Catalytic Fast Pyrolysis: A Review

Abstract
:1. Introduction
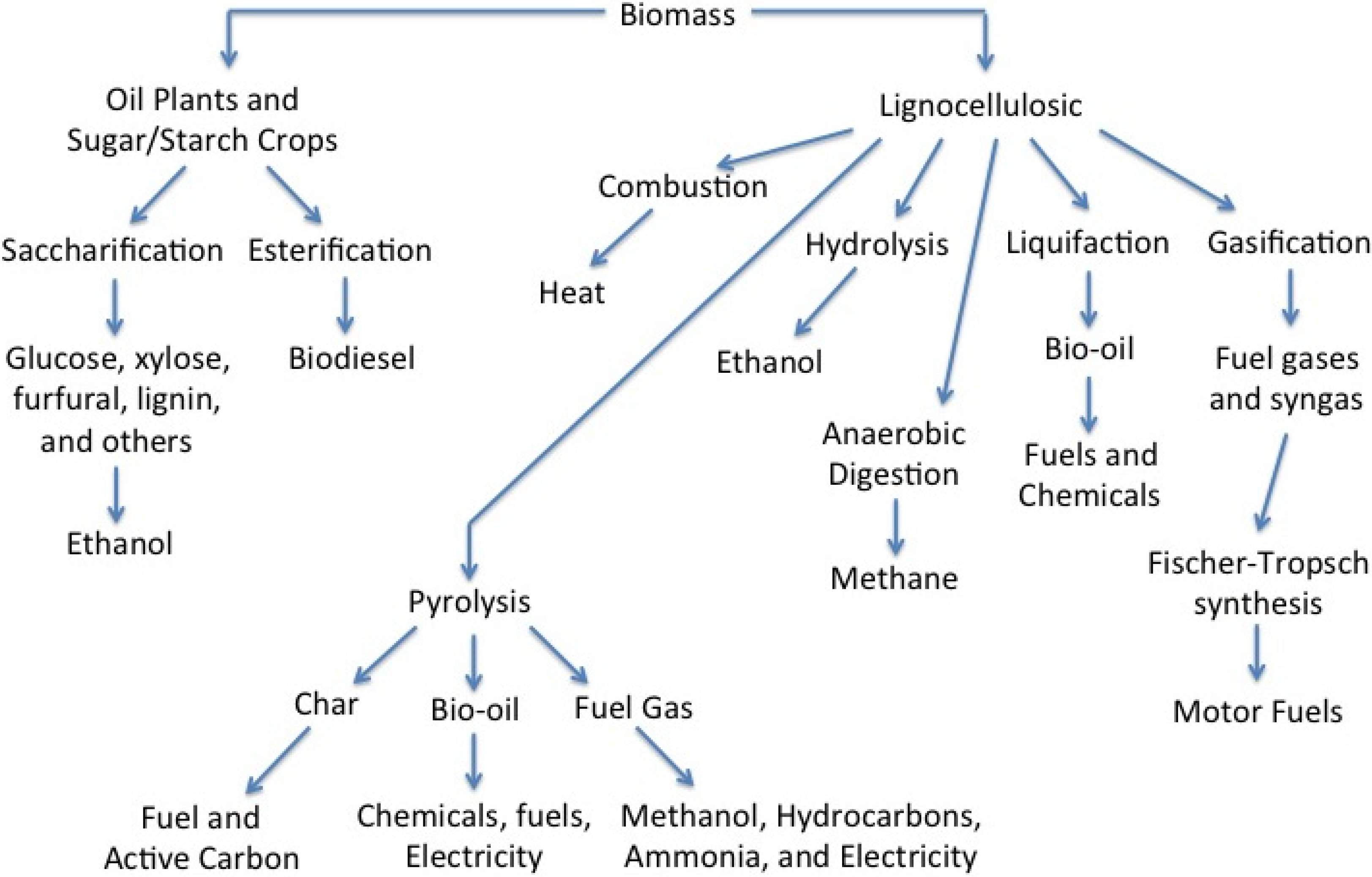
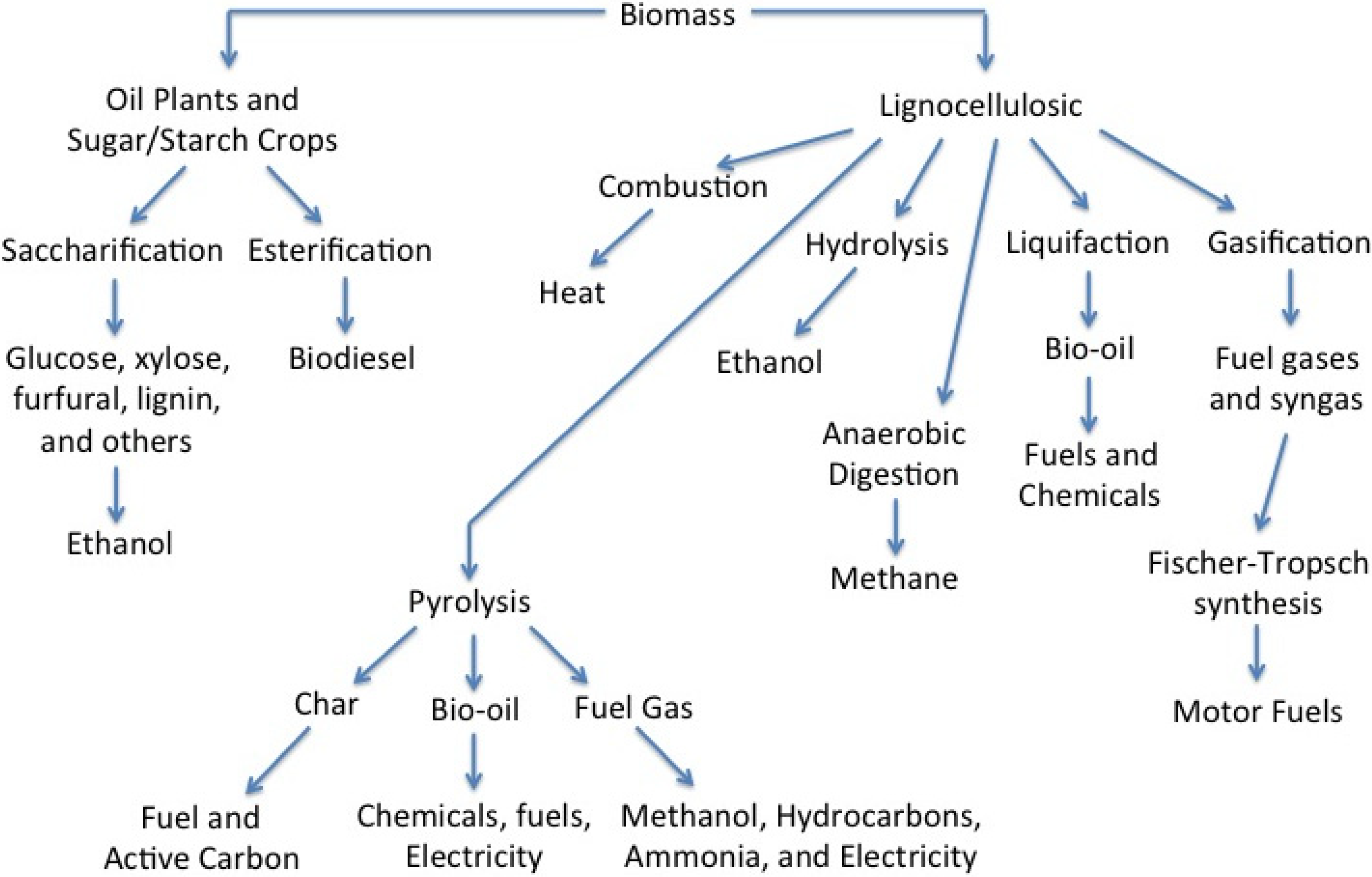
2. Pyrolysis
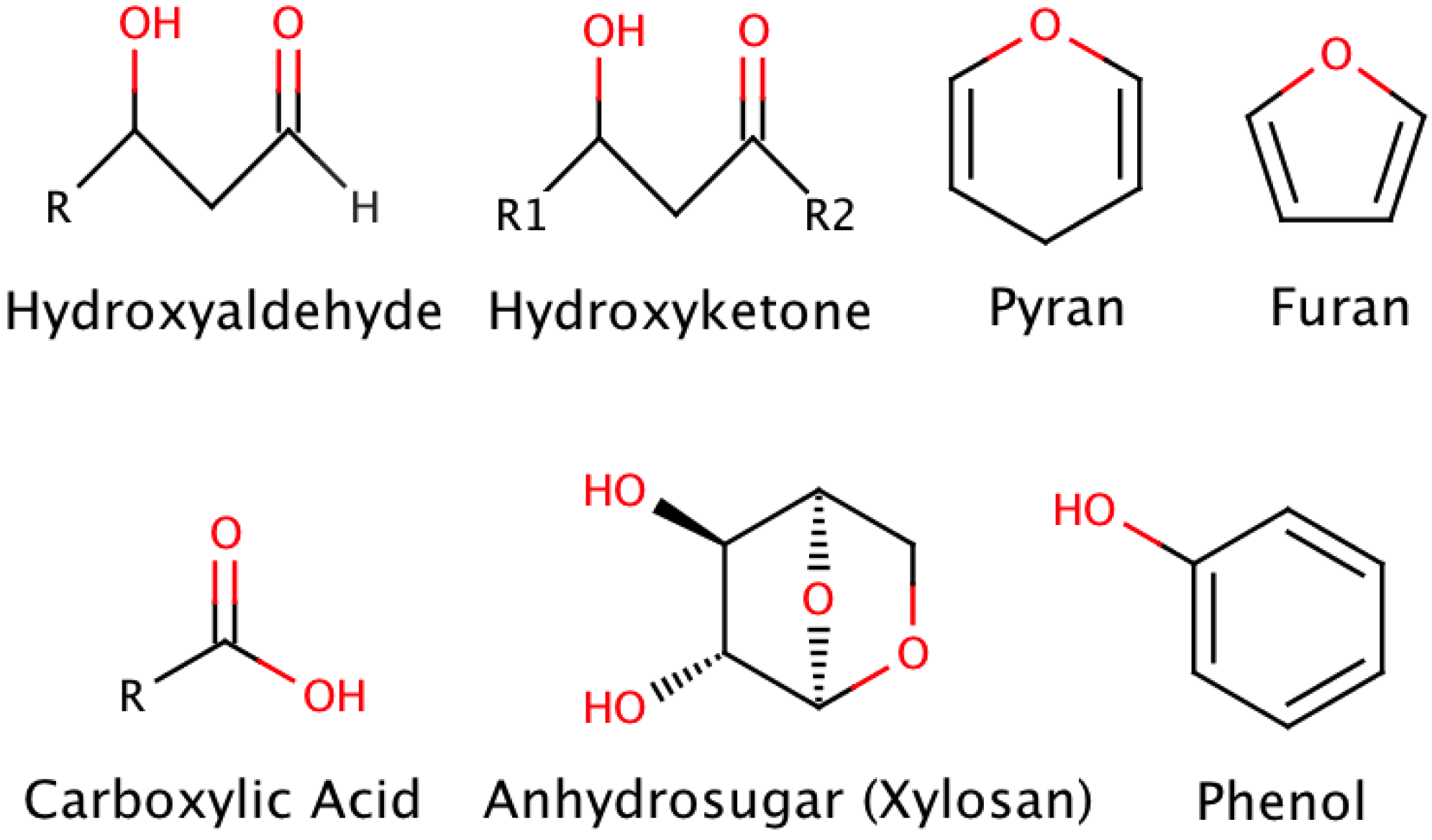
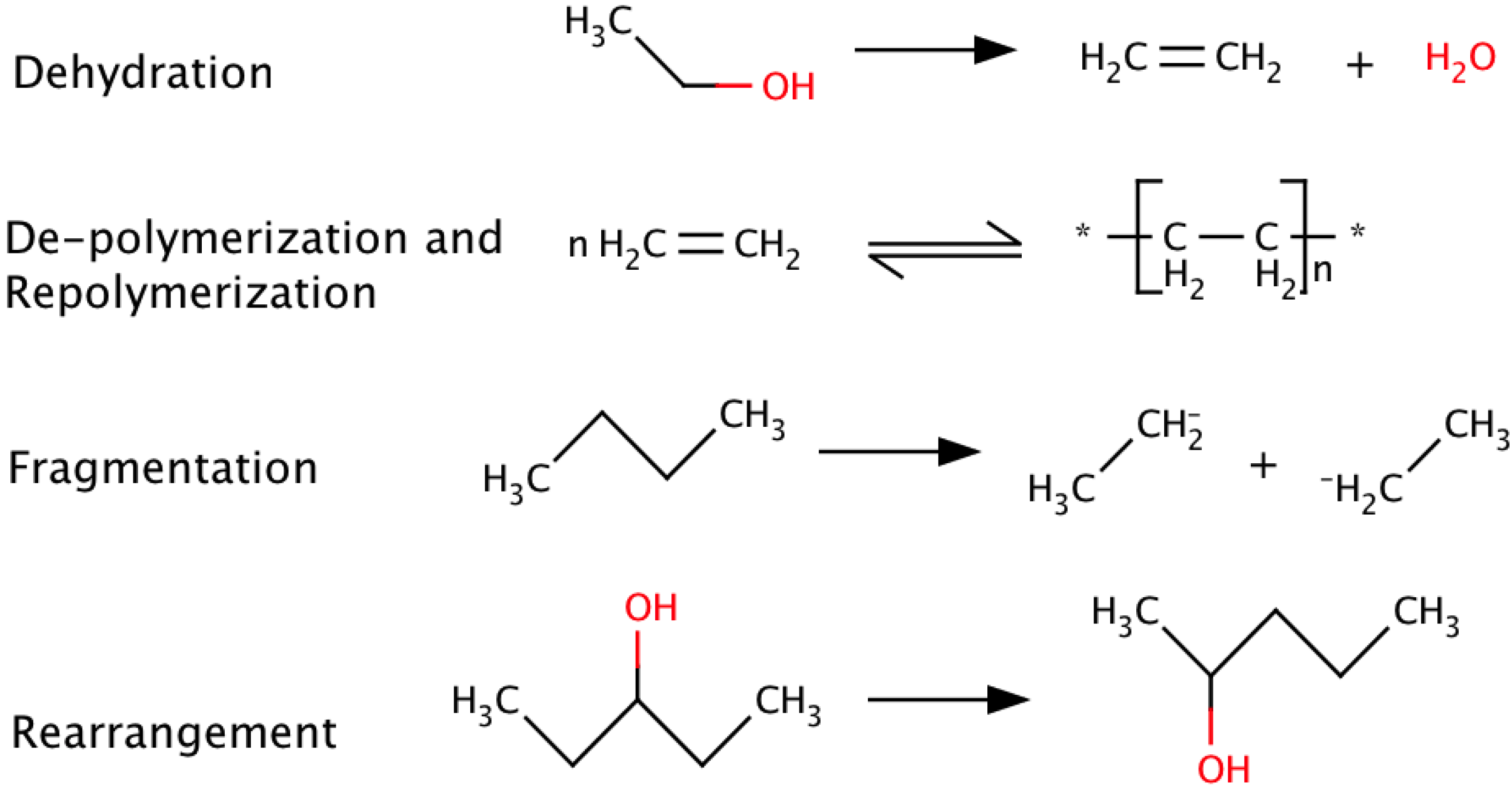

{kind=link}
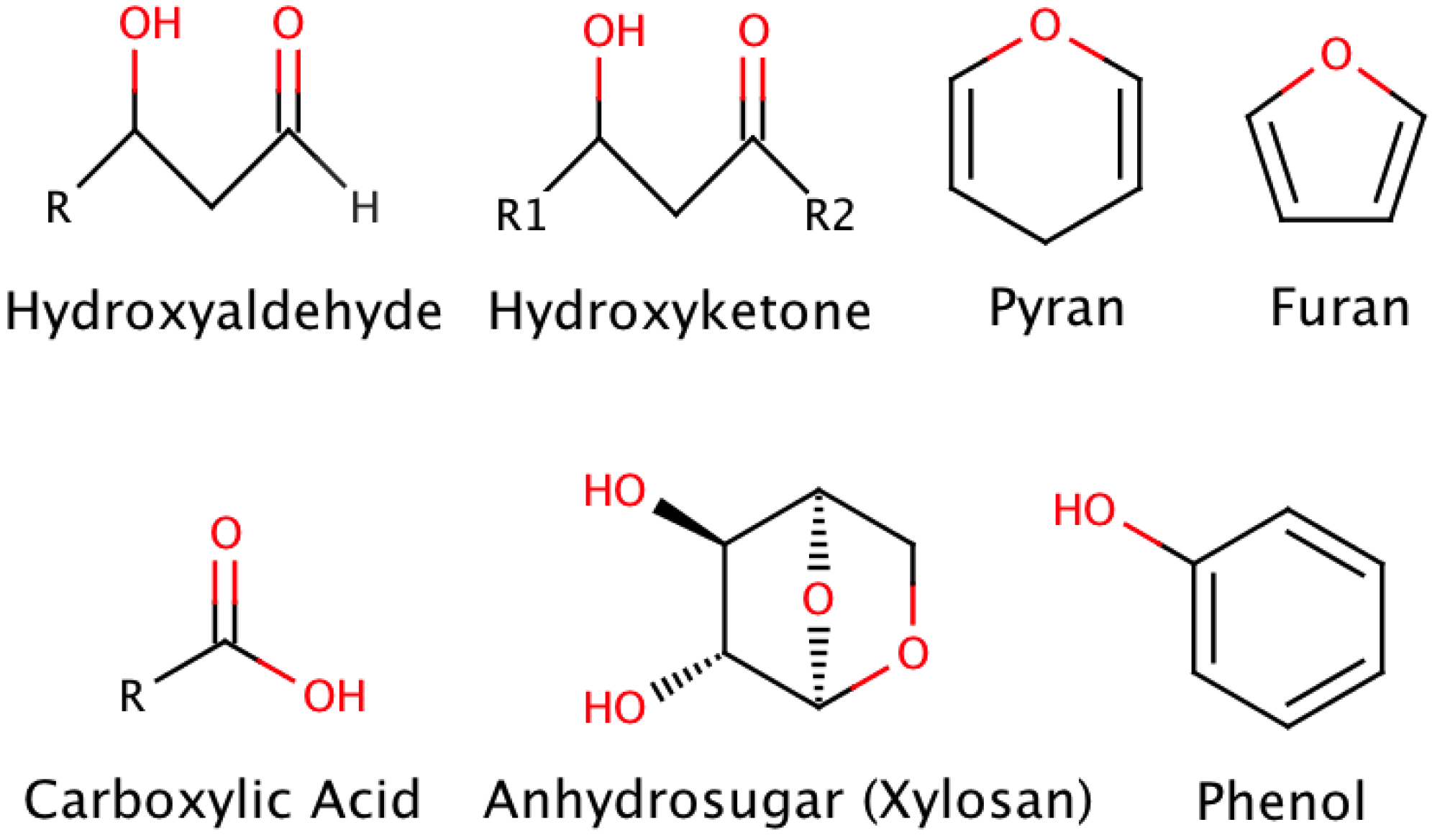{kind=link}
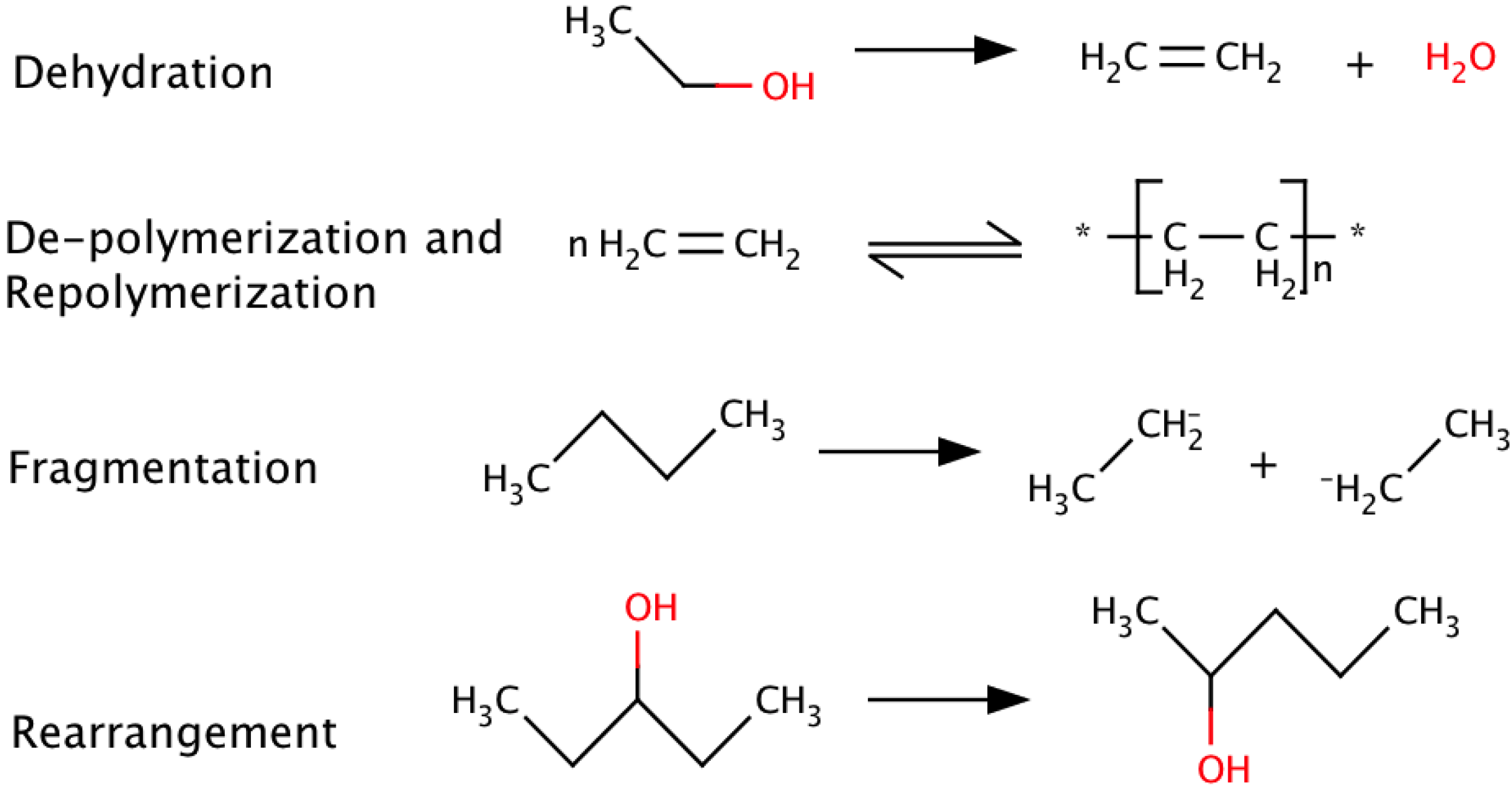{kind=link}
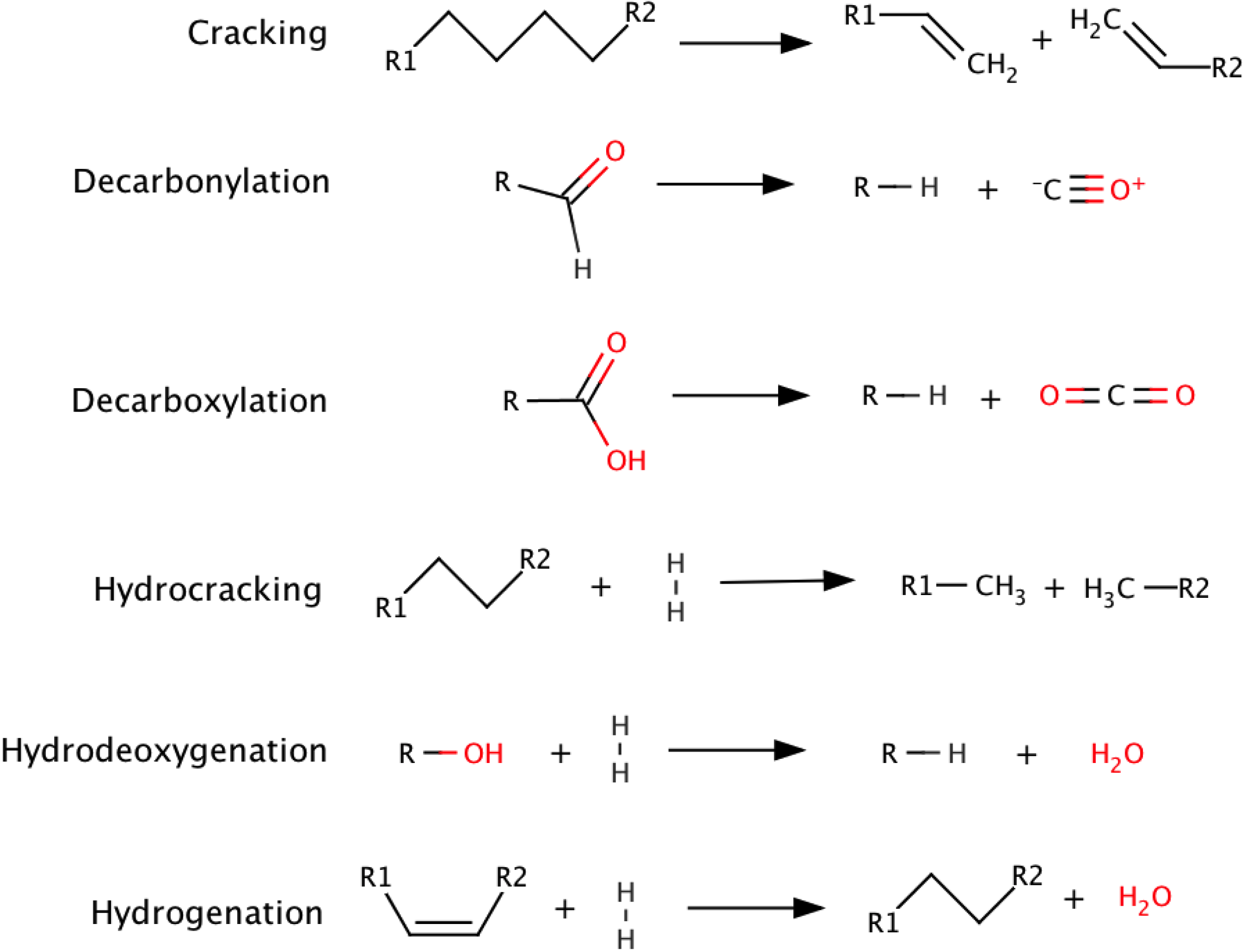{kind=link}
{kind=link}
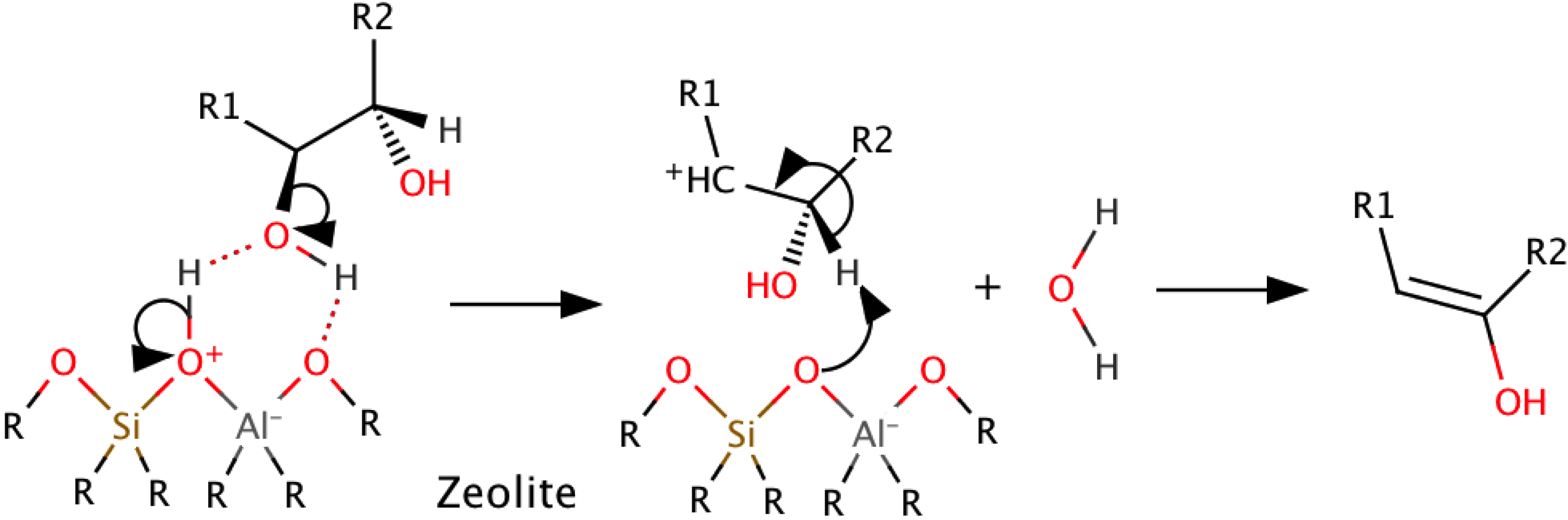{kind=link}
| Composition | Bio-oil | Crude Oil |
|---|---|---|
| Water (wt %) | 15–30 | 0.1 |
| pH | 2.8–3.8 | - |
| density (kg/L) | 1.05–1.25 | 0.86 |
| viscosity 50 °C (cP) | 40–100 | 180 |
| HHV (MJ/kg) | 16–19 | 44 |
| C (wt %) | 55–65 | 83-86 |
| O (wt %) | 28–40 | <1 |
| H (wt %) | 5–7 | 11–14 |
| S (wt %) | <0.05 | <4 |
| N (wt %) | <0.4 | <1 |
| Ash (wt %) | <0.2 | 0.1 |
| H/C | 0.9–1.5 | 1.5–2.0 |
| O/C | 0.3–0.5 | ≈0 |
3. Catalytic Upgrading
3.1. Introduction
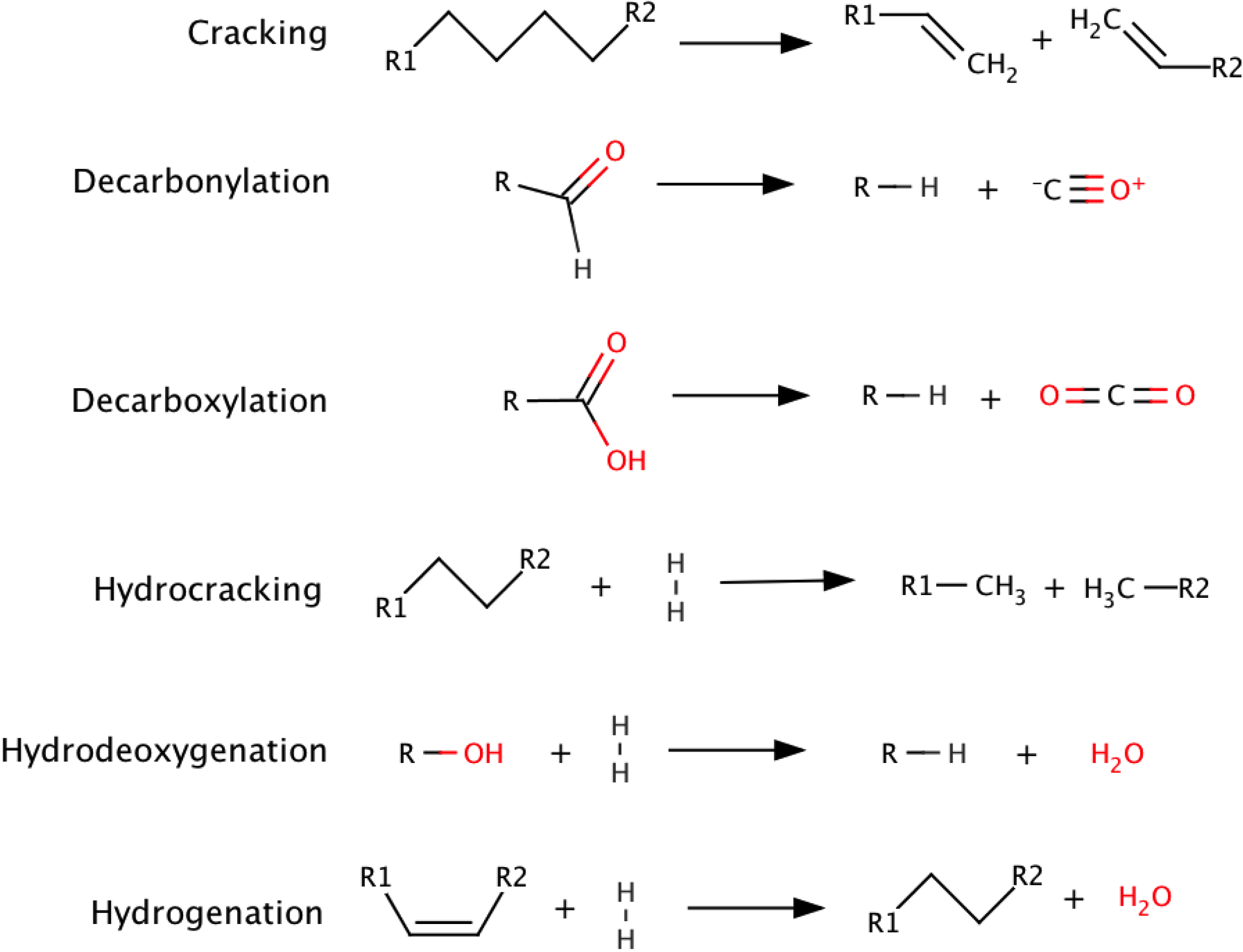
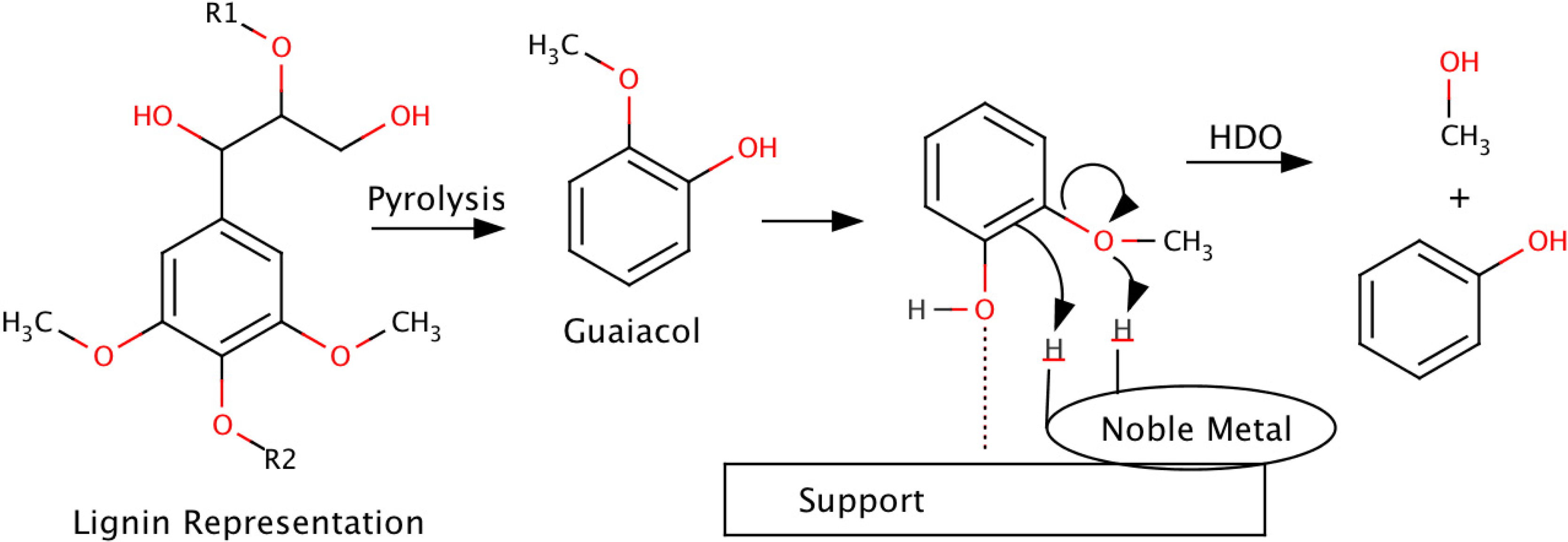
3.2. Hydrodeoxygenation
| Catalyst | Temp. (°C) | Pressure (bar) | DOD (%) | O/C | H/C | Oil Yield (wt %) | Ref. |
|---|---|---|---|---|---|---|---|
| Co-MoS2/Al2O3 | 350 | 200 | 81 | 0.8 | 1.3 | 26 | [58] |
| Co-MoS2/Al2O4 | 370 | 300 | 100 | 0 | 1.8 | 33 | [64] |
| Ni-MoS2/Al2O3 | 350 | 200 | 74 | 0.1 | 1.5 | 28 | [58] |
| Ni-MoS2/Al2O4 | 400 | 85 | 28 | - | - | 84 | [65] |
| Pd/C | 350 | 200 | 85 | 0.7 | 1.6 | 65 | [58] |
| Pd/C | 340 | 140 | 64 | 0.1 | 1.5 | 48 | [55] |
| Pd/ZrO2 | 300 | 80 | - | 0.1 | 1.3 | - | [56] |
| Pt/Al2O3/SiO2 | 400 | 85 | 45 | - | - | 81 | [65] |
| Pt/ZrO2 | 300 | 80 | - | 0.2 | 1.5 | - | [56] |
| Rh/ZrO2 | 300 | 80 | - | 0 | 1.2 | - | [56] |
| Ru/Al2O3 | 350 | 200 | 78 | 0.4 | 1.2 | 36 | [58] |
| Ru/C | 350–400 | 230 | 73 | 0.1 | 1.5 | 38 | [66] |
| Ru/C | 350 | 200 | 86 | 0.8 | 1.5 | 53 | [58] |
| Ru/TiO2 | 350 | 200 | 77 | 1 | 1.7 | 67 | [58] |
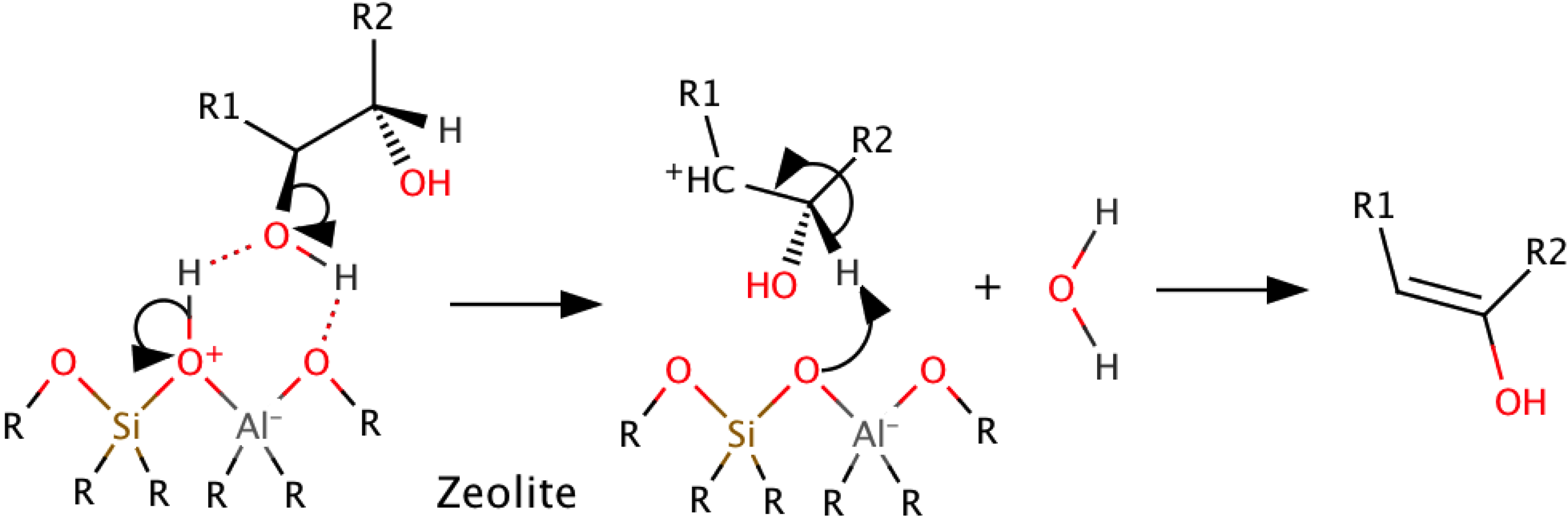
3.3. Catalytic Cracking with Zeolites
3.4. Catalyst Development
| Catalyst | Temp. (°C) | Feedstock | Catalyst Effects | Ref. |
|---|---|---|---|---|
| HZSM-5 with varying Si/Al2O3 ratios | 500–764 | Kraft Lignin | Decreasing the SiO2/Al2O3 ratio from 200/1 to 25/1 and increasing the catalyst-to-lignin ratio from 1:1 to 20:1 decreased the oxygenates and increased the aromatics. Aromatics yield increased from 500 to 650 °C and then decreased at higher temperatures. Under optimal reaction conditions, the aromatic yields were 2.0% (EHI 0.08) and 5.2% (EHI 0.35). | [88] |
| HZSM-5, Na/ZSM5, HBeta, and HUSY | 650 | Alkaline lignin | H-USY had the largest pore size and lowest Si/Al ratio (7) and had the best liquid yield of 75% and aromatic yield of 40%. | [89] |
| ZSM-5, Al/MCM-41, Al-MSU-F, ZnO, ZrO2, CeO2, Cu2Cr2O5, Criterion-534, alumina-stabilized ceria-MI-575, slate, char and ashes derived from char and biomass | 500 | Cassava rhizome | ZSM-5, Al/MCM-41, Al-MSU-F type, Criterion-534, alumina-stabilized ceria-MI-575, Cu2Cr2O5, and biomass-derived ash were selective to the reduction of most oxygenated lignin derivatives. ZSM-5, Criterion-534, and Al-MSU-F catalysts enhanced the formation of aromatic hydrocarbons and phenols. No single catalyst was found to reduce all carbonyl products but ZSM-5, Criterion-534 and MI-575 could reduce most of the carbonyl products that contained hydroxyl groups. ZSM-5, Criterion-534, Al/MCM-41, Al-MSU-F, copper chromite, char and ashes increased acetic, formic, and lactic acid. MI-575 did not increase acids. | [90] |
| Dolomite | 500–800 | Waste olive husks | Dolomite increased cracking and gas production. | [91] |
| HZSM-5, Al/MCM-41, Al-MSU-F, and alumina-stabilized ceria MI-575, pore sizes 5.5, 31, 15, and NA respectively | 500 | Cassava rhizome | HZSM-5 was the most effective catalyst for the production of aromatic hydrocarbons, phenols, and acetic acid and the reduction of oxygenated lignin-derived compounds and carbonyls containing side chain hydroxyl groups. Only MI-575 showed a decrease in acetic acid yields. MI-575 also showed the most increase in methanol with HZSM-5 a close second. | [81] |
3.4.1. Multistage Catalysis
3.4.2. Multifunctional Catalysts
| Catalyst | Temp. (°C) | Feedstock | Catalyst Effects | Ref. |
|---|---|---|---|---|
| Pt/HZSM-5 and HZSM-5 | 400–500 | Canola Oil | Pt/HZSM-5 increased isomerization and hydrogenation, increased gas yields, increased C4 iso/n-hydrocarbon ratio, and lowered organic liquid product (OLP) yield. Steam decreased the OLP yield. | [100] |
| Pretreatment with Na2CO3 | 300–450 | Chlorella algae | Na2CO3 lowered the initial degradation temperature. Catalyst also increased gas yield and decreased liquid yield. Resulting bio-oil had higher heating value, more aromatics, and lower acidity. | [101] |
| ZnCl2 impregnated in biomass | 250–500 | Corn cob, fir wood, bagasse, and rice husk | Enhanced charring and dehydration and promoted production of furfural (FF) and acetic acid (AA). Corn cob gave most FF (8%) at 340 °C with 15% ZnCl2 and a yield of 4% AA compared to a non-catalytic yield of FF 0.49%. | [102] |
| MgO at 5%, 10%, 15%, and 20% of raw material | 550 | Cotton seed | Increasing the amount of catalyst decreased the oil yield and increased the gas and char yields. MgO increased the oil quality by reducing oxygen levels from 9.56% to 4.90% and converting almost all of the long chain alkanes and alkenes to lower molecular weight hydrocarbons in the diesel range. | [103] |
| Boric Oxide mixed with biomass | 400 | Empty palm oil fruit bunch and oil palm fronds | Promoted deoxygenation, eliminated 50%–80% of the hydroxyl and methoxy groups, increased both water and char yields, and decreased gas yields. | [104] |
| Al/MCM-41, Al/MCM-48, HZSM-5, Meso-MFI, Pt/ HZSM-5 (0.5%), Pt/Meso-MFI (0.5%) | 450 | Miscanthus | Al/MCM-41, Al/MCM-48, and Meso-MFI produced more phenolics and reduced more oxygenates than HZSM-5. HZSM-5 and Meso-MFI produced aromatics due to their acidic sites. Meso-MFI zeolite, which has both mesopores and high acidity, performed the best overall. Pt enhanced deoxygenation and aromatization in both cases. | [105] |
| Meso-MFI and Pt/Meso-MFI (0.5%, ion exchanged) | 500 | Waste rice husk | Meso-MFI reduced oxygenates by 38%. Pt/Meso-MFI reduced oxygenates by 49%. Both converted heavy phenols to light phenols and aromatics. | [106] |
| Pt/Hbeta, Pt/SiO2, Hbeta | 400 | Anisole | Pt/Hbeta catalyzed both methyl transfer and hydrodeoxygenation at significantly higher rates than the monofunctional catalysts. Formed benzene, toluene, and xylenes with lower hydrogen consumption and a significant reduction in carbon losses. The rate of deactivation and coke deposition were moderately reduced. | [107] |
| Ga/HZSM-5 | 400–550 | Benzaldehyde | Ga/HZSM-5 catalyzed decarbonylation, producing benzene and CO in the absence of H2. In the presence of H2, it catalyzed toluene production. Addition of water increased benzene and reduced toluene. | [108] |
| Zn/HZSM-5 (0.5 and 1.5%) | 300–500 | Furfural | 1.5% Zn/H-ZSM-5 produced slightly more aromatics (~5%) than 0.5% Zn/HZSM-5. Zn/HZSM-5 catalysts yielded more aromatics and olefins and less furan and coke than HZSM-5. | [109] |
| Ce/HZSM-5 | 600 | Glucose | Increased oxygenated compounds and CO while reducing coke. | [110] |
| Hybrid Pt/HZSM-5 (mixture) and Pt/HZSM-5 | 350–450 | Pyrolysis gasoline | Hybrid Pt/HZSM-5 catalyst showed lower metal-support interaction but a higher catalytic activity. Pt/HZSM-5 increased C2+n-alkanes and decreased methane and hydrogen requirements. | [111] |
| Pd/HY, Pt/HY, Ir/HY, Ni/HY | 350–450 | Pyrolysis gasoline | Ir/HY showed better metal dispersion, acidity, hydrogen adsorption, and metal surface exposure than Pt/HY or Pd/HY. Ni/HY catalyzed less hydrogenation than the other three. Hydrogen pressure helped stabilize the catalysts. | [112] |
| (10%) Pd/C, (30%) Pd/C, Pd(OH)2/C, Pd(OAc)2, Pd-PEPPSI-iPr and Pd/Lindlar with Nafion SAC-13 used in every run. | 300 | Various lignin types and lignin model compounds | Various phenols such as guaiacol, pyrocatechol, and resorcinol were formed from lignin. Model compounds were hydrodeoxygenated, demethylated, and demethoxylated. Percentage yields were better than many other HDO techniques. Activity of catalysts was in the following order; Pd(OAc)2 < Pd-PEPPSI-IPr < Pd(OH)2/C < 10% Pd/C < Pd-Lindlar. | [113] |
| Ni/HZSM-5 (1%) | 450 | Bio-oil from Pinus insignis with 60% methanol | 90% conversion of the bio-oil in the feed with a selectivity for aromatics of 0.4 (benzene, toluene, xylenes (BTX) selectivity of 0.25). Rapid coke deposition was observed. | [114] |
| Ni/Al2O3, Ni/CeO2, and Ni/Al2O3-CeO2 with varying percentages of nickel. | 800 | Cellulose | Initial degradation at lower temp. All reduced tar. 30% Ni/CeO2 catalyst yielded least amount of tar and least CO. 30% Ni/Al2O3 produced maximum amount of H2 (43.5 vol % at 800 °C, 15 min residence time). | [115] |
| Ga/HZSM-5 | 600 | Furan with pinewood sawdust | Depending on preparation, Ga/HZSM-5 increased the rate of aromatics production. Ga seemed to increase the rate of decarbonylation and olefin aromatization, whereas HZSM-5 catalyzed other reactions such as oligomerization. 41% of the energy in the wood was converted into usable products. | [116] |
| NiCl2, HZSM-5, Ni/ZSM-5 | 700 | Kraft Lignin | HZSM-5 almost completely decomposed the aliphatic C-O bonds and carbonyl groups and eliminated 80% of the methoxy groups. It showed more deoxygenation than Ni/ZSM-5. NiCl2 reduced liquid yield while increasing the molecular weight and increasing the gas yield. It produced more aromatic carbons and less aliphatic carbons. | [117] |
| Al/MCM-41, Cu-Al/MCM-41, Fe-Al/MCM-41, Zn-Al/MCM-41 | 500 | Lignocel from beech wood and Miscanthus | Lignocel yielded more hydrocarbons and Miscanthus more phenols. All catalysts produced more phenols. A low Si/Al ratio increased product yields and improved final composition. Fe and Cu containing catalysts produced the most phenols. The presence of Al/MCM-41 reduced oxygenated compounds. Cu/MCM-41 promoted the largest increase of H2 in the gas yield. | [118] |
| 31 catalysts mixed with biomass. Included ZnO, CuO, Fe2O3. | 500 | Pine sawdust | A significant decrease in non-volatile fraction and slight decrease in bio-oil yield were obtained with ZnO (reduced the proportion of heavy fraction in the bio-oil with a limited decrease in its yield), CuO (exhibited the highest yields in semi-volatile compounds), Fe2O3, and mixed oxide catalysts containing Cu and Co. | [119] |
| HFer-20, Fe/HFer-20, HY-12, Fe/HY-12, HBeta-25, Fe/HBeta-25 | 400–450 | Pine wood | Iron modified zeolites increased coke and methyl substituted phenols, decreased methoxy substituted phenols, and didn't affect the CO to CO2 ratio. Beta zeolite was the most active in deoxygenation. All zeolites increased levoglucosan. | [120] |
| K2CO3 or Ca(OH)2 mixed with biomass | 700 | Pine wood | K2CO3 was more active producing no saccharides, aldehydes, or alcohols and substantially reducing the formation of acids, furans, and guaiacols. The yields of alkanes and phenols were increased. Ca(OH)2 reduced char, increased liquid, and increased alcohols, opposing the results from K2CO3. | [121] |
| MgO, CaO, TiO2, Fe2O3, NiO, and ZnO | 600 | Poplar Wood | ZnO showed no activity. CaO reduced heavy products including phenols and anhydrosugars and increased formation of cyclopentanones, hydrocarbons, and light products including acetaldehyde, 2-butanone, and methanol. CaO also reduced acids. Other catalysts were not as effective. Fe2O3 produced PAHs. | [13] |
| Ni/C mixed with biomass | 350 | Pubescens | Produced bio-oil with high content of phenols but low contents of acetic acid, furfural, and water. | [122] |
4. Conclusions
Acknowledgements
References
- Demirbas, A. Global renewable energy resources. Energy Sources Part A Recovery Util. Environ. Eff. 2006, 28, 779–792. [Google Scholar] [CrossRef]
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Ruiz, J.C.; Dumesic, J.A. Catalytic routes for the conversion of biomass into liquid hydrocarbon transportation fuels. Energy Environ. Sci. 2011, 4, 83–99. [Google Scholar] [CrossRef]
- Stephanidis, S.; Nitsos, C.; Kalogiannis, K.; Iliopoulou, E.F.; Lappas, A.A.; Triantafyllidis, K.S. Catalytic upgrading of lignocellulosic biomass pyrolysis vapours: Effect of hydrothermal pre-treatment of biomass. Catal. Today 2011, 167, 37–45. [Google Scholar] [CrossRef]
- Balat, M.; Balat, M.; Kirtay, E.; Balat, H. Main routes for the thermo-conversion of biomass into fuels and chemicals. Part 1: Pyrolysis systems. Energy Convers. Manag. 2009, 50, 3147–3157. [Google Scholar] [CrossRef]
- Bridgwater, A.; Peacocke, G. Fast pyrolysis processes for biomass. Renew. Sustain. Energy Rev. 2000, 4, 1–73. [Google Scholar] [CrossRef]
- Trimble, J.L.; Vanhook, R.I.; Folger, A.G. Biomass for energy: The environmental-issues. Biomass 1984, 6, 3–13. [Google Scholar] [CrossRef]
- Committee on Economic and Environmental Impacts of Increasing Biofuels Production. Renewable Fuel Standard: Potential Economic and Environmental Effects of U.S. Biofuel Policy; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Demirbas, F.M. Biorefineries for biofuel upgrading: A critical review. Appl. Energy 2009, 86, S151–S161. [Google Scholar] [CrossRef]
- Mohan, D.; Pittman, C.U., Jr.; Steele, P.H. Pyrolysis of wood/biomass for bio-oil: A critical review. Energy Fuels 2006, 20, 848–889. [Google Scholar] [CrossRef]
- Vispute, T.P.; Zhang, H.; Sanna, A.; Xiao, R.; Huber, G.W. Renewable chemical commodity feedstocks from integrated catalytic processing of pyrolysis oils. Science 2010, 330, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.P.; Kim, C.S. Lignin depolymerization and conversion: A review of thermochemical methods. Chem. Eng. Technol. 2011, 34, 29–41. [Google Scholar] [CrossRef]
- Lu, Q.; Zhang, Z.-F.; Dong, C.-Q.; Zhu, X.-F. Catalytic upgrading of biomass fast pyrolysis vapors with nano metal oxides: An analytical Py-GC/MS study. Energies 2010, 3, 1805–1820. [Google Scholar] [CrossRef]
- Soria, A.J.; McDonald, A.G.; Shook, S.R. Wood solubilization and depolymerization using supercritical methanol. Part 1: Process optimization and analysis of methanol insoluble components (bio-char). Holzforschung 2008, 62, 402–408. [Google Scholar]
- Soria, J.A. Unlocking hydrocarbons from biomass. Agroborealis 2010, 41, 39–40. [Google Scholar]
- Soria, A.J.; McDonald, A.G.; He, B.B. Wood solubilization and depolymerization by supercritical methanol. Part 2: Analysis of methanol soluble compounds. Holzforschung 2008, 62, 409–416. [Google Scholar]
- Speight, J. Synthetic Fuels Handbook: Properties, Process and Performance; McGraw-Hill Professional: New York, NY, USA, 2008. [Google Scholar]
- Lehmann, J.; Rillig, M.C.; Thies, J.; Masiello, C.A.; Hockaday, W.C.; Crowley, D. Biochar effects on soil biota: A review. Soil Biol. Biochem. 2011, 43, 1812–1836. [Google Scholar] [CrossRef]
- Roberts, K.G.; Gloy, B.A.; Joseph, S.; Scott, N.R.; Lehmann, J. Life cycle assessment of biochar systems: Estimating the energetic, economic, and climate change potential. Environ. Sci. Technol. 2010, 44, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Ozcimen, D.; Ersoy-Mericboyu, A. Characterization of biochar and bio-oil samples obtained from carbonization of various biomass materials. Renew. Energy 2010, 35, 1319–1324. [Google Scholar] [CrossRef]
- Nowakowski, D.J.; Bridgwater, A.V.; Elliott, D.C.; Meier, D.; de Wild, P. Lignin fast pyrolysis: Results from an international collaboration. J. Anal. Appl. Pyrolysis 2010, 88, 53–72. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Grunwaldt, J.D.; Jensen, P.A.; Knudsen, K.G.; Jensen, A.D. A review of catalytic upgrading of bio-oil to engine fuels. Appl. Catal. A Gen. 2011, 407, 1–19. [Google Scholar] [CrossRef]
- Freitas, S.R.; Soria, J.A. Development of a gasification system for utilizing fish processing waste and coastal small diameter wood in rural areas. Energy Fuels 2011, 25, 2292–2300. [Google Scholar] [CrossRef]
- Yaman, S. Pyrolysis of biomass to produce fuels and chemical feedstocks. Energy Conver. Manag. 2004, 45, 651–671. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Ross, J.R.H. Catalysis for conversion of biomass to fuels via pyrolysis and gasification: A review. Catal. Today 2011, 171, 1–13. [Google Scholar] [CrossRef]
- Faix, O.; Meier, D.; Fortmann, I. Thermal-degradation products of wood: A collection of electron-impact (EI) mass-spectra of monomeric lignin derived products. Holz als Roh- und Werkstoff 1990, 48, 351–354. [Google Scholar] [CrossRef]
- Faix, O.; Fortmann, I.; Bremer, J.; Meier, D. Thermal degradation products of wood. Eur. J. Wood Wood Prod. 1991, 49, 213–219. [Google Scholar] [CrossRef]
- Yanik, J.; Kommayer, C.; Saglam, M.; Yueksel, M. Fast pyrolysis of agricultural wastes: Characterization of pyrolysis products. Fuel Process. Technol. 2007, 88, 942–947. [Google Scholar] [CrossRef]
- Branca, C.; Giudicianni, P.; Di Blasi, C. GC/MS characterization of liquids generated from low-temperature pyrolysis of wood. Ind. Eng. Chem. Res. 2003, 42, 3190–3202. [Google Scholar] [CrossRef]
- Valtiner, S.M.; Bonn, G.K.; Huck, C.W. Characterization of different types of hay by solid-phase micro-extraction gas chromatography/mass spectrometry and multivariate data analysis. Phytochem. Anal. 2008, 19, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Ralph, J.; Hatfield, R.D. Pyrolysis-GC-MS characterization of forage materials. J. Agric. Food Chem. 1991, 39, 1426–1437. [Google Scholar] [CrossRef]
- Evans, R.J.; Milne, T.A. Molecular characterization of the pyrolysis of biomass. Energy Fuels 1987, 1, 123–137. [Google Scholar] [CrossRef]
- Marsman, J.H.; Wildschut, J.; Mahfud, F.; Heeres, H.J. Identification of components in fast pyrolysis oil and upgraded products by comprehensive two-dimensional gas chromatography and flame ionization detection. J. Chromatogr. A 2007, 1150, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Sipila, K.; Kuoppala, E.; Fagernas, L.; Oasmaa, A. Characterization of biomass-based flash pyrolysis oils. Biomass Bioenergy 1998, 14, 103–113. [Google Scholar] [CrossRef]
- Oasmaa, A.; Czernik, S. Fuel oil quality of biomass pyrolysis oils—State of the art for the end user. Energy Fuels 1999, 13, 914–921. [Google Scholar] [CrossRef]
- Zhang, Q.; Chang, J.; Wang, T.; Xu, Y. Review of biomass pyrolysis oil properties and upgrading research. Energy Conver. Manag. 2007, 48, 87–92. [Google Scholar] [CrossRef]
- French, R.; Czernik, S. Catalytic pyrolysis of biomass for biofuels production. Fuel Process. Technol. 2010, 91, 25–32. [Google Scholar] [CrossRef]
- Cheng, Y.T.; Huber, G.W. Chemistry of furan conversion into aromatics and olefins over HZSM-5: A model biomass conversion reaction. ACS Catal. 2011, 1, 611–628. [Google Scholar] [CrossRef]
- Zhang, H.; Cheng, Y.T.; Vispute, T.P.; Xiao, R.; Huber, G.W. Catalytic conversion of biomass-derived feedstocks into olefins and aromatics with ZSM-5: The hydrogen to carbon effective ratio. Energy Environ. Sci. 2011, 4, 2297–2307. [Google Scholar] [CrossRef]
- Samolada, M.C.; Baldauf, W.; Vasalos, I.A. Production of a bio-gasoline by upgrading biomass flash pyrolysis liquids via hydrogen processing and catalytic cracking. Fuel 1998, 77, 1667–1675. [Google Scholar] [CrossRef]
- Bridgwater, A.V. Production of high grade fuels and chemicals from catalytic pyrolysis of biomass. Catal. Today 1996, 29, 285–295. [Google Scholar] [CrossRef]
- Sekiguchi, Y.; Shafizadeh, F. The effect of inorganic additives on the formation, composition, and combustion of cellulosic char. J. Appl. Polym. Sci. 1984, 29, 1267–1286. [Google Scholar] [CrossRef]
- Raveendran, K.; Ganesh, A.; Khilar, K.C. Influence of mineral matter on biomass pyrolysis characteristics. Fuel 1995, 74, 1812–1822. [Google Scholar] [CrossRef]
- Muller-Hagedorn, M.; Bockhorn, H.; Krebs, L.; Muller, U. A comparative kinetic study on the pyrolysis of three different wood species. J. Anal. Appl. Pyrolysis 2003, 68–69, 231–249. [Google Scholar] [CrossRef]
- Scott, D.S.; Piskorz, J.; Radlein, D. Liquid products from the continuous flash pyrolysis of biomass. Ind. Eng. Chem. Process Des. Dev. 1985, 24, 581–588. [Google Scholar] [CrossRef]
- Richards, G.N.; Zheng, G.C. Influence of metal-ions and of salts on products from pyrolysis of wood: Applications to thermochemical processing of newsprint and biomass. J. Anal. Appl. Pyrolysis 1991, 21, 133–146. [Google Scholar] [CrossRef]
- Patwardhan, P.R.; Satrio, J.A.; Brown, R.C.; Shanks, B.H. Influence of inorganic salts on the primary pyrolysis products of cellulose. Bioresour. Technol. 2010, 101, 4646–4655. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.B.; Valkenburg, C.; Walton, C.W.; Elliott, D.C.; Holladay, J.E.; Stevens, D.J.; Kinchin, C.; Czernik, S. Production of Gasoline and Diesel From Biomass via Fast Pyrolysis, Hydrotreating and Hydrocracking: A Design Case; Pacific Northwest National Laboratory: Richland, WA, USA, 2009. [Google Scholar]
- Marquevich, M.; Czernik, S.; Chornet, E.; Montane, D. Hydrogen from biomass: Steam reforming of model compounds of fast-pyrolysis oil. Energy Fuels 1999, 13, 1160–1166. [Google Scholar] [CrossRef]
- Grange, P.; Laurent, E.; Maggi, R.; Centeno, A.; Delmon, B. Hydrotreatment of pyrolysis oils from biomass: Reactivity of the various categories of oxygenated compounds and preliminary techno-economical study. Catal. Today 1996, 29, 297–301. [Google Scholar] [CrossRef]
- Elliott, D.C. Historical developments in hydroprocessing bio-oils. Energy Fuels 2007, 21. [Google Scholar] [CrossRef]
- Thangalazhy-Gopakumar, S.; Adhikari, S.; Gupta, R.B.; Tu, M.; Taylor, S. Production of hydrocarbon fuels from biomass using catalytic pyrolysis under helium and hydrogen environments. Bioresour. Technol. 2011, 102, 6742–6749. [Google Scholar] [CrossRef] [PubMed]
- Wildschut, J.; Melian-Cabrera, I.; Heeres, H.J. Catalyst studies on the hydrotreatment of fast pyrolysis oil. Appl. Catal. B Environ. 2010, 99, 298–306. [Google Scholar] [CrossRef]
- Su-Ping, Z.; Yong-Jie, Y.; Zhengwei, R.; Tingchen, L. Study of hydrodeoxygenation of bio-oil from the fast pyrolysis of biomass. Energy Sources 2003, 25, 57–65. [Google Scholar] [CrossRef]
- Elliott, D.C.; Hart, T.R.; Neuenschwander, G.G.; Rotness, L.J.; Zacher, A.H. Catalytic hydroprocessing of biomass fast pyrolysis bio-oil to produce hydrocarbon products. Environ. Prog. Sustain. Energy 2009, 28, 441–449. [Google Scholar] [CrossRef]
- Gutierrez, A.; Kaila, R.K.; Honkela, M.L.; Slioor, R.; Krause, A.O.I. Hydrodeoxygenation of guaiacol on noble metal catalysts. Catal. Today 2009, 147, 239–246. [Google Scholar] [CrossRef]
- Damartzis, T.; Zabaniotou, A. Thermochemical conversion of biomass to second generation biofuels through integrated process design- a review. Renew. Sustain. Energy Rev. 2011, 15, 366–378. [Google Scholar] [CrossRef]
- Wildschut, J.; Mahfud, F.H.; Venderbosch, R.H.; Heeres, H.J. Hydrotreatment of fast pyrolysis oil using heterogeneous noble-metal catalysts. Ind. Eng. Chem. Res. 2009, 48, 10324–10334. [Google Scholar] [CrossRef]
- Xiong, W.-M.; Fu, Y.; Zeng, F.-X.; Guo, Q.-X. An in situ reduction approach for bio-oil hydroprocessing. Fuel Process. Technol. 2011, 92, 1599–1605. [Google Scholar] [CrossRef]
- Kunkes, E.L.; Simonetti, D.A.; West, R.M.; Serrano-Ruiz, J.C.; Gartner, C.A.; Dumesic, J.A. Catalytic conversion of biomass to monofunctional hydrocarbons and targeted liquid-fuel classes. Science 2008, 322, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Panagiotopoulou, P.; Kondarides, D.I. A comparative study of the water-gas shift activity of Pt catalysts supported on single (MOx) and composite (MOx/Al2O3, MOx/TiO2) metal oxide carriers. Catal. Today 2007, 127, 319–329. [Google Scholar] [CrossRef]
- Fisk, C.A.; Morgan, T.; Ji, Y.; Crocker, M.; Crofcheck, C.; Lewis, S.A. Bio-oil upgrading over platinum catalysts using in situ generated hydrogen. Appl. Catal. A Gen. 2009, 358, 150–156. [Google Scholar] [CrossRef]
- Oasmaa, A.; Kuoppala, E.; Ardiyanti, A.; Venderbosch, R.H.; Heeres, H.J. Characterization of hydrotreated fast pyrolysis liquids. Energy Fuels 2010, 24, 5264–5272. [Google Scholar] [CrossRef]
- Baldauf, W.; Balfanz, U.; Rupp, M. Upgrading of flash pyrolysis oil and utilization in refineries. Biomass Bioenergy 1994, 7, 237–244. [Google Scholar] [CrossRef]
- Sheu, Y.H.E.; Anthony, R.G.; Soltes, E.J. Kinetic-studies of upgrading pine pyrolytic oil by hydrotreatment. Fuel Process. Technol. 1988, 19, 31–50. [Google Scholar] [CrossRef]
- Venderbosch, R.H.; Ardiyanti, A.R.; Wildschut, J.; Oasmaa, A.; Heeresb, H.J. Stabilization of biomass-derived pyrolysis oils. J. Chem. Technol. Biotechnol. 2010, 85, 674–686. [Google Scholar] [CrossRef]
- Aho, A.; Kumar, N.; Eranen, K.; Salmi, T.; Hupa, M.; Murzin, D.Y. Catalytic pyrolysis of biomass in a fluidized bed reactor: Influence of the acidity of H-beta zeolite. Process Saf. Environ. Prot. 2007, 85, 473–480. [Google Scholar] [CrossRef]
- Gayubo, A.G.; Aguayo, A.T.; Atutxa, A.; Aguado, R.; Bilbao, J. Transformation of oxygenate components of biomass pyrolysis oil on a HZSM-5 zeolite. I. Alcohols and phenols. Ind. Eng. Chem.Res. 2004, 43, 2610–2618. [Google Scholar] [CrossRef]
- Gayubo, A.G.; Aguayo, A.T.; Atutxa, A.; Prieto, R.; Bilbao, J. Deactivation of a HZSM-5 zeolite catalyst in the transformation of the aqueous fraction of biomass pyrolysis oil into hydrocarbons. Energy Fuels 2004, 18, 1640–1647. [Google Scholar] [CrossRef]
- Mante, O.D.; Agblevor, F.A.; Oyama, S.T.; McClung, R. The influence of recycling non-condensable gases in the fractional catalytic pyrolysis of biomass. Bioresour. Technol. 2012, 111, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Horne, P.A.; Nugranad, N.; Williams, P.T. Catalytic coprocessing of biomass-derived pyrolysis vapors and methanol. J. Anal. Appl. Pyrolysis 1995, 34, 87–108. [Google Scholar] [CrossRef]
- Chen, N.Y.; Walsh, D.E.; Koenig, L.R. Fluidized-bed upgrading of wood pyrolysis liquids and related-compounds. In Pyrolysis Oils from Biomass; Soltes, J., Milne, T.A., Eds.; American Chemical Society: Washington, DC, USA, 1988; pp. 277–289. [Google Scholar]
- Evans, R.J.; Milne, T. Molecular-beam, mass-spectrometric studies of wood vapor and model compounds over an HZSM-5 catalyst. In Pyrolysis Oils from Biomass; Soltes, J., Milne, T.A., Eds.; American Chemical Society: Washington, DC, USA, 1988; pp. 311–327. [Google Scholar]
- Gayubo, A.G.; Aguayo, A.T.; Atutxa, A.; Aguado, R.; Olazar, M.; Bilbao, J. Transformation of oxygenate components of biomass pyrolysis oil on a HZSM-5 zeolite. II. Aldehydes, ketones, and acids. Ind. Eng. Chem. Res. 2004, 43, 2619–2626. [Google Scholar] [CrossRef]
- Zhu, X.; Mallinson, R.G.; Resasco, D.E. Role of transalkylation reactions in the conversion of anisole over HZSM-5. Appl. Catal. A Gen. 2010, 379, 172–181. [Google Scholar] [CrossRef]
- Peralta, M.A.; Sooknoi, T.; Danuthai, T.; Resasco, D.E. Deoxygenation of benzaldehyde over CsNax zeolites. J.Mol. Catal. A Chem. 2009, 312, 78–86. [Google Scholar] [CrossRef]
- Santikunaporn, M.; Herrera, J.E.; Jongpatiwut, S.; Resasco, D.E.; Alvarez, W.E.; Sughrue, E.L. Ring opening of decalin and tetralin on HY and Pt/HY zeolite catalysts. J. Catal. 2004, 228, 100–113. [Google Scholar] [CrossRef]
- Adjaye, J.D.; Bakhshi, N.N. Production of hydrocarbons by catalytic upgrading of a fast pyrolysis bio-oil. Part 1. Conversion over various catalysts. Fuel Process. Technol. 1995, 45, 161–183. [Google Scholar] [CrossRef]
- Adjaye, J.D.; Bakhshi, N.N. Upgrading of a wood-derived oil over various catalysts. Biomass Bioenergy 1994, 7, 201–211. [Google Scholar] [CrossRef]
- Aho, A.; Kumar, N.; Eranen, K.; Salmi, T.; Hupa, M.; Murzin, D.Y. Catalytic pyrolysis of woody biomass in a fluidized bed reactor: Influence of the zeolite structure. Fuel 2008, 87, 2493–2501. [Google Scholar] [CrossRef]
- Pattiya, A.; Titiloye, J.O.; Bridgwater, A.V. Fast pyrolysis of cassava rhizome in the presence of catalysts. J. Anal. Appl. Pyrolysis 2008, 81, 72–79. [Google Scholar] [CrossRef]
- Samolada, M.C.; Papafotica, A.; Vasalos, I.A. Catalyst evaluation for catalytic biomass pyrolysis. Energy Fuels 2000, 14, 1161–1167. [Google Scholar] [CrossRef]
- Corma, A.; Huber, G.W.; Sauvanaud, L.; O’Connor, P. Processing biomass-derived oxygenates in the oil refinery: Catalytic cracking (FCC) reaction pathways and role of catalyst. J. Catal. 2007, 247, 307–327. [Google Scholar] [CrossRef]
- Lappas, A.A.; Samolada, M.C.; Iatridis, D.K.; Voutetakis, S.S.; Vasalos, I.A. Biomass pyrolysis in a circulating fluid bed reactor for the production of fuels and chemicals. Fuel 2002, 81, 2087–2095. [Google Scholar] [CrossRef]
- Williams, P.T.; Nugranad, N. Comparison of products from the pyrolysis and catalytic pyrolysis of rice husks. Energy 2000, 25, 493–513. [Google Scholar] [CrossRef]
- Murata, K.; Liu, Y.; Inaba, M.; Takahara, I. Catalytic fast pyrolysis of jatropha wastes. J. Anal. Appl. Pyrolysis 2012, 94, 75–82. [Google Scholar] [CrossRef]
- Jae, J.; Tompsett, G.A.; Lin, Y.-C.; Carlson, T.R.; Shen, J.; Zhang, T.; Yang, B.; Wyman, C.E.; Conner, W.C.; Huber, G.W. Depolymerization of lignocellulosic biomass to fuel precursors: Maximizing carbon efficiency by combining hydrolysis with pyrolysis. Energy Environ. Sci. 2010, 3, 295–303. [Google Scholar] [CrossRef]
- Li, X.; Su, L.; Wang, Y.; Yu, Y.; Wang, C.; Li, X.; Wang, Z. Catalytic fast pyrolysis of Kraft lignin with HZSM-5 zeolite for producing aromatic hydrocarbons. Front. Environ. Sci. Eng. 2012, 6, 295–303. [Google Scholar] [CrossRef]
- Ma, Z.; Troussard, E.; van Bokhoven, J.A. Controlling the selectivity to chemicals from lignin via catalytic fast pyrolysis. Appl. Catal. A Gen. 2012, 423–424, 130–136. [Google Scholar] [CrossRef]
- Pattiya, A.; Titiloye, J.O.; Bridgwater, A.V. Evaluation of catalytic pyrolysis of cassava rhizome by principal component analysis. Fuel 2010, 89, 244–253. [Google Scholar] [CrossRef]
- Encinar, J.M.; Gonzalez, J.F.; Martinez, G.; Roman, S. Catalytic pyrolysis of exhausted olive oil waste. J. Anal. Appl. Pyrolysis 2009, 85, 197–203. [Google Scholar] [CrossRef]
- Kuen-Song, L.; Wang, H.P.; Chang, N.B.; Jou, C.; Hsiao, M. Synthesis of ZSM-type zeolites from biowaste gasification ashes. Energy Sources 2003, 25, 565–576. [Google Scholar] [CrossRef]
- Muradov, N.; Fidalgo, B.; Gujar, A.C.; Garceau, N.; T-Raissi, A. Production and characterization of lemna minor bio-char and its catalytic application for biogas reforming. Biomass Bioenergy 2012, 42, 123–131. [Google Scholar] [CrossRef]
- Gagnon, J.; Kaliaguine, S. Catalytic hydrotreatment of vacuum pyrolysis oils from wood. Ind. Eng. Chem. Res. 1988, 27, 1783–1788. [Google Scholar] [CrossRef]
- Wang, Y.; He, T.; Liu, K.; Wu, J.; Fang, Y. From biomass to advanced bio-fuel by catalytic pyrolysis/hydro-processing: Hydrodeoxygenation of bio-oil derived from biomass catalytic pyrolysis. Bioresour. Technol. 2012, 108, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Yan, Y.J.; Ren, Z.W. Online upgrading of organic vapors from the fast pyrolysis of biomass. J. Fuel Chem. Technol. 2008, 36, 666–671. [Google Scholar] [CrossRef]
- Alyani, M.; Towfighi, J.; Sadrameli, S.M. Effect of process variables on product yield distribution in thermal catalytic cracking of naphtha to light olefins over Fe/HZSM-5. Korean J. Chem. Eng. 2011, 28, 1351–1358. [Google Scholar] [CrossRef]
- Song, H.; Tan, B.; Ozkan, U.S. Novel synthesis techniques for preparation of Co/CeO2 as ethanol steam reforming catalysts. Catal. Lett. 2009, 132, 422–429. [Google Scholar] [CrossRef]
- Butler, E.; Devlin, G.; Meier, D.; McDonnell, K. A review of recent laboratory research and commercial developments in fast pyrolysis and upgrading. Renew. Sustain. Energy Rev. 2011, 15, 4171–4186. [Google Scholar] [CrossRef]
- Katikaneni, S.P.R.; Adjaye, J.D.; Bakhshi, N.N. Conversion of canola oil to various hydrocarbons over Pt/HZSM-5 bifunctional catalyst. Can. J. Chem. Eng. 1997, 75, 391–401. [Google Scholar] [CrossRef]
- Babich, I.V.; van der Hulst, M.; Lefferts, L.; Moulijn, J.A.; O'Connor, P.; Seshan, K. Catalytic pyrolysis of microalgae to high-quality liquid bio-fuels. Biomass Bioenergy 2011, 35, 3199–3207. [Google Scholar] [CrossRef]
- Lu, Q.; Wang, Z.; Dong, C.-Q.; Zhang, Z.-F.; Zhang, Y.; Yang, Y.-P.; Zhu, X.-F. Selective fast pyrolysis of biomass impregnated with ZnCl2: Furfural production together with acetic acid and activated carbon as by-products. J. Anal. Appl. Pyrolysis 2011, 91, 273–279. [Google Scholar] [CrossRef]
- Putun, E. Catalytic pyrolysis of biomass: Effects of pyrolysis temperature, sweeping gas flow rate and MgO catalyst. Energy 2010, 35, 2761–2766. [Google Scholar] [CrossRef]
- Lim, X.Y.; Andresen, J.M. Pyro-catalytic deoxgenated bio-oil from palm oil empty fruit bunch and fronds with boric oxide in a fixed-bed reactor. Fuel Process. Technol. 2011, 92, 1796–1804. [Google Scholar] [CrossRef]
- Park, H.J.; Park, K.-H.; Jeon, J.-K.; Kim, J.; Ryoo, R.; Jeong, K.-E.; Park, S.H.; Park, Y.-K. Production of phenolics and aromatics by pyrolysis of miscanthus. Fuel 2012, 97, 379–384. [Google Scholar] [CrossRef]
- Jeon, M.-J.; Kim, S.-S.; Jeon, J.-K.; Park, S.H.; Kim, J.M.; Sohn, J.M.; Lee, S.-H.; Park, Y.-K. Catalytic pyrolysis of waste rice husk over mesoporous materials. Nanoscale Res. Lett. 2012, 7, 18:1–18:5. [Google Scholar] [CrossRef]
- Zhu, X.; Lobban, L.L.; Mallinson, R.G.; Resasco, D.E. Bifunctional transalkylation and hydrodeoxygenation of anisole over a Pt/Hbeta catalyst. J. Catal. 2011, 281, 21–29. [Google Scholar] [CrossRef]
- Ausavasukhi, A.; Sooknoi, T.; Resasco, D.E. Catalytic deoxygenation of benzaldehyde over gallium-modified ZSM-5 zeolite. J. Catal. 2009, 268, 68–78. [Google Scholar] [CrossRef]
- Fanchiang, W.-L.; Lin, Y.-C. Catalytic fast pyrolysis of furfural over HZSM-5 and Zn/HZSM-5 catalysts. Appl. Catal. A Gen. 2012, 419–420, 102–110. [Google Scholar] [CrossRef]
- Neumann, G.T.; Hicks, J.C. Effects of cerium and aluminum in cerium-containing hierarchical HZSM-5 catalysts for biomass upgrading. Top. Catal. 2012, 55, 196–208. [Google Scholar] [CrossRef]
- Castano, P.; Gutierrez, A.; Villanueva, I.; Pawelec, B.; Bilbao, J.; Arandes, J.M. Effect of the support acidity on the aromatic ring-opening of pyrolysis gasoline over Pt/HZSM-5 catalysts. Catal. Today 2009, 143, 115–119. [Google Scholar] [CrossRef]
- Castano, P.; Pawelec, B.; Fierro, J.L.G.; Arandes, J.M.; Bilbao, J. Aromatics reduction of pyrolysis gasoline (pygas) over HY-supported transition metal catalysts. Appl. Catal. A Gen. 2006, 315, 101–113. [Google Scholar] [CrossRef]
- Liguori, L.; Barth, T. Palladium-nafion sac-13 catalysed depolymerisation of lignin to phenols in formic acid and water. J. Anal. Appl. Pyrolysis 2011, 92, 477–484. [Google Scholar] [CrossRef]
- Valle, B.; Gayubo, A.G.; Aguayo, A.T.; Olazar, M.; Bilbao, J. Selective production of aromatics by crude bio-oil valorization with a nickel-modified HZSM-5 zeolite catalyst. Energy Fuels 2010, 24, 2060–2070. [Google Scholar] [CrossRef]
- Chattopadhyay, J.; Son, J.E.; Pak, D. Preparation and characterizations of Ni-alumina, Ni-ceria and Ni-alumina/ceria catalysts and their performance in biomass pyrolysis. Korean J. Chem. Eng. 2011, 28, 1677–1683. [Google Scholar] [CrossRef]
- Cheng, Y.-T.; Jae, J.; Shi, J.; Fan, W.; Huber, G.W. Production of renewable aromatic compounds by catalytic fast pyrolysis of lignocellulosic biomass with bifunctional Ga/ZSM-5 catalysts. Angew. Chem. Int. Ed. 2012, 124, 1416–1419. [Google Scholar] [CrossRef]
- Ben, H.; Ragauskas, A.J. Pyrolysis of Kraft lignin with additives. Energy Fuels 2011, 25, 4662–4668. [Google Scholar] [CrossRef]
- Antonakou, E.; Lappas, A.; Nilsen, M.H.; Bouzga, A.; Stocker, M. Evaluation of various types of Al-MCM-41 materials as catalysts in biomass pyrolysis for the production of bio-fuels and chemicals. Fuel 2006, 85, 2202–2212. [Google Scholar] [CrossRef]
- Torri, C.; Reinikainen, M.; Lindfors, C.; Fabbri, D.; Oasmaa, A.; Kuoppala, E. Investigation on catalytic pyrolysis of pine sawdust: Catalyst screening by Py-GC-MIP-AED. J. Anal. Appl. Pyrolysis 2010, 88, 7–13. [Google Scholar] [CrossRef]
- Aho, A.; Kumar, N.; Lashkul, A.V.; Eranen, K.; Ziolek, M.; Decyk, P.; Salmi, T.; Holmbom, B.; Hupa, M.; Murzin, D.Y. Catalytic upgrading of woody biomass derived pyrolysis vapours over iron modified zeolites in a dual-fluidized bed reactor. Fuel 2010, 89, 1992–2000. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, F.; Cao, J.; Wang, J. Pyrolysis of pine wood in a slowly heating fixed-bed reactor: Potassium carbonate versus calcium hydroxide as a catalyst. Fuel Process. Technol. 2010, 91, 942–950. [Google Scholar] [CrossRef]
- Luo, J.; Li, J.; Shen, D.; He, L.; Tong, D.; Hu, C. Catalytic pyrolysis of pubescens to phenols over Ni/C catalyst. Sci. China Chem. 2010, 53, 1487–1491. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dickerson, T.; Soria, J. Catalytic Fast Pyrolysis: A Review. Energies 2013, 6, 514-538. https://doi.org/10.3390/en6010514
Dickerson T, Soria J. Catalytic Fast Pyrolysis: A Review. Energies. 2013; 6(1):514-538. https://doi.org/10.3390/en6010514
Chicago/Turabian StyleDickerson, Theodore, and Juan Soria. 2013. "Catalytic Fast Pyrolysis: A Review" Energies 6, no. 1: 514-538. https://doi.org/10.3390/en6010514
APA StyleDickerson, T., & Soria, J. (2013). Catalytic Fast Pyrolysis: A Review. Energies, 6(1), 514-538. https://doi.org/10.3390/en6010514