1. Introduction
In 1934, a Texoma Natural Gas Company Engineer named E.G. Hammerschmidt wrote “Solid compounds, resembling snow or ice in appearance, are formed with methane, ethane, propane and isobutane in the presence of water at elevated pressures and temperatures” [
1]. Those solids which resembled snow or ice turned out to be gas hydrates and the petroleum industry has since spent vast sums of money trying to avoid their formation [
2]. Gas hydrates are crystalline compounds in which cage networks of water molecules trap gas molecules. Depending on the gas molecules involved, gas hydrates that are found in nature will usually be found in one of three crystal forms: sI (cubic), sII (cubic) or sH (hexagonal). Smaller molecules such as methane or ethane will form sI hydrates, whereas propane and other similar sized molecules will form sII hydrates. sH hydrates require the presence of both a large molecule and a smaller “helper” molecule and as such, they are only very rarely found in nature [
2].
Since Hammerschmidt’s seminal paper, the flow assurance industry has been established to ensure that petroleum fluids can flow without being obstructed by gas hydrates, waxes or asphaltenes [
3]. Historically, flow assurance as applied to gas hydrates has involved injecting large quantities of thermodynamic hydrate inhibitors, such as methanol, into pipelines. The addition of these compounds extends the thermodynamic limits for hydrate-free operation. However, thermodynamic inhibitors are expensive and they are not environmentally benign. Additionally, as offshore oil and gas production moves into deeper water, the higher pressures push the required amount of methanol past the point of being economically feasible [
4].
Parallel to field developments in hydrate prevention and management, the last thirty years have also seen the development of computer tools to predict the formation and dissociation of hydrate pipeline plugs. Modelling the formation and dissociation of a hydrate pipeline plug requires the simultaneous solution of the heat/mass/momentum balance equations as well as the appropriate thermodynamic and kinetic equations. As such, these problems are computationally-intensive and it has only been since the late 1980s that computers have been powerful enough to tackle even the most simplistic cases.
Over the last thirty years several groups tackled the problem of predicting hydrate plug formation [
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15]; however, less attention has been paid to predicting hydrate plug dissociation. The first serious attempt to model the dissociation of a gas hydrate plug came from Ullerich et al. [
16] who proposed a 1D moving boundary heat transfer model. Their model [
16] considered the hydrate to be a rectangular 1D semi-infinite solid, in which a constant heat flux is applied at one of the boundaries. Additionally, their model treated the dissociating hydrate as an ablation problem; in an ablation problem the evolved gas blows the water away from the dissociating hydrate surface. The model [
16] was solved analytically and the results were compared to experimental results [
16] for hydrate mass loss measurements. They [
16] were able to predict the quantitative behavior of the dissociating hydrate plug; however, they consistently underpredicted the mass of the collected gas by up to 10%. It should be noted that in 1987, not all of the thermophysical properties of methane hydrates were known and, as such, errors of 10% were acceptable. Even though this work presented no attempt at addressing hydrate dissociation by pressure reduction it was an important first step towards modelling hydrate pipeline plug dissociation.
Two years later, Jamaluddin et al. [
17] modelled the dissociation of a semi-infinite gas hydrate plug by combining 1D heat transfer with the kinetics of hydrate dissociation. As was done by Ullerich et al. [
16], their model [
17] treated the dissociating hydrate as an ablation problem. However, the inclusion of dissociation kinetics resulted in a boundary condition that was non-linear in temperature. Thus, a numerical solution was necessary. The results of the simulations [
17] predicted that changes in the system pressure were sufficient to transition hydrate dissociation from a heat-transfer-controlled process to a kinetics-controlled process. In the region of kinetics control, it was further seen that the value of the activation energy and the hydrate surface roughness have a significant effect on the observed rate of hydrate dissociation. Although these results predicted a transition from heat transfer control to kinetic control, the authors were not able to compare their predictions to experimental data.
Kelkar et al. [
18] derived a heat-conduction-based model for gas hydrate plug dissociation via double-sided depressurization. In their model, the radial dissociation of the hydrate plug was modelled as a 1D moving boundary heat conduction problem in rectangular coordinates. Since they used rectangular coordinates and since they assumed that there was no axial hydrate dissociation, Kelkar et al. [
18] were able to derive an analytical expression for the position of the dissociating hydrate interface, as a function of time. Their [
18] results predicted that a rapid double-sided depressurization of the hydrate plug could result in the hydrate plug becoming coated by a layer of ice. Interestingly, Kelkar et al. [
18] also predicted that the time for complete plug dissociation went through a minimum at −5 °C. Unfortunately, Kelkar et al. [
18] were not able to compare their predictions to any experimental data.
While Kelkar’s model [
18] suffered from several questionable assumptions, such as the dissociating hydrate being surrounded by a stagnant layer of water, it became the starting point for several subsequent attempts at modelling hydrate plug dissociation. In 2003, Bollavaram and Sloan [
19] presented experimental results and modelling results for hydrate dissociation by single-sided and double-sided depressurization. In the experiments, hydrate plugs were formed in 36” by 1” cylindrical vessels and then subsequently dissociated by either single- or double-sided depressurization. In modelling the results for the double-sided depressurization, Bollavaram and Sloan [
19] transformed Kelkar’s model [
18] to cylindrical coordinates. A key observation of their work [
19] was to confirm Kelkar’s [
18] assumption that axial dissociation is negligible during double-sided depressurization.
Bollavaram and Sloan [
19] also described a model for single-sided hydrate dissociation; however, in their 2003 work [
19] they only described the equations in words. The full set of equations for single-sided depressurization was eventually presented by Davies et al. [
20] in 2006. Their model [
20] consists of four successive steps: (1) the computation of the axial pressure profile, (2) the computation of the gas flow velocity through the hydrate plug, (3) the computation of the axial temperature profile and (4) the successive radial dissociation of small sections of the hydrate plug. When used to correlate experimental data [
20], the model can capture the trend of the single-sided depressurization process. Additionally, Davies et al. [
20] also correlated experimental results for the case of hydrate pipeline plug dissociation via electrical heating. Like the case of double-sided dissociation, they [
20] observed that dissociation via electrical heating primarily proceeded in the radial direction.
Despite the successes of their model [
20], the derivation of the model leads to two issues that warrant mention. First, it was assumed [
20] that each of the axial slices of the hydrate plug are at a constant temperature. This gives rise to a temperature discontinuity at the hydrate–pipe interface, which in turn can create numerical challenges when implementing the model. Second, as each of the axial slices dissociates, it was assumed that they were surrounded by a stagnant layer of water. This assumption is questionable since the pressure gradient will likely cause the water to flow downstream. Finally, it should also be noted that Davies et al. [
20] mentioned that the effect of kinetics should be studied; however, they [
20] did not provide any insight into how kinetics should be incorporated into their model.
Since 2006 there have been no further studies, at least in the open literature, that attempt to model the dissociation of a hydrate pipeline plug. However, there have been a handful of related studies that bear mentioning in the context of the current work. The first is that of Sean et al. [
21] who proposed a kinetic model to describe the dissociation of a spherical pellet of methane hydrate suspended in a vertical downward flow of water. This model [
21] was not presented in the context of hydrate pipeline plug dissociation; however, it illustrated a method to couple hydrate dissociation kinetics and transport equations. In their model, Sean et al. [
21] expressed the driving force for hydrate dissociation in terms of the Gibbs energy difference between the hydrate and the liquid phases. A limitation of expressing the driving force in this manner is that it requires knowledge of the liquid phase mole fraction of methane, at equilibrium and at the system conditions; the mole fraction of methane at the system conditions was not known and thus, was fit to the experimental data. Subsequently the model was verified with experimental data in which a 1 cm diameter methane hydrate pellet was suspended in a stream of flowing water and dissociated at a pressure of 9 MPa and temperatures ranging from (276 to 284) K.
In 2007, Nazridoust and Ahmadi [
22] performed a computational fluid dynamics (CFD) simulation of methane hydrate dissociation within a sandstone core. In their model [
22] the energy balance on the dissociating hydrate is coupled with the dissociation kinetics model of Kim et al. [
23]. It was noted [
22] that the rate of dissociation is affected by the heat transfer from the environment as well as by the temperature/pressure conditions in the core holder. Unfortunately, in 2007, the experimental data needed to verify the modelling results were not available.
Also in 2007, Sean et al. [
24] extended their earlier work [
21] to dissociation by both depressurization and by temperature increase. Their generalized model [
24] was used to correlate experimental data on the dissociation of a hydrate-encrusted methane bubble. While the model was able to correlate the results, it suffered from the same limitations as their previous work [
22]; specifically, the dissociation driving force was expressed in terms of quantities that had to be fit to the experimental data.
Kumar et al. [
25], in 2013, proposed a model to describe the dissociation of CO
2 hydrates formed in a bed of glass beads. Their model [
25] combined hydrate dissociation with heat and mass flow in porous media. Subsequently, the model was used to correlate lab data on the dissociation of CO
2 hydrates formed in glass beads. In their model [
25], the kinetics of hydrate dissociation were described by the model of Kim et al. [
23]. While this work presented a methodology for including hydrate dissociation kinetics in modelling the dissociation of in situ hydrates, it did not address the question of whether the kinetics were significant. In other words, the results [
25] with the kinetics were not compared to the case of dissociation as a heat-transfer-controlled process. Almost a half-decade after the work of Kumar et al. [
25], Yu et al. [
26] used CFD to study the flow of methane through a porous mixture of sediment and dissociating methane hydrate. Their proposed model included the hydrate dissociation kinetics term proposed by Sean et al. [
21]. Of possible relevance to the current study is their observation that the inclusion of kinetics becomes important as the porosity decreases.
Finally, in 2021, Li et al. [
27] proposed an integral heat conduction model with Neumann boundary conditions. Of relevance to the current study, Li et al. [
27] presented the Stefan boundary conditions in a generalized vector form. The model was subsequently applied to a semi-infinite hydrate reservoir and it was seen that the model was able to track the location of the dissociating interface. The authors [
27] did not present any results for hydrate dissociation by depressurization; however, they did allude to how their model could be extended to such cases.
In the current study, a 2D axisymmetric model for hydrate plug dissociation is developed and implemented with COMSOL (Version 6.2) [
28], a commercially available CFD software package. Like the models of Bollavaram and Sloan [
19] and Davies et al. [
20], the current study considers the solid hydrate plug to be porous. However, the current model also includes the additional resistances of the kinetics of dissociation and the heat conduction through the wall of the pipe. Furthermore, the current study solves the 2D partial differential equations of heat/mass/momentum using a fully coupled solver whereas the previous works [
19,
20] computed the pressure, velocity, and temperature profiles sequentially.
2. Theory
This section presents a 2D axisymmetric model for the dissociation of a gas hydrate plug in a natural gas pipeline.
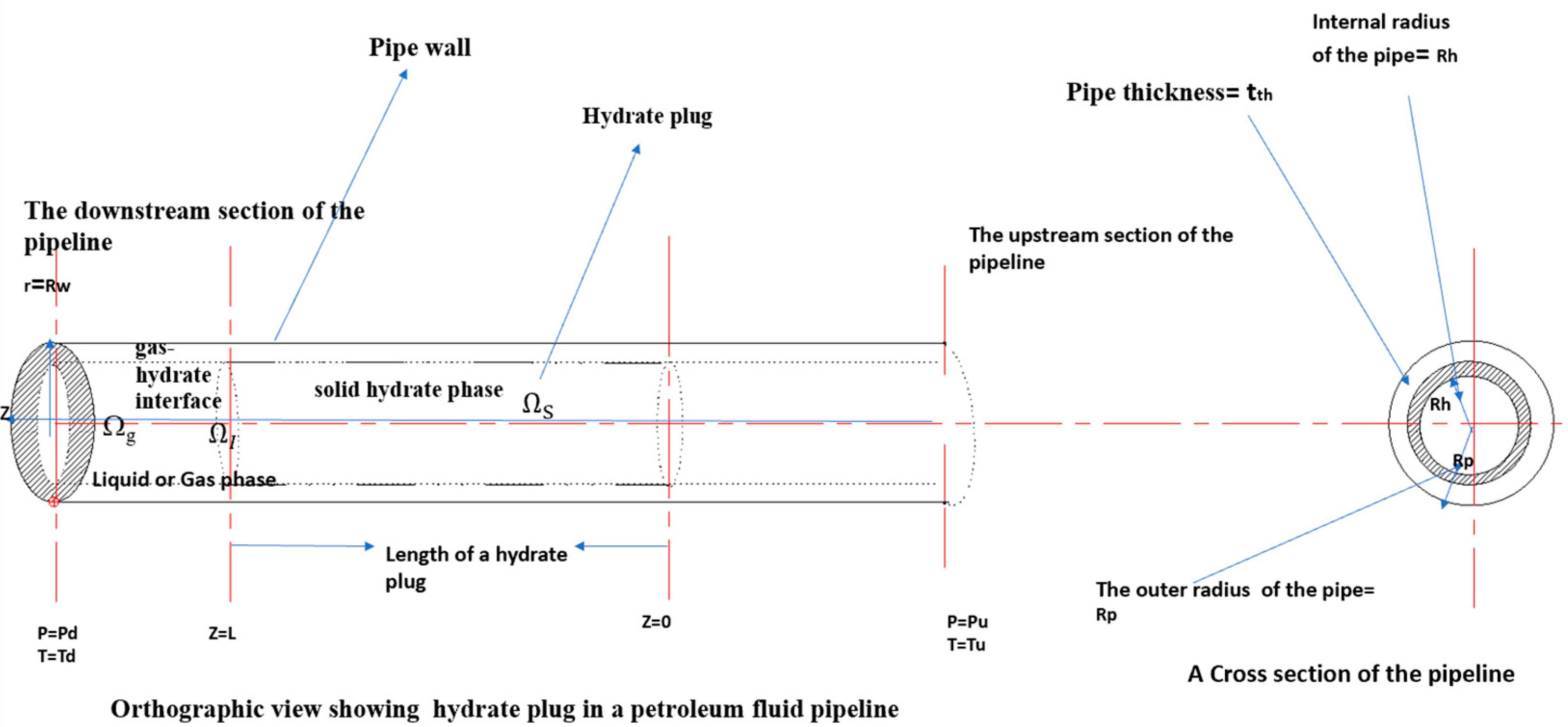
Figure 1 shows a schematic of a dissociating hydrate plug in a gas pipeline. In
Figure 1, the region containing the solid gas hydrate is denoted as
Ωh, the region containing the solid pipeline wall is denoted as
, the region that forms the interface between the hydrate phase and the gas phase is denoted as
ΩI and the region downstream of the hydrate plug is denoted as
Ωg.
At t = 0, a region of the pipeline of length L is filled with a porous gas hydrate plug. It is also assumed that there is no free liquid water present before dissociation begins. Hydrate dissociation can proceed by either single-sided or double-sided depressurization and the dissociation will result in a moving boundary problem. At the dissociating hydrate interface, it will be assumed that the hydrate surface temperature is equal to the three-phase equilibrium temperature at the downstream pressure. Furthermore, radiative effects, viscous dissipation and the work done by pressure changes are assumed to be negligible.
The model will compute the relevant temperature, pressure and phase fraction profiles by solving the coupled continuity, heat transfer, momentum transfer and hydrate dissociation kinetic equations. The equations will be solved using COMSOL (Version 6.2) [
28], which is a commercially available finite element method (FEM) solver. In the following sections, the equations for continuity, momentum transfer, heat transfer and hydrate dissociation kinetics will be presented, for each of the domains.
2.1. Continuity Equation
The continuity equation describes the conservation of mass within a given region. Within the hydrate region, the hydrate plug has a finite porosity and permeability, which is assumed to remain constant with respect to both time and position. As noted by Bollavaram and Sloan [
19], the single-sided depressurization of a sufficiently porous plug can result in gas flow through the plug. Even though gas may be able to flow through the plug it is assumed that free water can only be liberated at the hydrate–gas interface. Hence, there is no need to explicitly write a continuity equation for water flow within the hydrate. Within the porous solid hydrate, the conservation of mass equation for compressible gas flow can be written as follows:
where
u is the fluid velocity vector,
is the gas density and
is the volumetric flow rate of gas that is evolved from the dissociating hydrate. The volumetric flow rate of gas evolved from the dissociating hydrate is computed with the hydrate dissociation kinetic model of Clarke and Bishnoi [
29], which will be detailed in a later section. The gas density is computed with the Peng Robinson Equation of State [
30].
At the dissociating hydrate surface, it is assumed that the evolved gas ‘blows’ the water away from the hydrate, and that the pressure gradient subsequently carries the gas and the water away from the hydrate. As noted earlier, this assumption was also invoked by Jamaluddin et al. [
17] and by Ullerich et al. [
16]. For the sake of simplicity, it is assumed that the flow characteristics in this region can be approximated as a single-phase gas. In future works, the presence of additional fluids (oils and non-aqueous liquids) and solids may be considered. In such cases it will be necessary to perform a rigorous multi-phase flow simulation in this region; however, in the current work, the continuity equation in this region is simply the following:
In the initial state, the gas velocity is zero since it is assumed that there is no pressure gradient along the length of the hydrate plug.
2.2. Momentum Conservation for the Gas Flowing through the Hydrate Plug
During dissociation by depressurization, gas can flow through the porous hydrate plug if a pressure gradient exists. It is assumed that radial pressure gradients are negligible compared to the axial pressure gradient. In the model of Bollavaram and Sloan [
19] it was assumed that the gas flow through the porous hydrate plug was slow enough as to obey Darcy’s law. However, in the current study this assumption is relaxed and the full Brinkman equation [
31] will instead be used to describe the gas flow within the porous hydrate plug:
where
F is the body force vector or external force on gas per unit volume,
I is the identity matrix,
ρ is the gas density,
μ is the dynamic viscosity of the gas,
is the pressure,
is a mass source per unit volume of the porous medium and
K is the stress tensor, which is given by the following:
2.3. Momentum Conservation for the Free-Flowing Gas
As previously noted, the water and gas that are evolved from the dissociating hydrate are assumed to flow downstream due to the downstream pressure gradient. This assumption is consistent with the works of Ullerich et al. [
16] and Jamaluddin et al. [
17], who treated hydrate dissociation as an ablation problem. This assumption is also in contrast to what was done by Bollavaram and Sloan [
19], who assumed that the evolved water forms a stagnant liquid layer surrounding the dissociating hydrate. In the current work, the result for the momentum balance for this region is as follows:
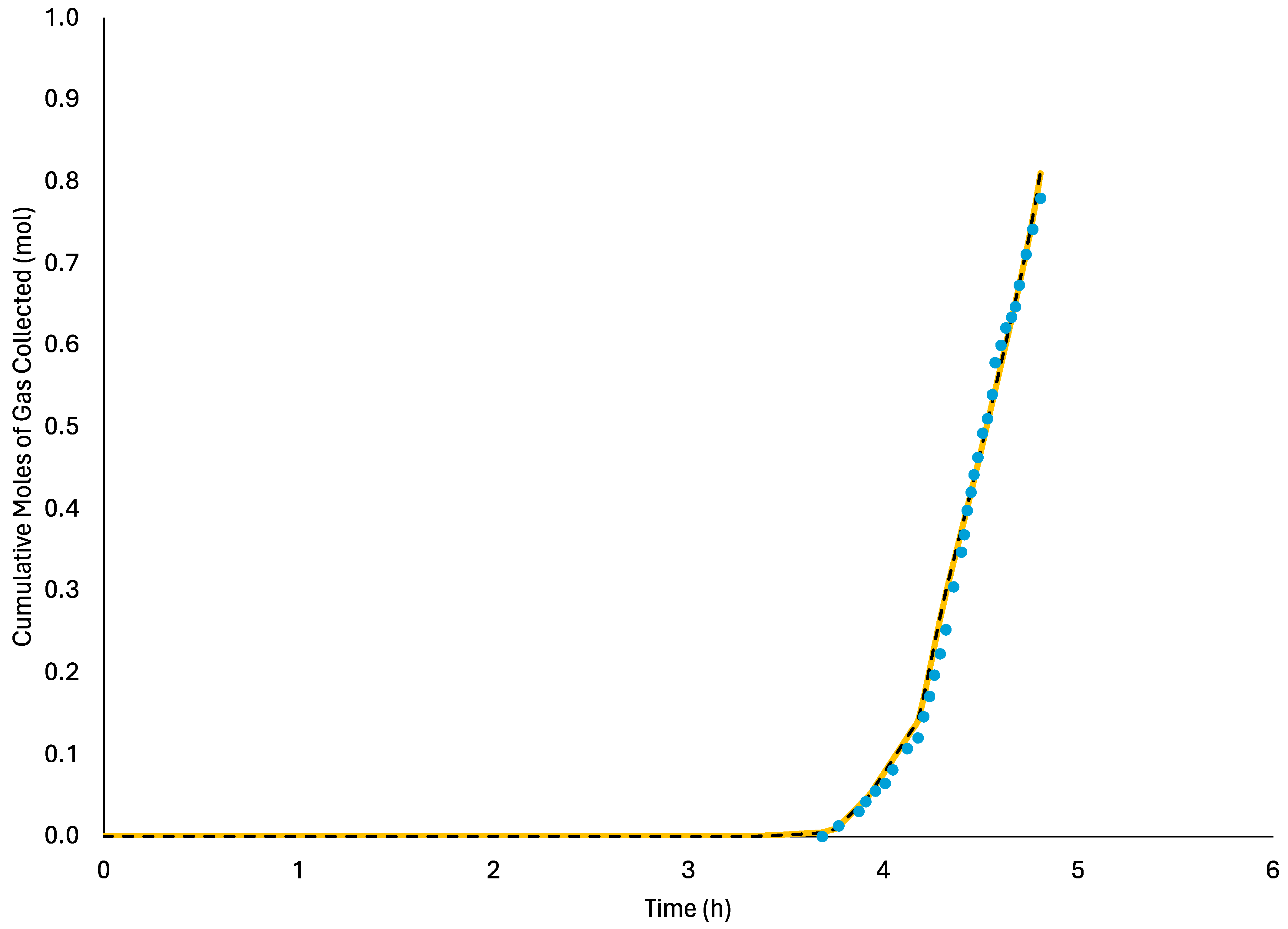
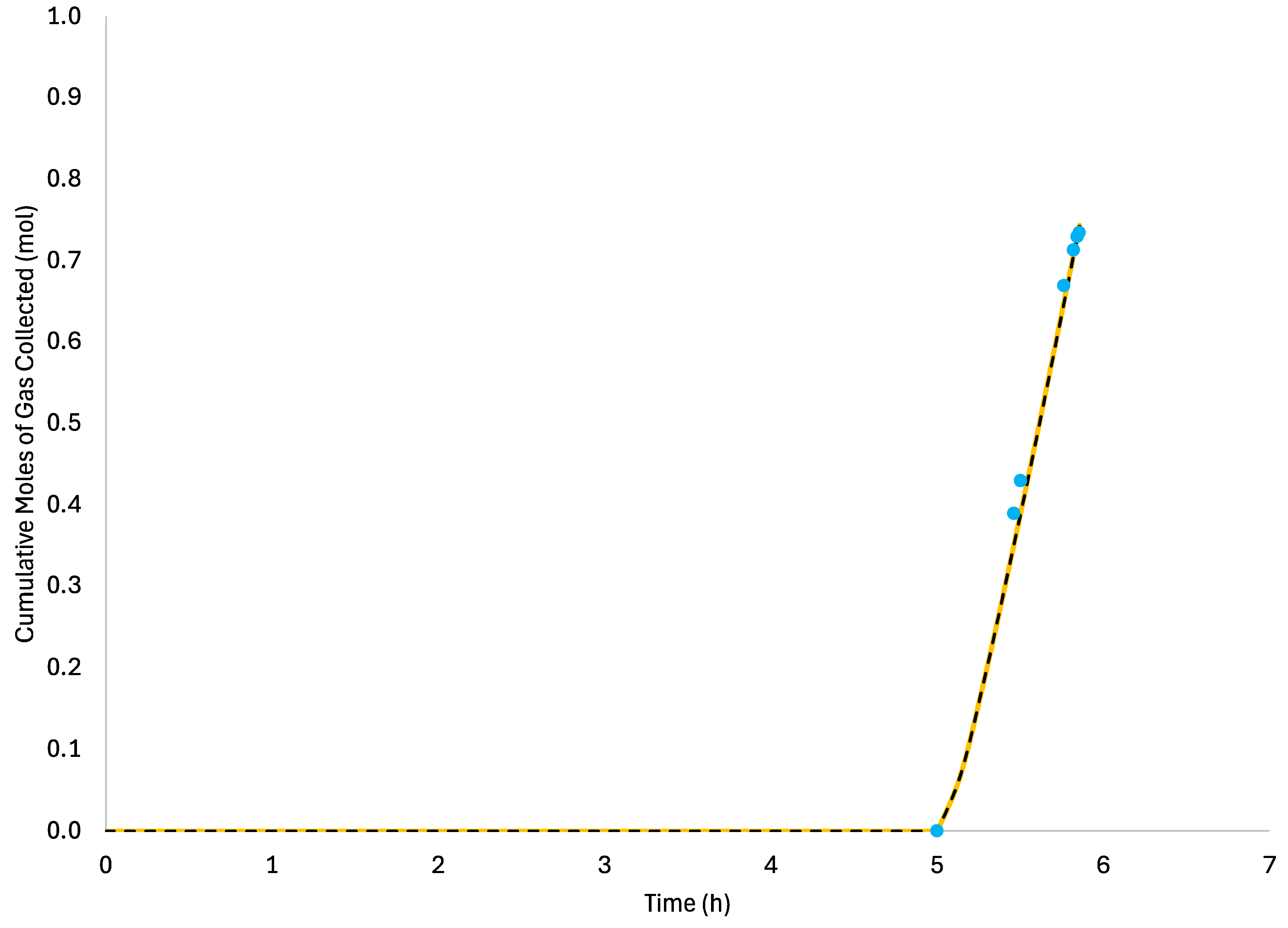
In this region, the initial gas velocity is taken to be zero since the pressure is assumed to be constant. For model verification, it will be assumed that the pressure is changed in step changes since the experimental pressure changes [
19,
20] are approximately step changes. It should be noted, however, that the current model can also consider other upstream and downstream pressure change functions by changing the appropriate pressure boundary conditions.
2.4. Energy Balance within the Solid Hydrate Plug
At
t = 0, it is assumed that the temperature of the hydrate and its surroundings (i.e., the pipe wall) are uniform. Once the downstream pressure is reduced, the resulting pressure gradient gives rise to a temperature gradient. Within the hydrate there may be heat transfer between the pipe wall and the hydrate as well as between the hydrate and the flowing gas. As noted by Bollavaram and Sloan [
19], the one-sided depressurization problem is effectively forced convection flow through a porous medium. Based on the experimental observations of Bollavaram and Sloan [
19], this work assumes that local thermal equilibrium exists between the gas and the hydrate phase. As a result, the energy balance for the hydrate phase and for the gas phase reduces to the following single equation:
where
At the interface between the dissociating hydrate and the gas, the temperature of the hydrate is assumed to be equal to the three-phase equilibrium temperature, at the gas pressure.
2.5. Energy Balance within the Free-Flowing Gas Region
In the case of dissociation by depressurization, gas can flow downstream from the dissociating hydrate plug. This region was not considered by Bollavaram and Sloan [
19] but it is being considered in this work because the temperature gradient at the interface may affect the rate of hydrate dissociation. In this region, the energy balance is given by Equation (9):
where the subscript g refers to the free-flowing gas,
is the specific heat capacity at a constant pressure of the gas,
is the thermal conductivity of gas,
is density of gas and
is temperature of gas. At the interface between the dissociating hydrate and the free-flowing gas, the temperature of the gas is the same on both sides of the interface.
2.6. Energy Balance at the Dissociating Hydrate Interface
At the interface between the dissociating hydrate phase and the gas, the energy balance is given by the Stefan equation [
32]:
where
is the velocity vector of the dissociating hydrate interface,
is the unit normal vector,
K is the thermal conductivity,
T is the temperature,
λ is the heat of dissociation and the subscripts
h and
g represent the hydrate and gas phases, respectively. When Equation (10) is used in conjunction with Equations (1)–(9) it becomes possible to track the location of the dissociating hydrate interface, as a function of time.
To account for the kinetics of hydrate dissociation at the interface, the work of Jamaluddin et al. [
1] will be generalized to a 2D hydrate. The rate of hydrate dissociation is given by the model of Kim et al. [
23]:
where
A is the surface area of the dissociating hydrate,
Kd is the hydrate dissociation rate constant,
f is the fugacity and the subscripts
eq,
g and
j refer to the three-phase equilibrium condition, the gas phase and component
j, respectively. In the current work, the model of Kim et al. [
23] is chosen due to the availability of rate constants for a relatively large number of compounds and due to the driving force being expressed in terms of quantities that are easily computed. As noted by Kim et al. [
23] and by Clarke and Bishnoi [
29], the rate constants for hydrate dissociation follow an Arrhenius relationship, as given in Equation (13):
where
Ko is the intrinsic rate constant of hydrate dissociation,
E is the activation energy of hydrate dissociation and
R is the universal gas constant. In the current study, only hydrate dissociation at temperatures above 273.15 K will be studied since intrinsic rate constants and activation energies are currently only available at those conditions.
To relate the molar rate of hydrate dissociation to the energy balance at the hydrate–gas interface, Jamaluddin et al. [
17] performed a mass balance around the 1D dissociating hydrate surface. When generalized to a 2D hydrate plug, the mass balance of Jamaluddin et al. [
17] becomes the following:
where
Ageo is the geometric area of the dissociating hydrate plug. By combining Equations (13), (11) and (10), the Stefan boundary condition can be re-written in terms of the hydrate dissociation rate parameters:
where the surface roughness,
, is an adjustable parameter which is defined as follows:
2.7. Energy Conservation within the Pipe Wall
In the experimental model of Bollavaram and Sloan [
19], heat conduction through the pipeline wall was not considered. In the current work, this additional resistance is considered to avoid the potential for encountering temperature discontinuities at the interface between the hydrate and the pipe inner wall. The energy balance within the pipe is given by the following equation:
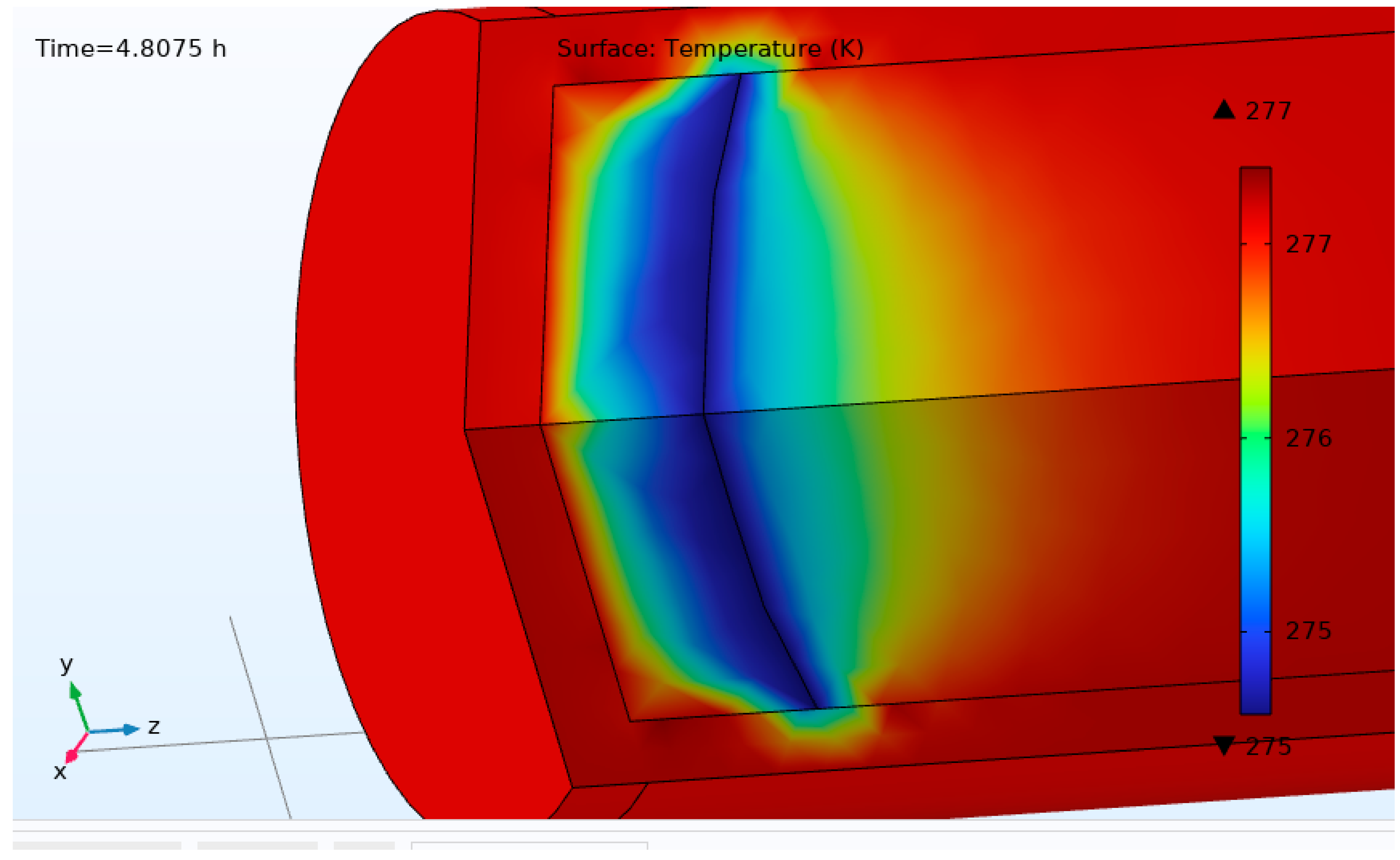
At the interface between the hydrate and the inner pipe wall, the temperature on both sides of the interface must be equal. Similarly, at the interface between the inner pipe wall and the free gas phase, the temperature also must be equal on both sides of the interface. The temperature boundary condition on the outer pipe wall can be set to either a constant temperature, a time varying temperature or a constant heat flux, depending on the scenario under consideration.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}