protective coating films or nanolubricants and are used in several systems [
4,
5,
6,
7,
8]. In addition to the homogeneous structure, recent studies have found that heterostructures made by the stacking of different 2D atomic layer crystals, such as graphene/
h-BN and graphene/MoS
2, also exhibit unique electronic structures [
9,
10] and robust interfacial superlubricity [
11,
12]. The weak vdW interactions and naturally occurring lattice mismatch between two contacting structures is a key ingredient for this superlubricity phenomenon. All above discussions support that layered materials are not only excellent lubricants from the macroscale to the microscale, but also ideal models for investigations of the mechanism of friction.
Electronic structure calculation based on density functional theory (DFT) has become an effective method to study the tribological properties of solid interfaces [
13,
14,
15,
16,
17,
18,
19]. The earlier first-principles theory that tried to solve the friction problem was developed by Zhong et al., who studied the stick-slip motion of a single Pd atom over graphite [
13]. The model assumes that the potential energy is completely dissipated, but the dissipating process is not considered. Therefore, the maximum-friction model only can obtain major static frictional parameters, such as the maximal potential barrier, friction force, and coefficient of friction (COF). Wolloch et al. further presented a quasistatic model to obtain an approximation of nanofriction based on DFT [
14]. In their model, energy dissipation is considered by atomic relaxations. Zilibotti et al. provided a method to calculate the ideal interfacial shear strength by constructing the potential energy surface (PES) that describes the variation of the adhesion energy between two surfaces in contact as a function of their relative lateral position [
15]. Recently, Restuccia et al. released a computational protocol to calculate the intrinsic tribological properties of a solid interface from first principles [
16], and the high-throughput
ab initio approach can be employed to easily and effectively calculate friction. Based on these models, DFT calculations have achieved great progress in friction research of solid materials. Cahangirov et al. determined the frictional figures of merit for a pair of layered honeycomb nanostructures by carrying out
ab initio calculations [
17]. They explained the ultralow interlayer friction in the view of critical stiffness. Gao et al. investigated the interlayer sliding potential of multilayer
h-BN and graphene. They found that interlayer sliding constraints can be employed to tune the contribution of electrostatic interactions and dispersive forces to the sliding energy profile [
18]. Reguzzoni et al. investigated the microscopic origin of the increase of friction under load by DFT calculations [
19]. They found that pressure-induced charge transfer from the interlayer region toward the near-layer regions is responsible for the increase of friction. More recently, Wolloch et al. constructed a connection between the intrinsic tribological properties and the electronic properties of a solid interface [
20]. They showed that the adhesion and frictional forces are dictated by the charge redistribution occurring due to the relative displacements of the two surfaces in contact. All of the abovementioned research works indicated that the electronic structure is a decisive factor in friction, and that DFT calculation can effectively reveal the frictional mechanism.
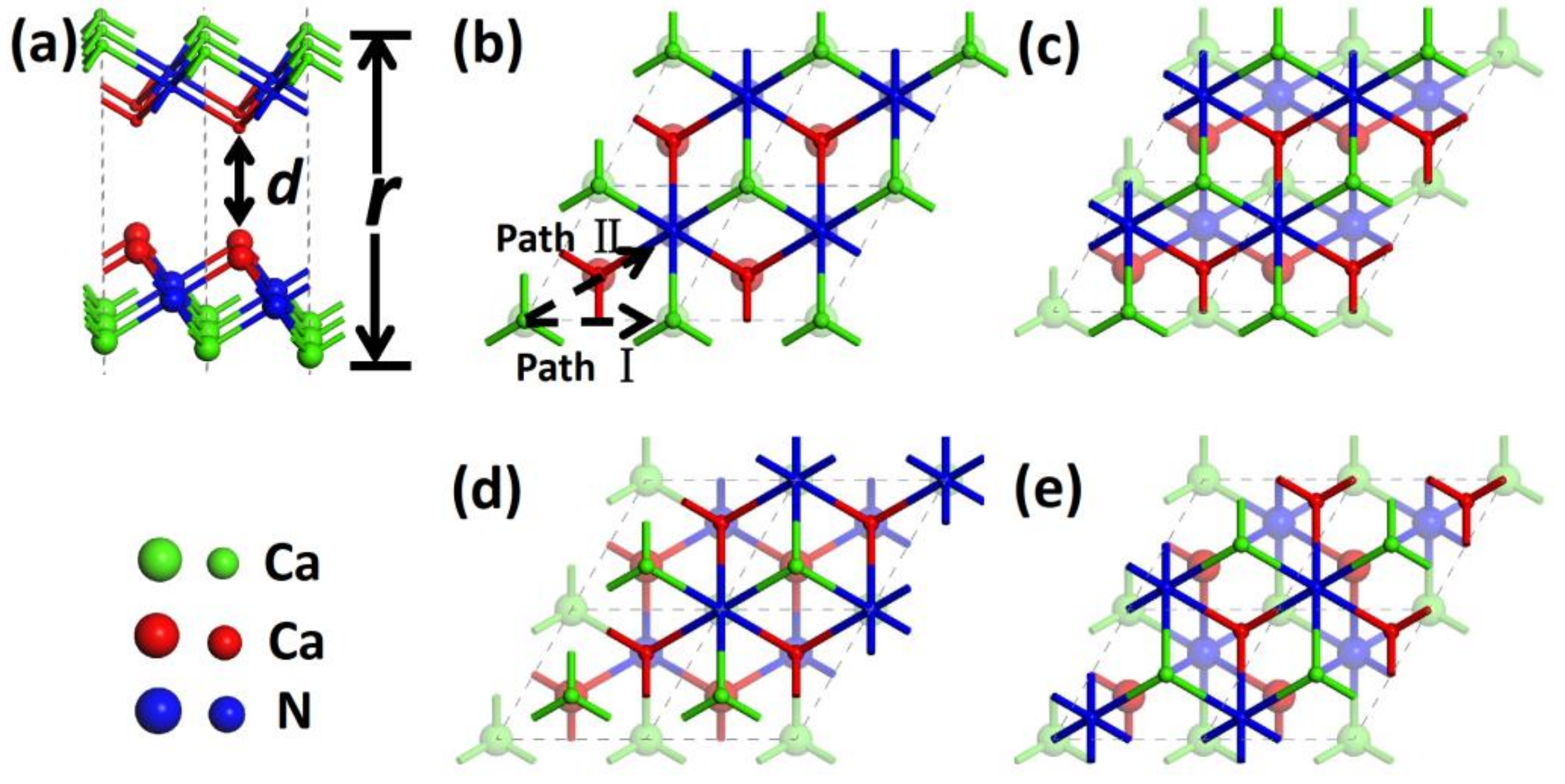
Recent studies reported a new type of 2D-layered material of dicalcium nitride (Ca
2N), which has a hexagonal layered structure in the
space group with high
c/a ratio, where
a and
c are in-plane and out-of-plane unit cell dimensions, respectively [
21,
22,
23,
24,
25,
26]. The layered unit (Ca-N-Ca) is closely packed, and adjacent units have a large separation of about 4 Å (
Figure 1a). Electronic structure calculations show that Ca
2N is a kind of 2D electride with a formula of [Ca
2N]
+·e
−, in which the [Ca
2N]
+ unit acts as a positively charged ionic slab and the residual electrons trapped in the interlayer space serve as anions [
23,
24]. Therefore, Ca
2N can be regarded as a crystalline structure combined by the Coulomb force between [Ca
2N]
+ and e
−. Experimental and theoretical investigations further found that Ca
2N exhibits excellent 2D transport characteristics with a high electron mobility (520 cm
2 V
−1 S
−1) and long mean scattering time (0.6 picoseconds) with a mean free path of 0.12 micrometers [
23]. The delocalized homogeneous electrons in the 2D interlayer space are responsible for the excellent transport characteristics, which mainly conduct through the 2D space instead of the [Ca
2N]
+ layer [
23,
24]. Lee et al. also calculated the mechanical properties of the structure. They found that the in-plane stiffness is sufficient to support a Ca
2N monolayer and thus proposed the possibility of obtaining a Ca
2N monolayer via mechanical exfoliation [
24]. Zhao et al. and Guan et al. further investigated the electronic and phononic structures of monolayer and few-layer Ca
2N, respectively [
25,
26]. They confirmed that the monolayer and few-layer Ca
2N samples are stable and have great potential in nanotechnology. It is well known that for a layered structure, larger interlayer distance and high in-plane stiffness are the basic requirements for a 2D solid lubricant [
27,
28]. Considering that Ca
2N possesses layered geometry, in-plane mechanical properties, and interlayer distance similar to those of the traditional layered lubricant graphite, excellent friction behaviors are expected. However, the interlayer binding styles are dramatically different for the two kinds of layered materials. Therefore, the special interlayer friction mechanisms of Ca
2N need to be revealed. However, to our knowledge, there is no experimental or theoretical investigation on the friction properties of Ca
2N and other alkaline earth subnitrides, such as Sr
2N and Ba
2N. Thus, elucidating the tribology properties is important to extend Ca
2N applications.
In the study, we investigated the interlayer friction properties between two layers of Ca2N by applying first-principles calculations. An interesting finding was that the ionic crystal Ca2N has comparable friction force with that of the traditional layered lubricants bonded by vdW interactions. The results will contribute to understanding atomic scale friction and extending the range of 2D lubricants.
3. Results and Discussion
The calculated in-plane lattice constants and layer thickness of Ca
2N monolayer are 3.566 and 2.515 Å, respectively. These values are very close to the experimental values of the corresponding bulk structure (3.66 and 2.51 Å) and the other calculation results of the monolayer Ca
2N (3.562 and 2.516 Å) [
21,
22,
23,
24,
25,
26]. Based on the optimized structure, two sheets of Ca
2N monolayer were placed to slide against each other along two high symmetrical paths to model the friction process, as shown in
Figure 1. Along path I, the two sheets of Ca
2N alternately encountered the top and bridge stackings. Meanwhile, the top, hollow, and bridge stackings appeared on path II periodically. Therefore, the top, hollow, and bridge stackings are the three key configurations for friction calculations.
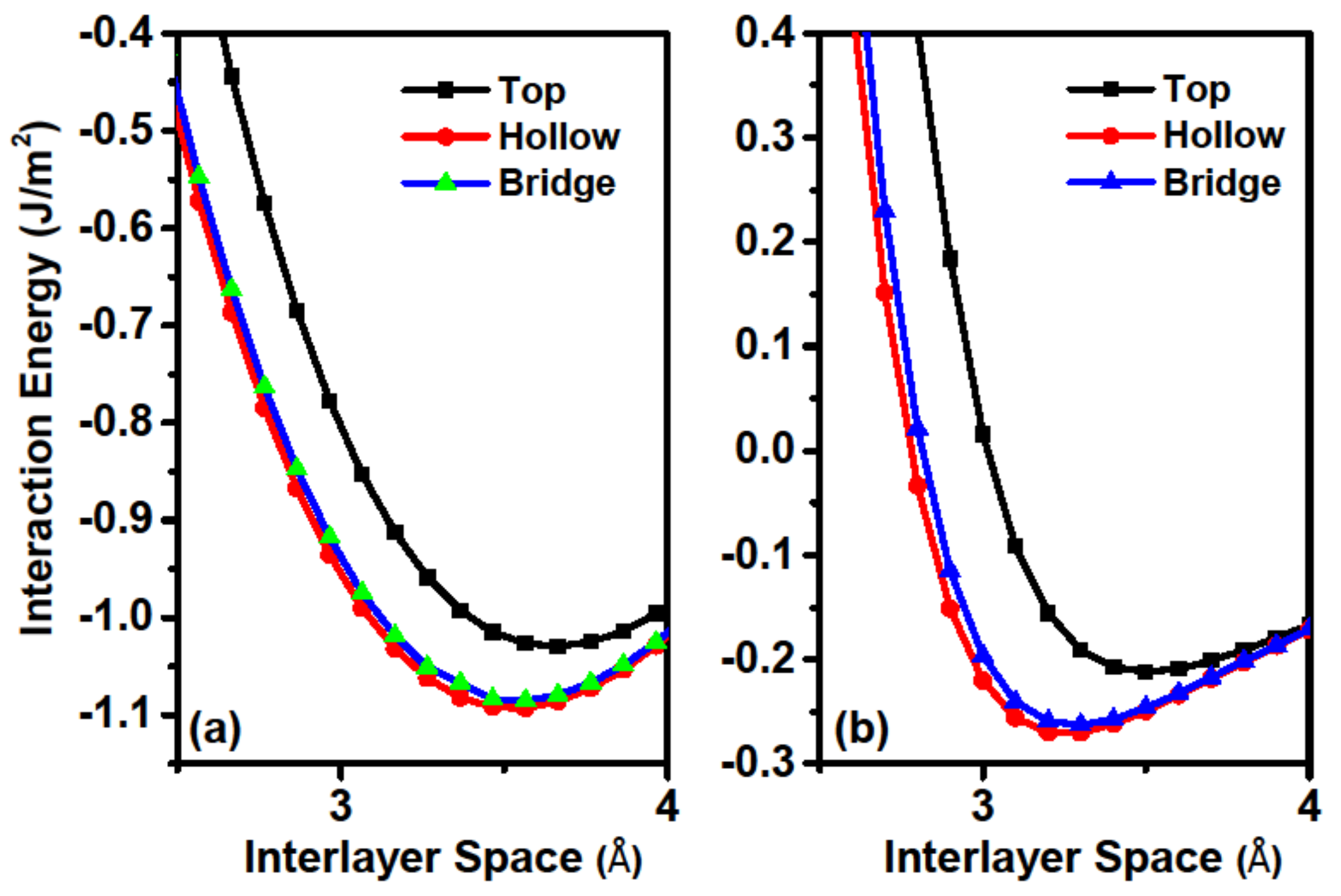
As friction is closely related to the interfacial interaction, we firstly calculated the interaction energy (IE) of the sliding system. IE is defined as the energy of the combined system minus the energy sum of the component films, and was calculated by using the formula
, where
is the total energy of the two contacting films at the distance of
r and
is the energy of the separate film. The vertical distance between the bottom and top Ca layers is defined as
r, as shown in
Figure 1a. The load effect was applied by setting different
r values. For each interlayer distance
r, only the Ca atoms in the bottom layer of the lower slab and the topmost layer of the upper slab were kept frozen, whereas all other atoms were relaxed in all of our calculations. According to the definition of IE, a more negative IE indicates better stability. The calculated IEs as a function of
r for top, bridge, and hollow stackings are shown in
Figure 2a. For comparison, the IEs for the bilayer graphene system are also presented in
Figure 2b. Comparisons of IEs in different stackings reveal that both systems have the weakest interaction at the top stacking, followed by the bridge one, while the hollow stacking has the strongest values. This behavior occurs because interfacial Ca atoms exhibit large spaces for movement and avoid repulsive forces at both the hollow and bridge stackings. These results indicate that the hollow stacking has the largest binding energy (the absolute value of the minimum IE) and is the most stable stacking configuration. To reveal the difference of interlayer binding style between the two systems, we compared the IEs for the two systems. The most striking difference between the two systems is that the IEs for the Ca
2N system (approximately 1 J/m
2) is about five times larger than that of the graphene systems (typical physical adsorption of about 0.1 J/m
2). Therefore, the chemical interlayer bonding character of the Ca
2N system is dramatically different than the vdW interaction within the graphite system [
34]. We next considered the relative difference of IEs between the top and hollow stackings for the two systems. The IE difference at the equilibrium adsorption position is about 0.063 J/m
2 for Ca
2N system, which is comparable with that of the graphene system (about 0.058 J/m
2). We also examined the influence of vdW on the IE in Ca
2N, and found that the IE difference is virtually unchanged if PBE is used without vdW corrections. These results illustrate that although the two systems have different bonding energies, the difference of binding energy among different stackings is comparable. Interfacial space
d (as shown in
Figure 1a) is another important value for friction calculations, and the calculated values are 3.57 Å and 3.25 Å for Ca
2N and graphene, respectively. From the comparisons we can see that the Ca
2N system has a larger interfacial space than that of the graphene system. Therefore, excellent friction behaviors are expected for the Ca
2N system in the view of its IE and interfacial space.
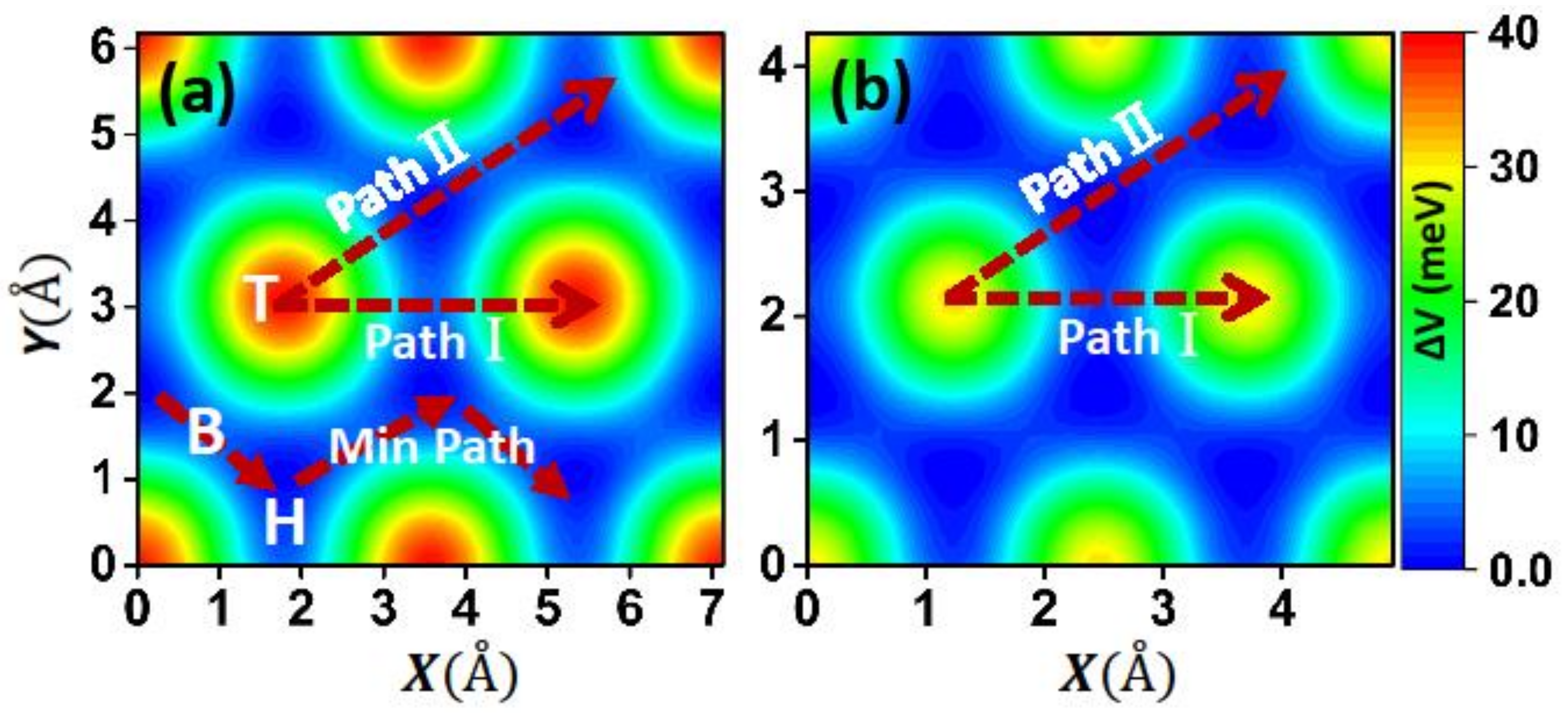
To gain an overall view of friction, we also constructed the static PES by calculating the interlayer IE for different relative lateral positions of the two films at the equilibrium distance [
14], as shown in
Figure 3. From the figure we can see that the two systems exhibit a similar pattern of PES, with the maximum at the top and minimum at the hollow sites. The maximum is about 40 meV for the Ca
2N system, which is 1.6 times larger than that of the graphene system (26 meV). However, the length of the sliding path and cell area of the Ca
2N system are 1.5 and 2 times larger than those of the graphene system, respectively. Therefore, the difference of the lateral force (
experienced by a surface unit cell during its displacement along the direction α) between the two systems should be subtler. We also presented the lowest energy sliding path, as shown in
Figure 3a, and the minimum barrier is only about 4 meV, which is comparable to that of the graphene system (about 3.2 meV). This indicates that Ca
2N could be considered a candidate for lubricant materials.
We next investigated the detailed frictional properties along the two chosen directions by Zhong’s method [
13], and more details of the calculating process can be found in our previous works [
34,
35,
36].
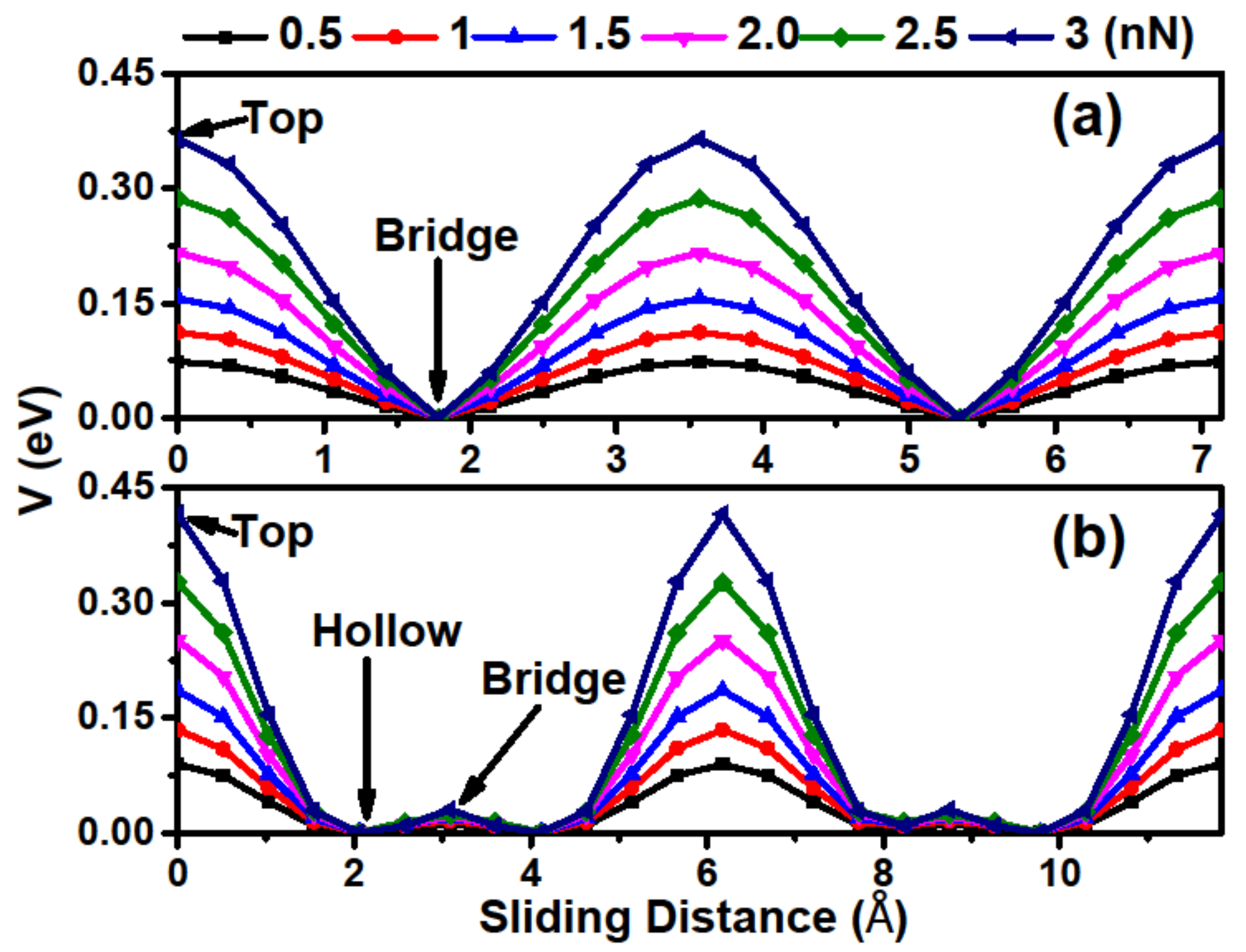
Figure 4 exhibits the potential barrier
V along the sliding path, in which the hollow stacking at path I and the bridge stacking at path II are set to 0 eV. From the figure, one can see that the relative
V curves periodically fluctuate along the sliding directions, with a maximum at the top stacking and a minimum at the bridge and hollow stackings for paths I and II, respectively. We also considered the load effect, which can be obtained by differentiating the fitted interaction energy function with respect to the interlayer distance [
34]. It was found that
V increases and its curve becomes steeper with increasing loads, indicating that relative sliding is difficult to achieve under high loads. Another important characteristic is that the disparity of the potential barrier amplitude between two paths is not obvious, suggesting that Ca
2N has an isotropous mean interlayer friction behavior.
The mean friction force <
f> was calculated by dividing the maximum potential barrier by the distance between the two nearest neighbor maximum energies in the sliding direction, as shown in
Figure 5. First of all, the curves of <
f> for both paths are almost coincident throughout the calculated normal forces, which further illustrates that the interfacial friction is isotropous for the Ca
2N system. By comparison, we also calculated the interfacial <
f> of the bilayer graphene system. From the comparisons of <
f> in
Figure 5, it can be seen that the Ca
2N system has lower friction than the graphene system for all chosen loads, which indicates that layered materials with stronger binding can have lower friction, and that friction is closely related to the IEs difference for different stackings but not the absolute IE. It should be noticed that Zhong’s method can successfully deal with the vdW binding layered materials, but it underestimates frictional forces in metallic systems [
14]. It is doubtful whether this method is suitable for the calculation of larger adsorption layered systems. However, as the constructed PBS of the Ca
2N system has the same tendency as the friction calculation by Zhong’s method, the calculated results here are reliable. It also should be emphasized that, although these results reveal static friction at 0 K, and the effects of velocity and temperature must affect the quantitative estimate, such effects will not break the significant ground state mechanism of atomic-level friction. Therefore, bilayer or multilayer Ca
2N systems may be promising candidates for nanolubricant or coating materials. Additionally, first-principles calculations have also been employed to predict two other 2D electride alkaline earth subnitrides, Sr
2N and Ba
2N [
21,
22], and it was found that they are stable down to monolayer thickness [
23]. As these materials have structures similar to that of Ca
2N, we can infer that this class of materials has excellent friction properties.
The vdW interlayer interaction is the main mechanism of traditional solid lubricants. Due to the different interlayer bonding styles, the ultralow friction mechanism of the Ca
2N system may be fundamentally different from that of the vdW layered lubricants. To reveal the ultralow friction mechanism, we calculated the electronic structures of the Ca
2N system, as shown in
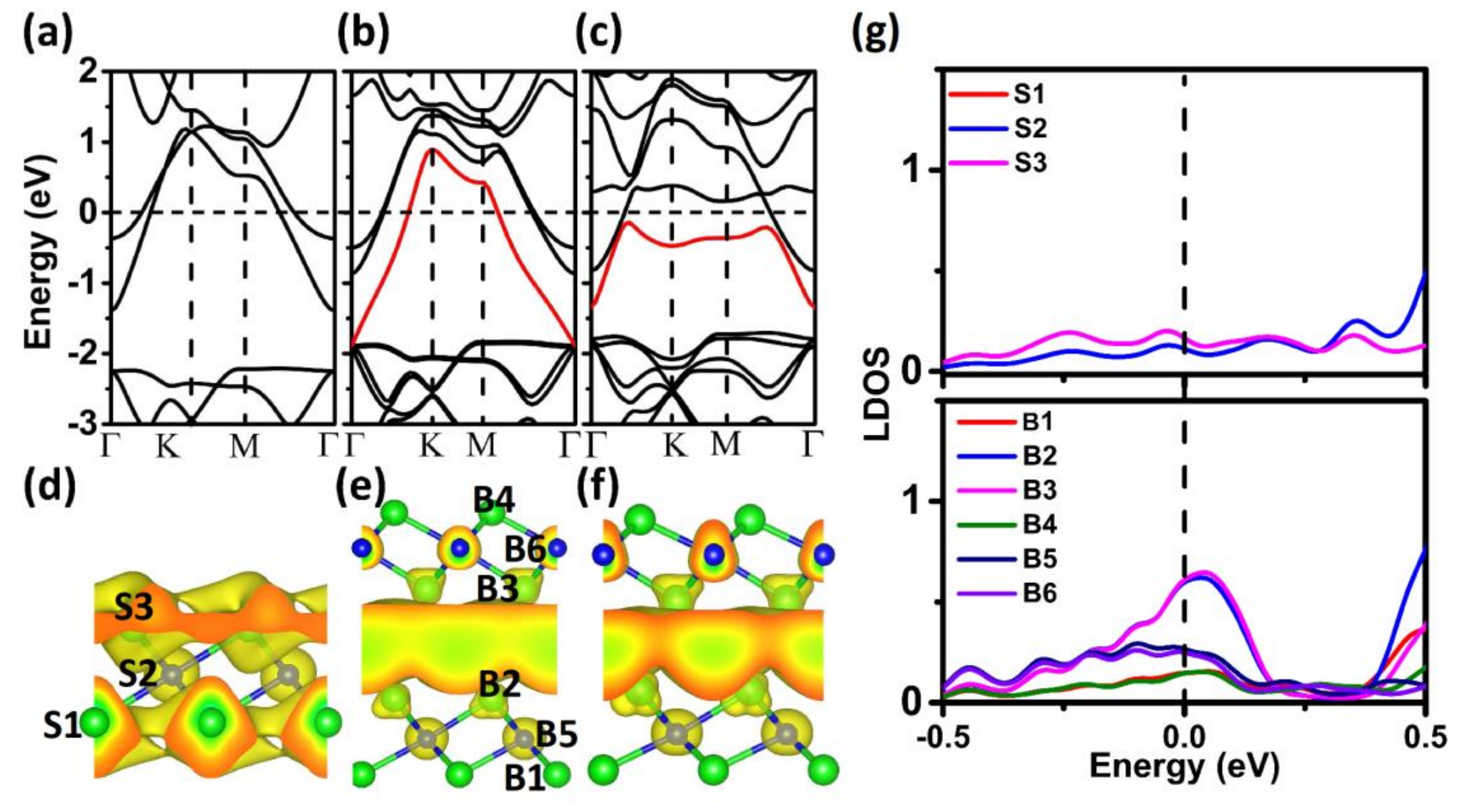
Figure 6. The monolayer Ca
2N is metallic with two bands crossing the Fermi level (
Figure 6a), and the corresponding partial electron density shows that the two bands are from the 2D-confined electron layers residing at the two sides of [Ca
2N]
+ (
Figure 6d). However, when the two sheets are joined, an additional band induced by the coupling effect between the two layers appears, which is mainly from the 2D electron layers confined in the interlayer regions (
Figure 6b). As the additional band also crosses the Fermi level, the electrons confined in the interlayer regions are loosely bound conduction electrons, suggesting that the delocalized 2D charge is distributed uniformly in the interlayer space of the Ca
2N system (
Figure 6d), which yields similar IEs at different stackings and smooth
V. To obtain more information about charge distribution, we plotted the local density of states (LDOS) for monolayer and bilayer Ca
2N samples. Compared to the monolayer system, there is a clear peak at the Fermi level in the bilayer system, which comes from the two Ca atoms at the interface (B2 and B3 in
Figure 6e). These results indicate that electrons confined in the interlayer regions are loosely bound conduction electrons, which is consistent with the above charge redistribution. Therefore, uniform interfacial charge distribution is the main cause of the ultralow friction mechanism of the layered Ca
2N. For a larger load, the additional band becomes flat and drops below the Fermi level (
Figure 6c,f). These results indicate that the confined effect becomes stronger and the homogeneity of the interfacial charge distribution decreases with the increase of the load, which can explain the larger
V and friction under the larger load. It should be noted that our results are in contrast to the findings of Wolloch et al., as larger adhesion corresponds to larger interlayer friction [
20]. This controversy may be attributed to the unique geometric and electronic structures of the Ca
2N system. The large interfacial space, strong adsorption, and unique 2D uniform interfacial charge distribution make it dramatically different from any other material in their calculations [
20].


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





