
Pyrolysis of Cyclohexane and 1-Hexene at High Temperatures and Pressures—A Photoionization Mass Spectrometry Study

Abstract
:1. Introduction
2. Methods
2.1. Experiments
2.2. Simulations
3. Results
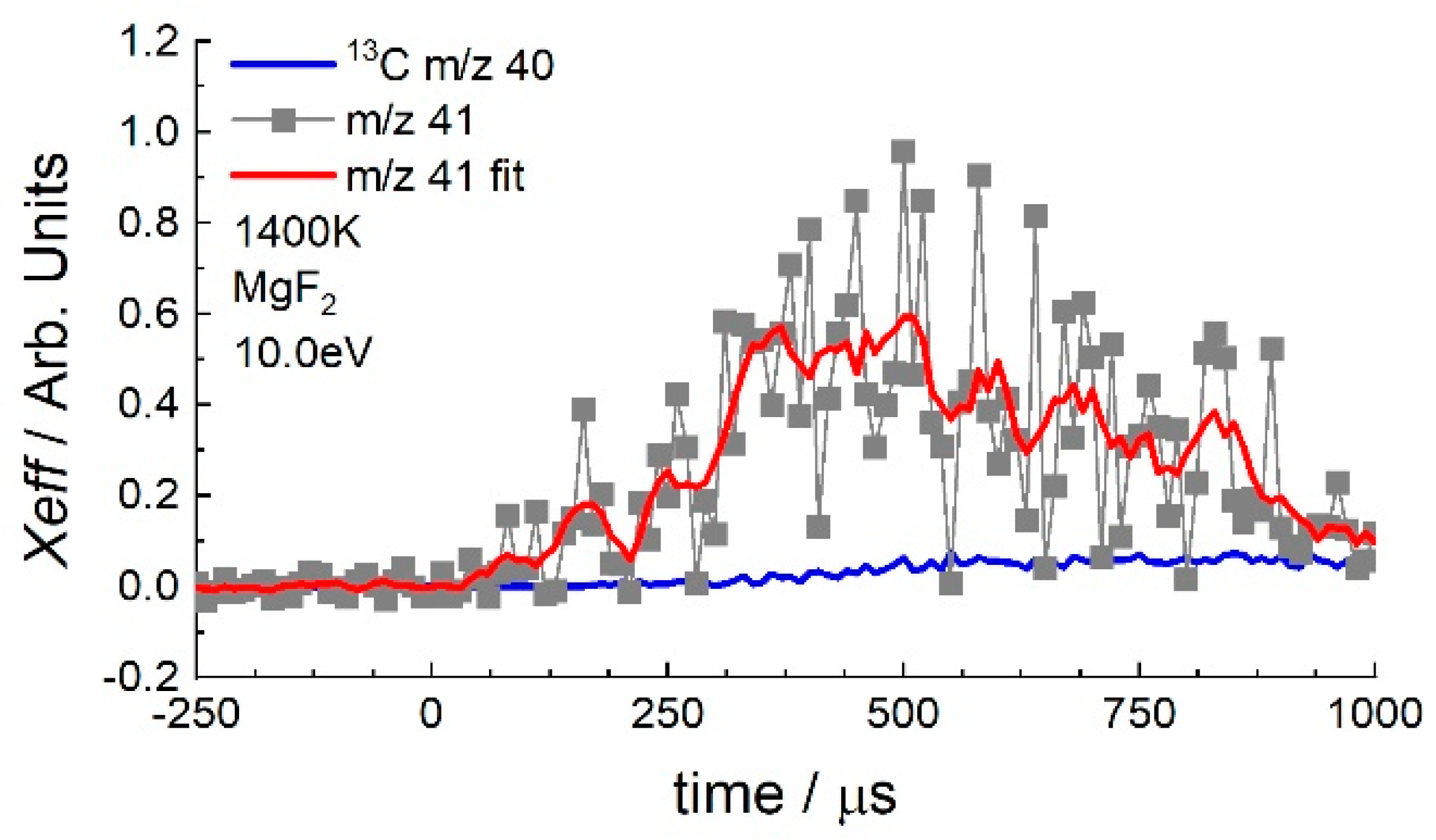
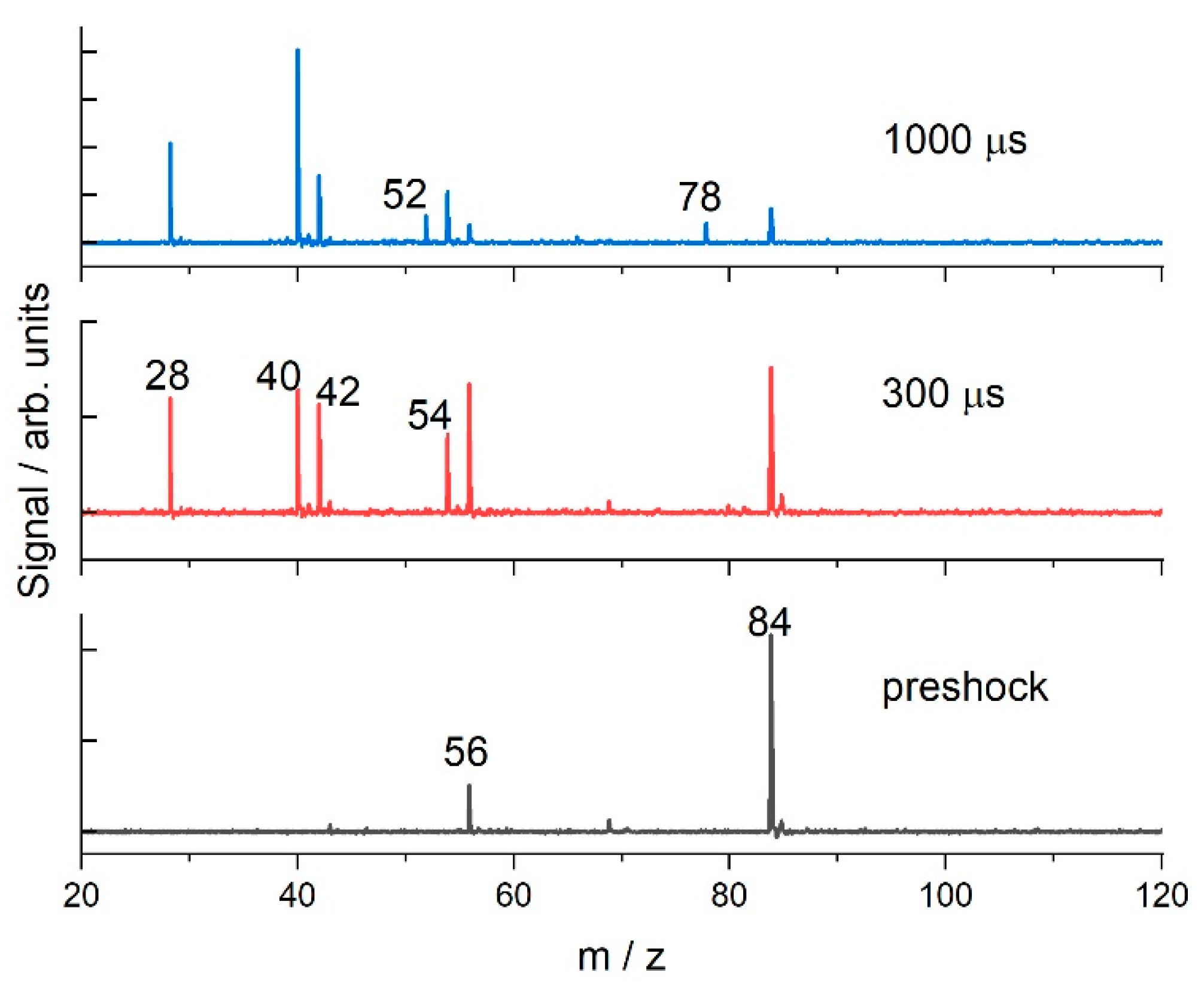
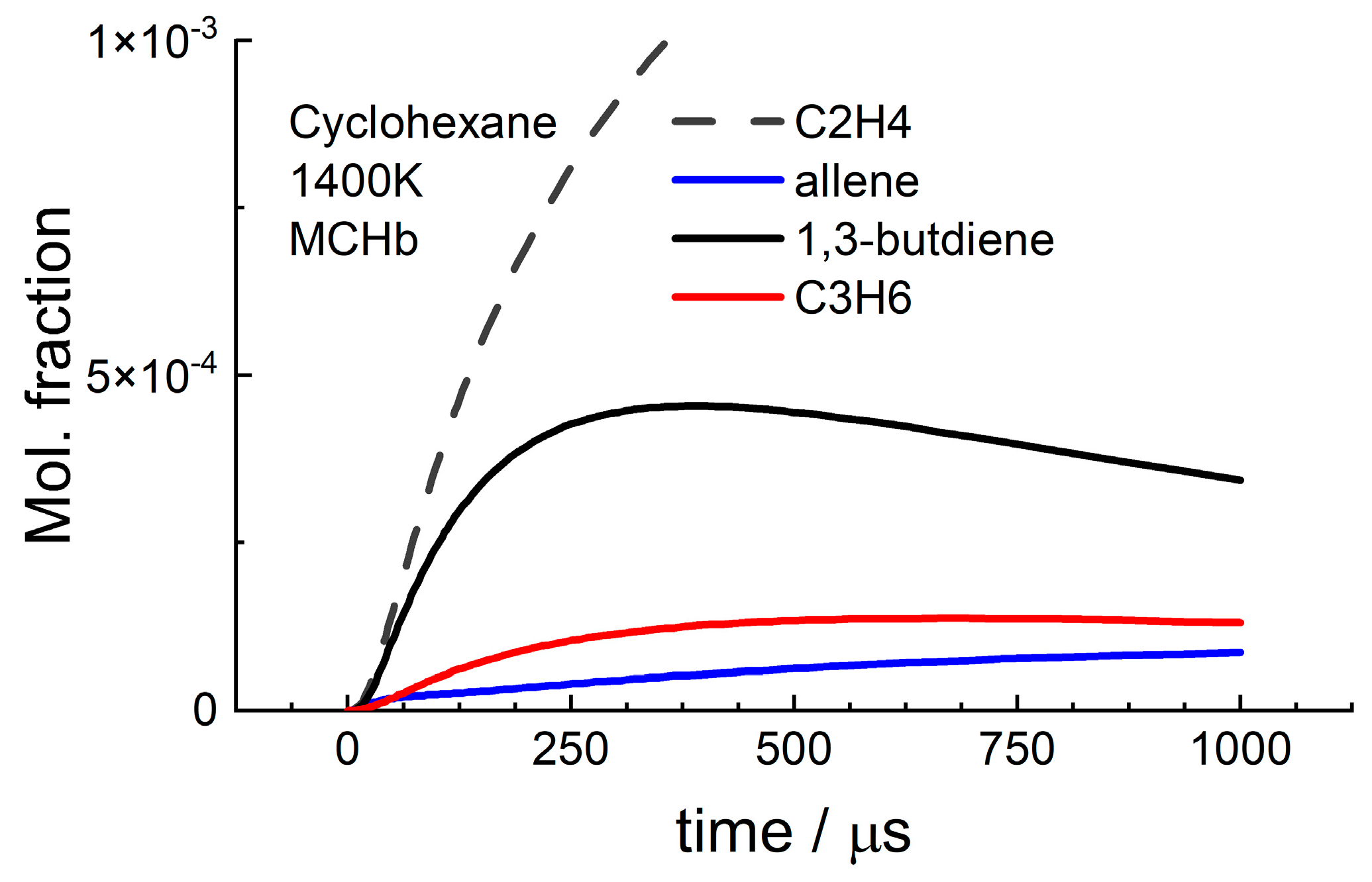
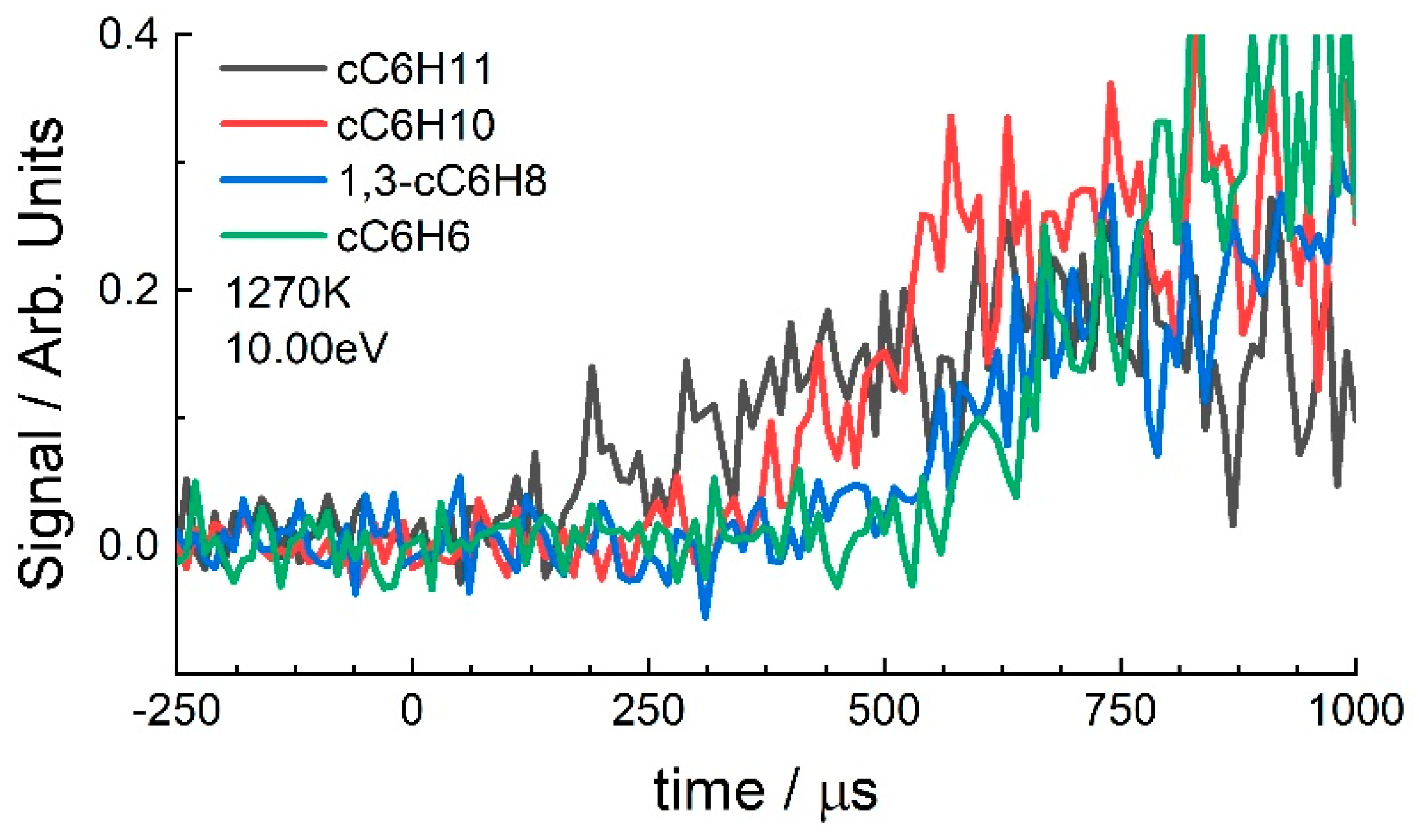
3.1. Cyclohexane
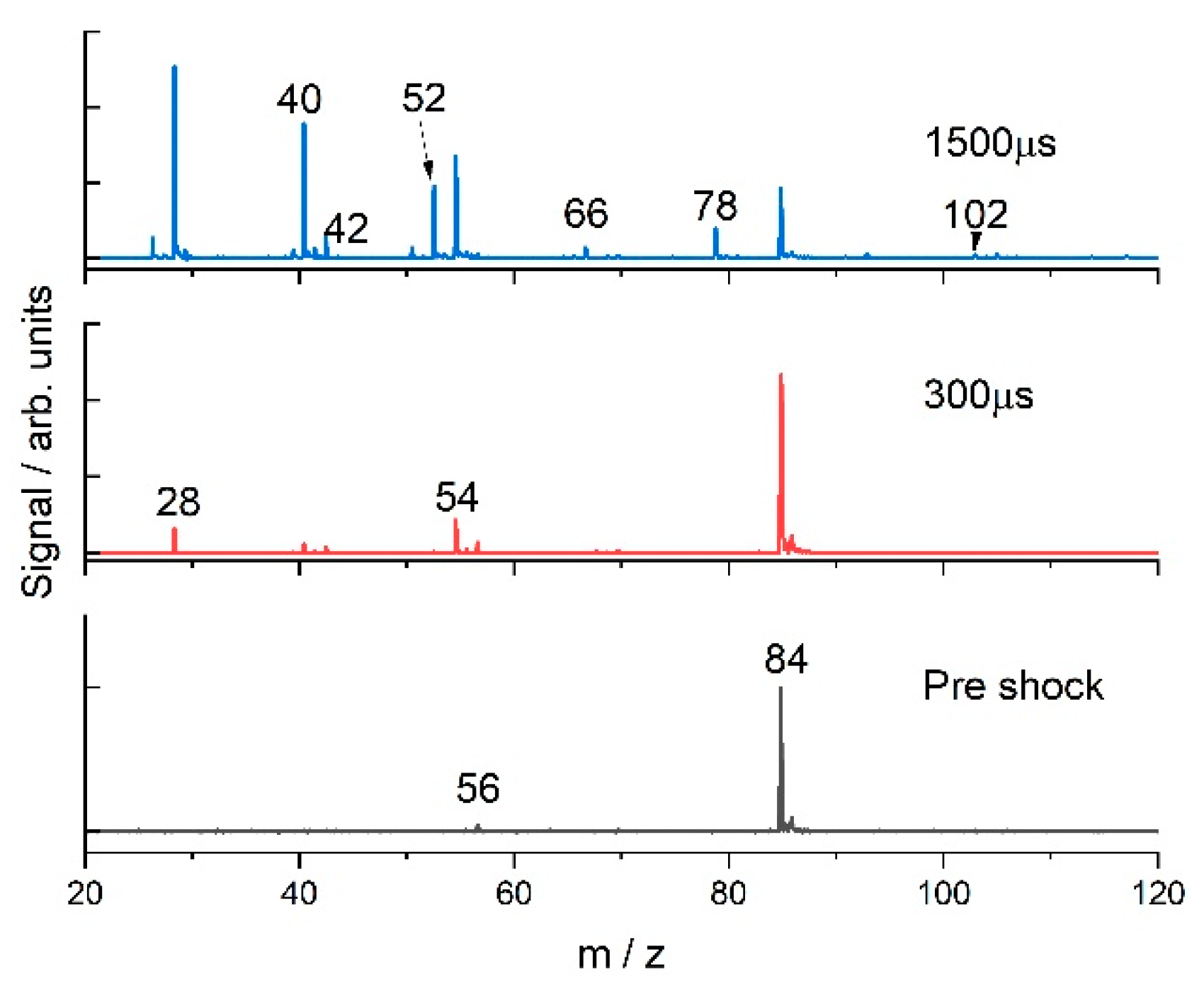
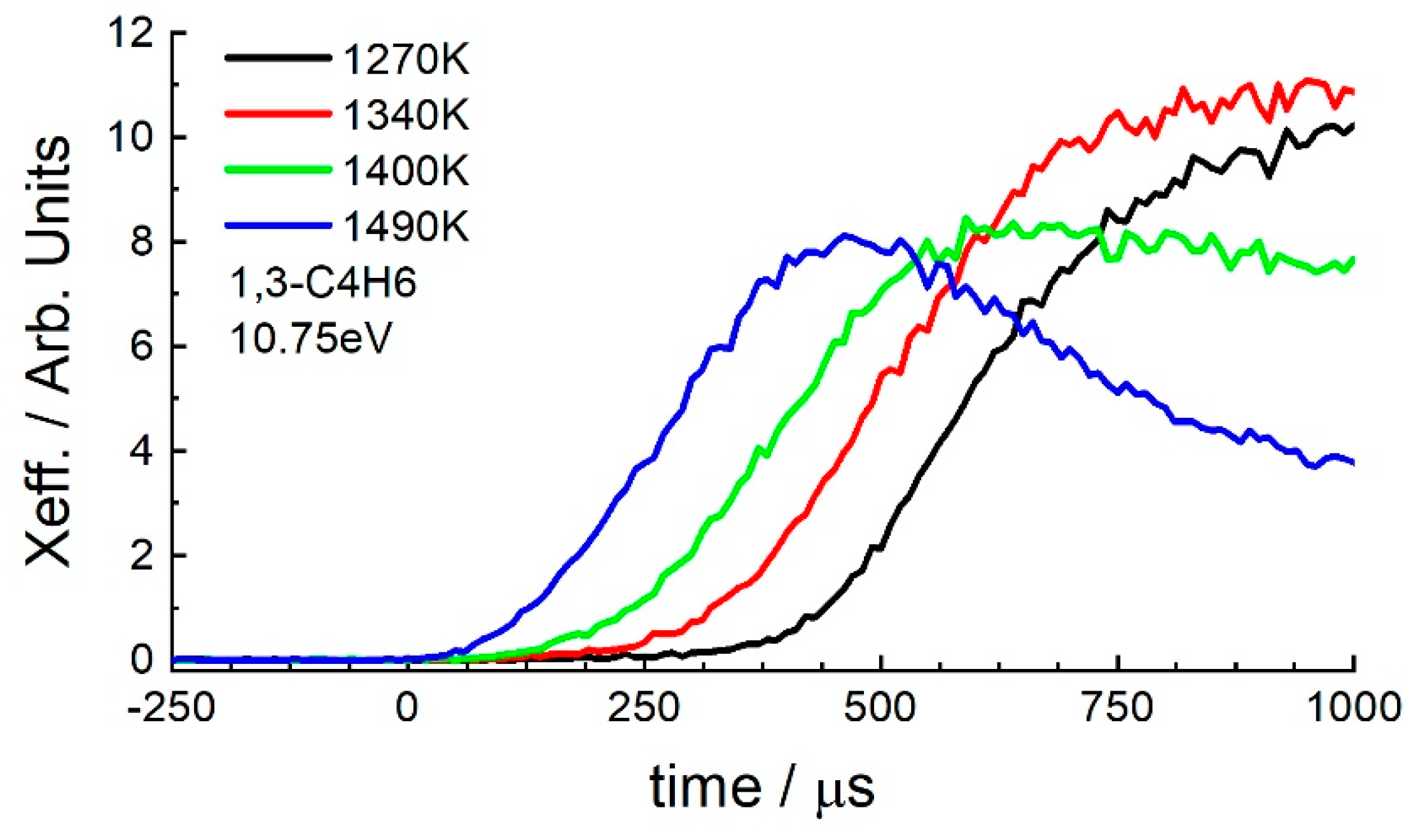
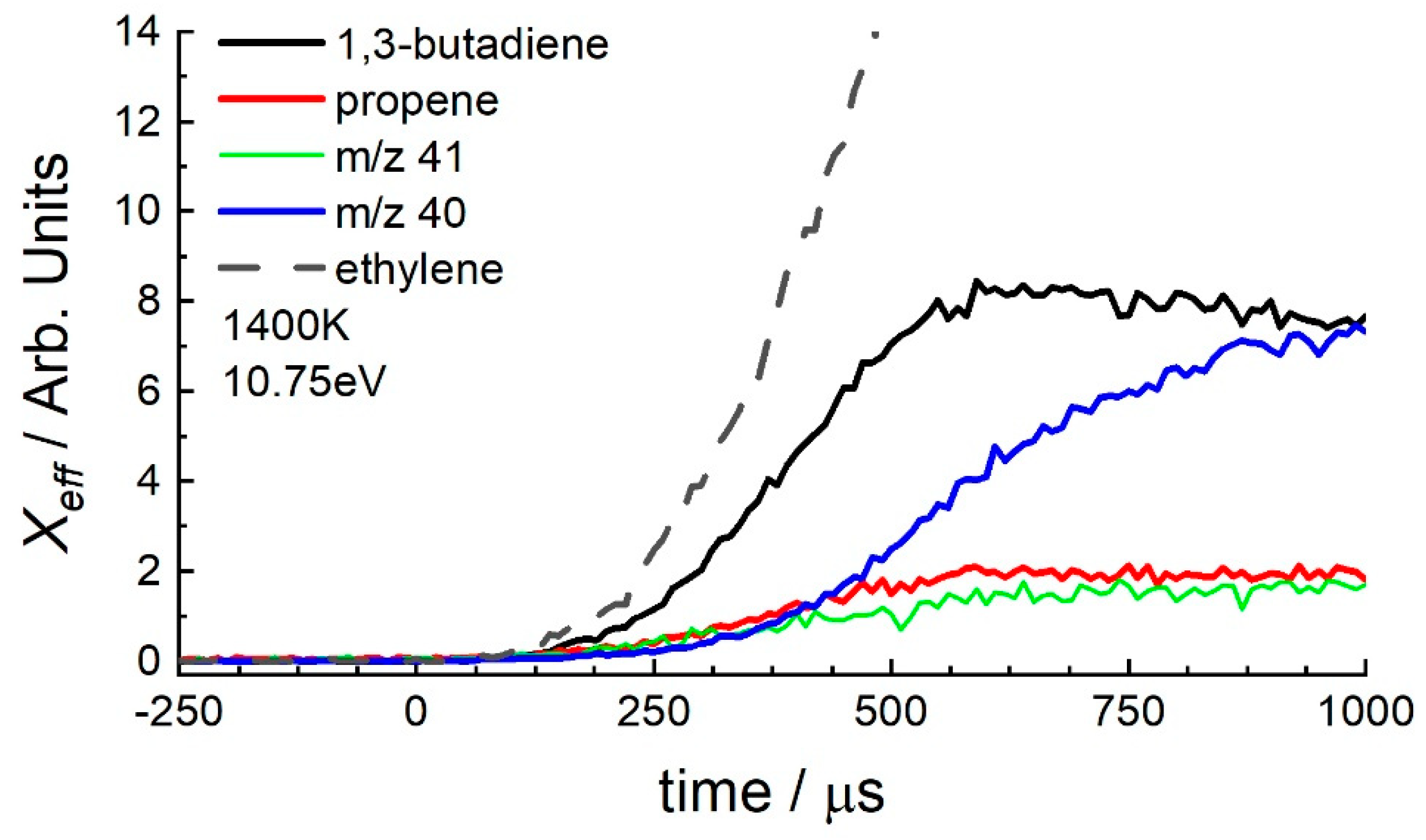
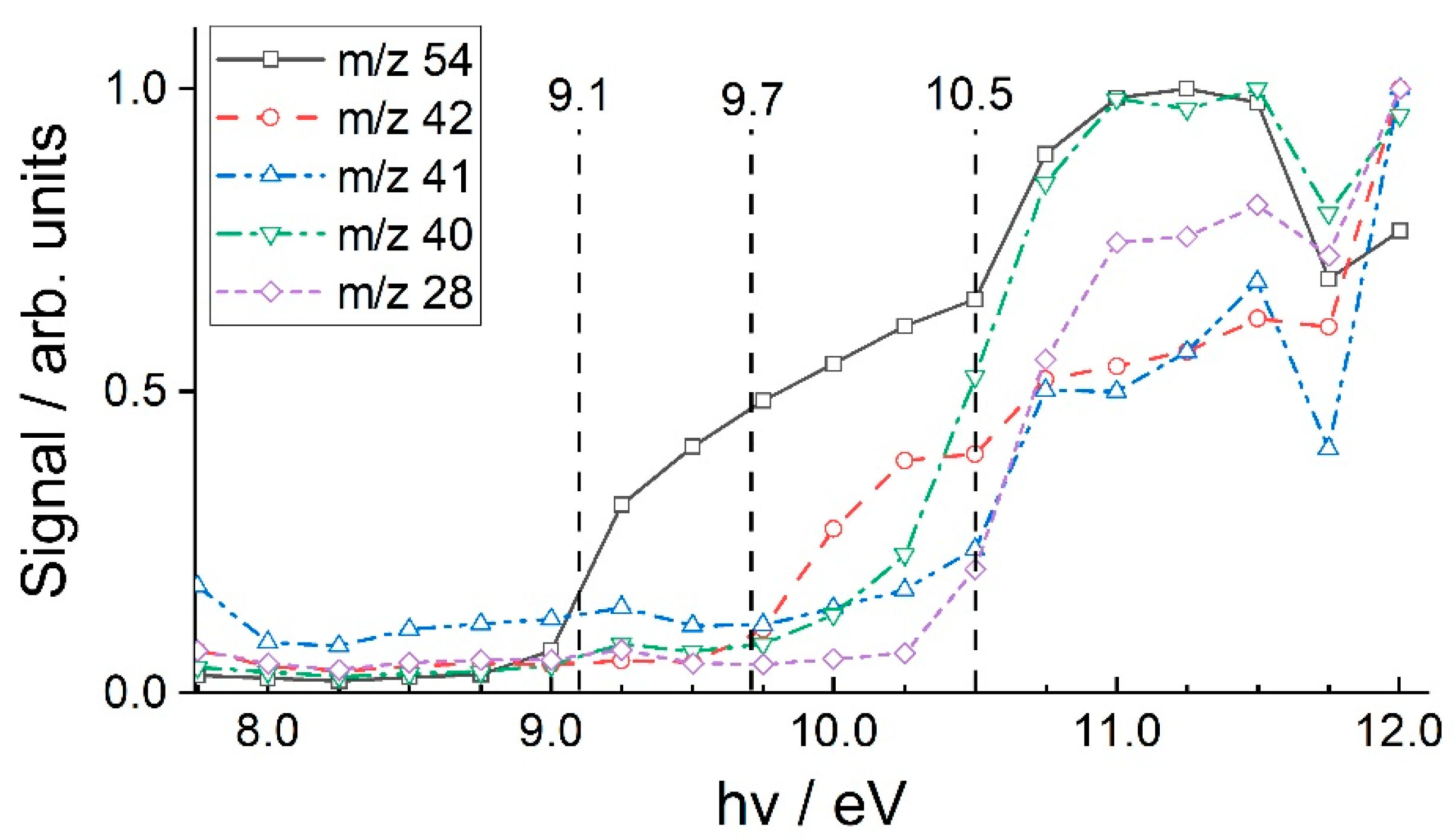
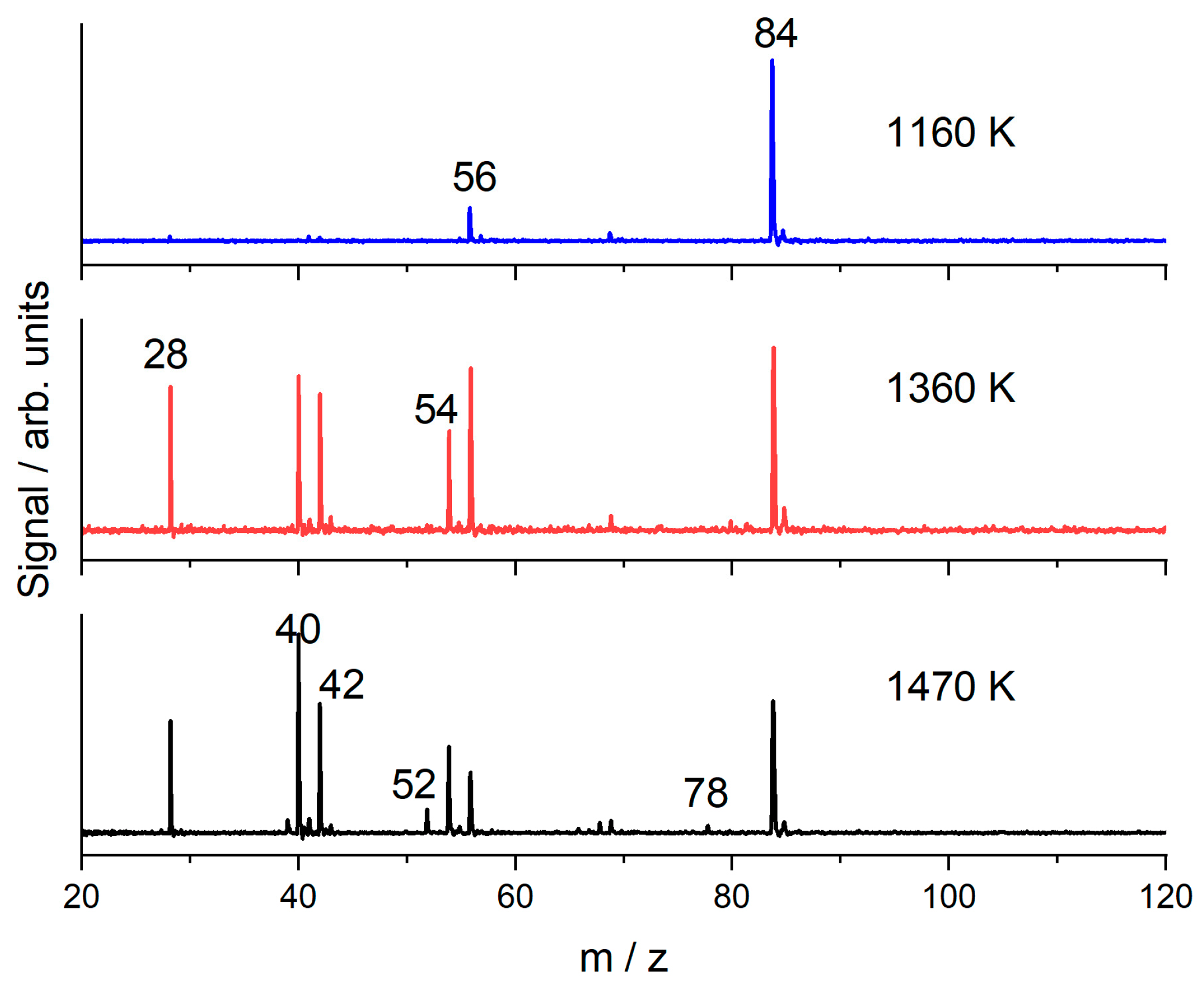
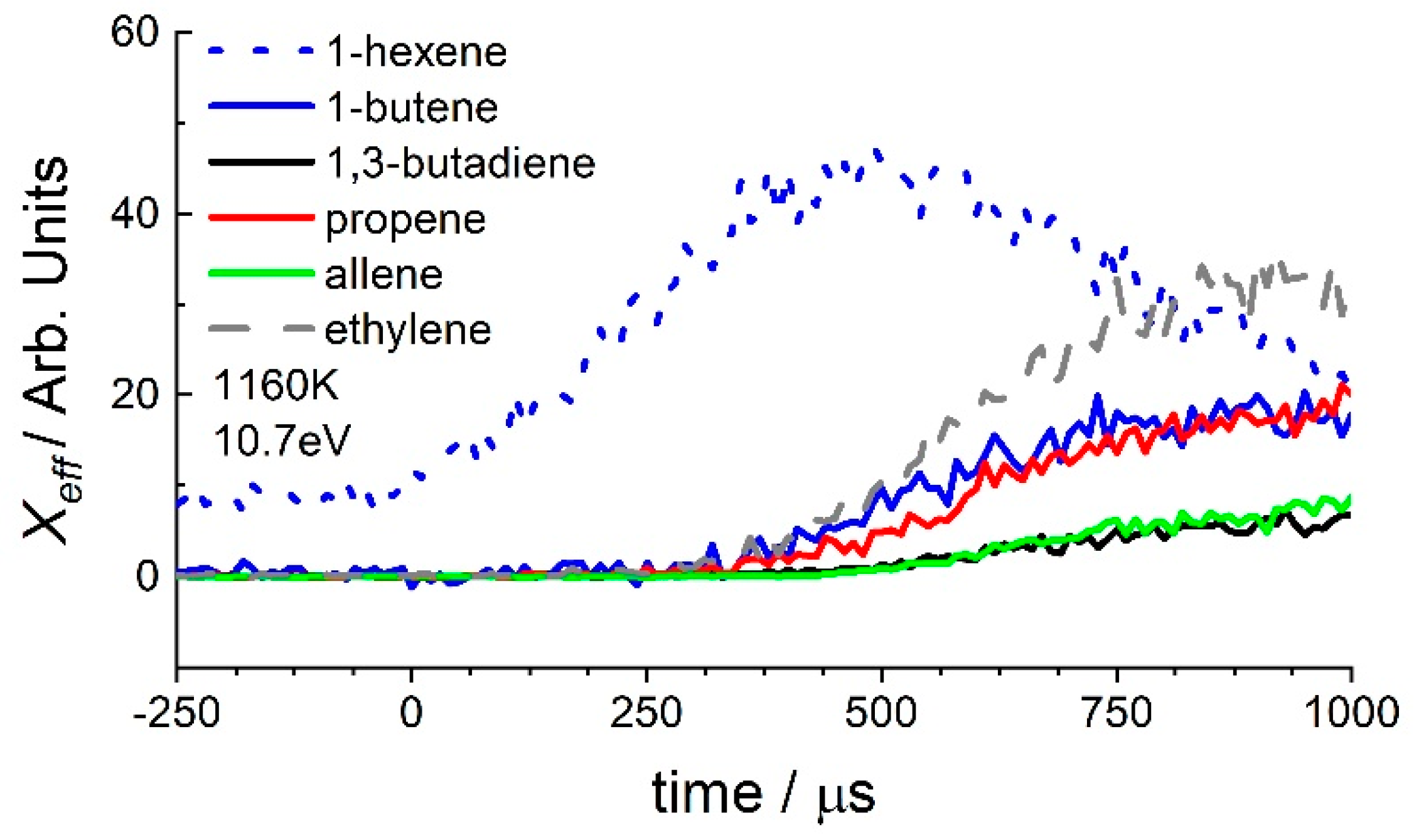
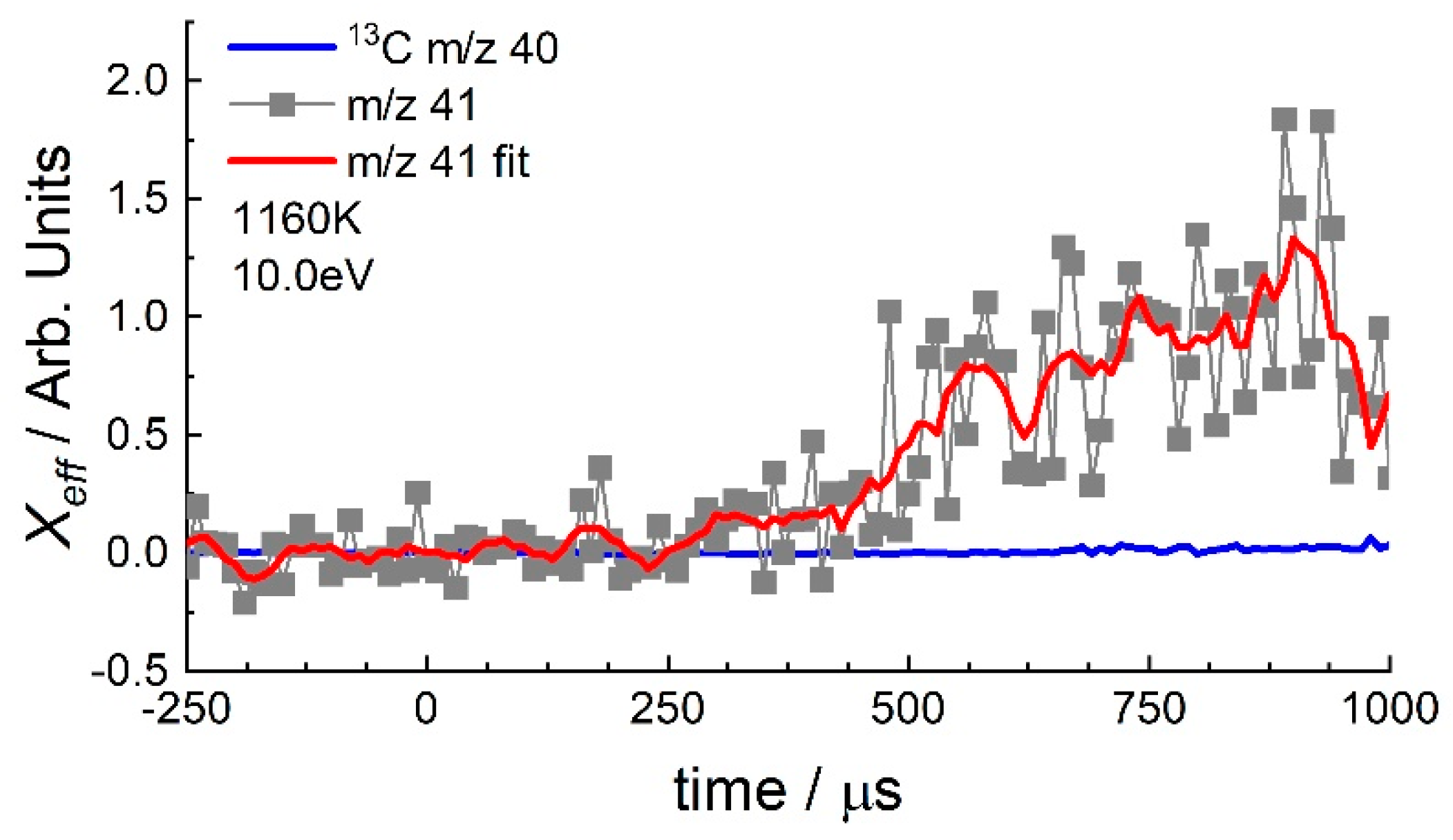
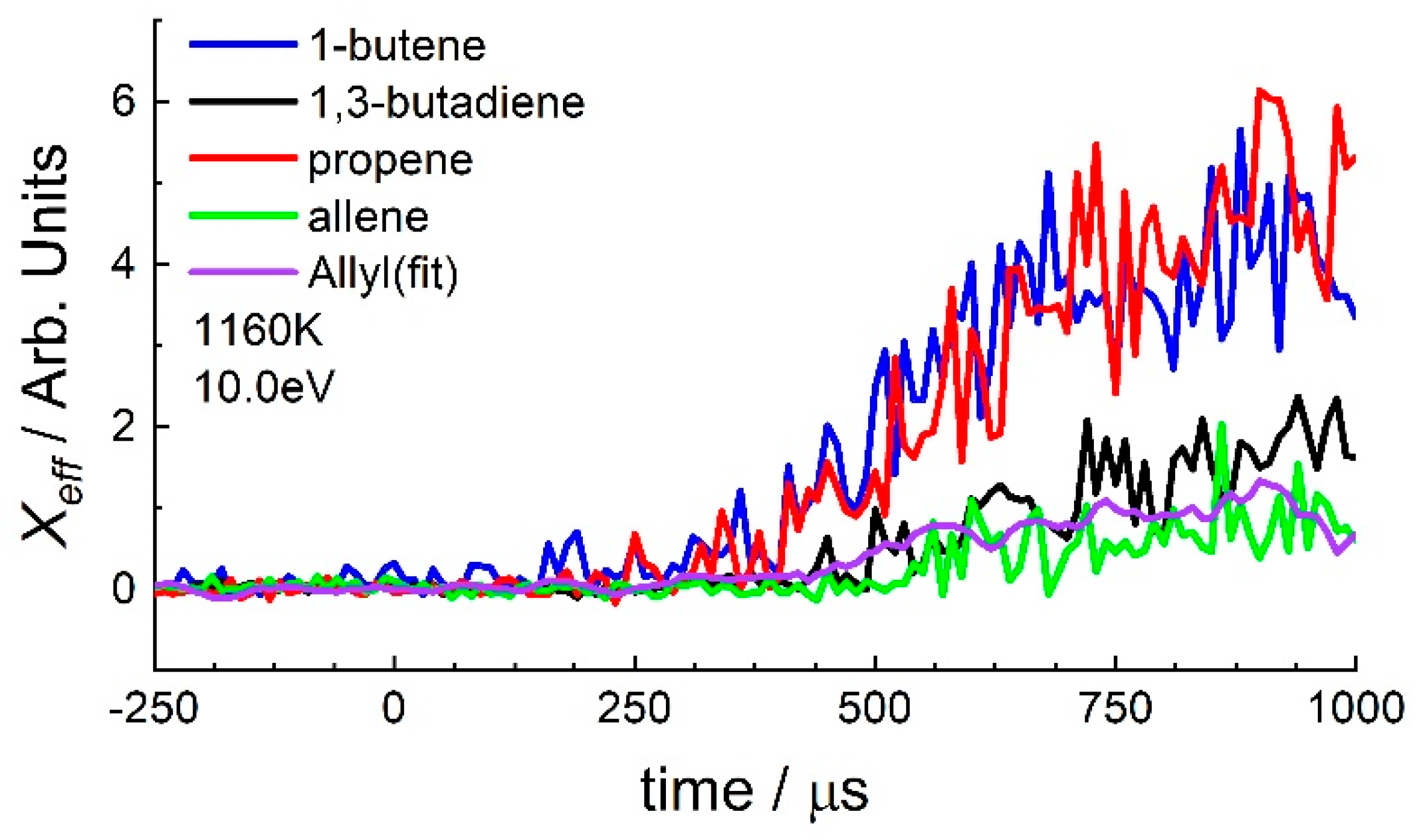
3.2. 1-Hexene
4. Discussion
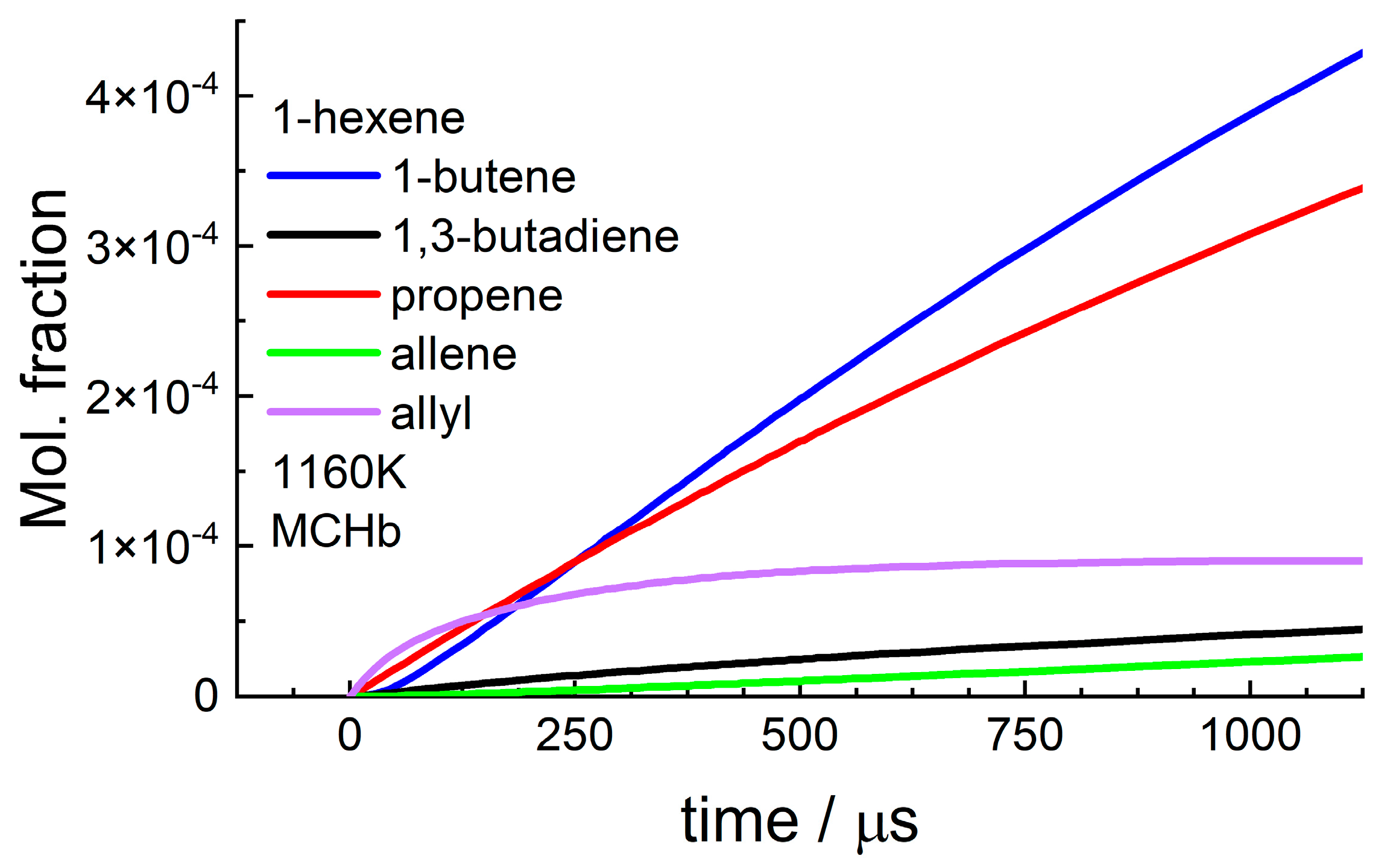
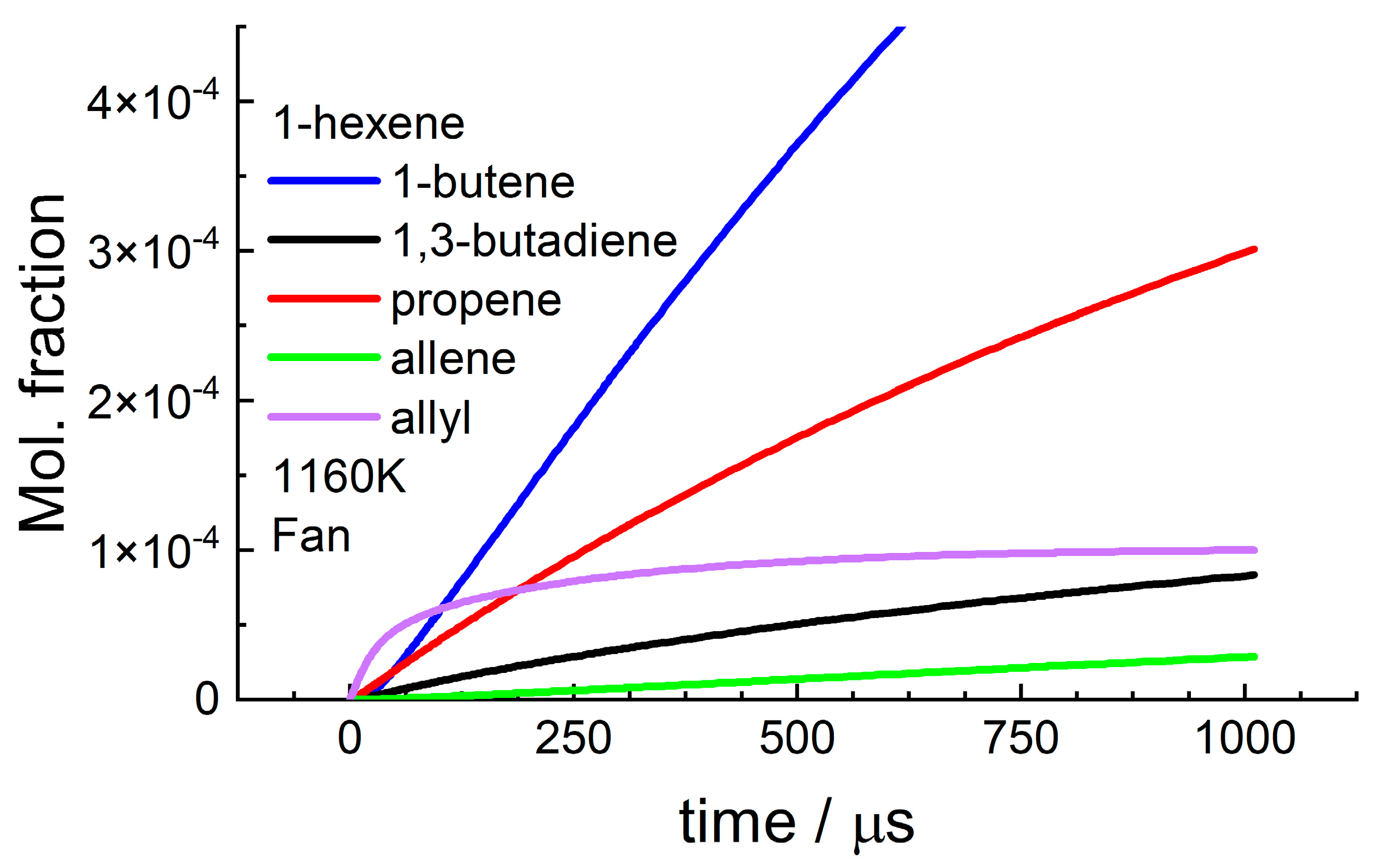
4.1. 1-Hexene
4.2. Cyclohexane
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, H.; Qiu, Y.; Wu, Z.; Wang, S.; Lu, X.; Huang, Z. Ignition delay of diisobutylene-containing multicomponent gasoline surrogates: Shock tube measurements and modeling study. Fuel 2019, 235, 1387–1399. [Google Scholar] [CrossRef]
- Ren, S.; Kokjohn, S.L.; Wang, Z.; Liu, H.; Wang, B.; Wang, J. A multi-component wide distillation fuel (covering gasoline, jet fuel and diesel fuel) mechanism for combustion and PAH prediction. Fuel 2017, 208, 447–468. [Google Scholar] [CrossRef]
- Pitz, W.J.; Mueller, C.J. Recent progress in the development of diesel surrogate fuels. Prog. Energy Combust. Sci. 2011, 37, 330–350. [Google Scholar] [CrossRef]
- Tsang, W. Thermal Stability of Cyclohexane and 1-Hexene. Int. J. Chem. Kinet. 1978, 10, 1119–1138. [Google Scholar] [CrossRef]
- Brown, T.C.; King, K.D.; Nguyen, T.T. Kinetics of primary processes in the pyrolysis of cyclopentanes and cyclohexanes. J. Phys. Chem. 1986, 90, 419–424. [Google Scholar] [CrossRef]
- Kiefer, J.H.; Gupte, K.S.; Harding, L.B.; Klippenstein, S.J. Shock tube and theory investigation of cyclohexane and 1-hexene decomposition. J. Phys. Chem. A 2009, 113, 13570–13583. [Google Scholar] [CrossRef] [PubMed]
- Peukert, S.; Naumann, C.; Braun-Unkhoff, M.; Riedel, U. The reaction of cyclohexane with H-atoms: A shock tube and modeling study. Int. J. Chem. Kinet. 2012, 44, 130–146. [Google Scholar] [CrossRef]
- Wang, Z.; Cheng, Z.; Yuan, W.; Cai, J.; Zhang, L.; Zhang, F.; Qi, F.; Wang, J. An experimental and kinetic modeling study of cyclohexane pyrolysis at low pressure. Combust. Flame 2012, 159, 2243–2253. [Google Scholar] [CrossRef]
- Khandavilli, M.V.; Djokic, M.; Vermeire, F.H.; Carstensen, H.-H.; Van Geem, K.M.; Marin, G.B. Experimental and Kinetic Modeling Study of Cyclohexane Pyrolysis. Energy Fuels 2018, 32, 7153–7168. [Google Scholar] [CrossRef]
- Liszka, M.K.; Brezinsky, K. Variable high-pressure and concentration study of cyclohexane pyrolysis at high temperatures. Int. J. Chem. Kinet. 2019, 51, 49–73. [Google Scholar] [CrossRef]
- Fan, X.; Wang, G.; Li, Y.; Wang, Z.; Yuan, W.; Zhao, L. Experimental and kinetic modeling study of 1-hexene combustion at various pressures. Combust. Flame 2016, 173, 151–160. [Google Scholar] [CrossRef]
- Sirjean, B.; Glaude, P.A.; Ruiz-Lopez, M.F.; Fournet, R. Detailed Kinetic Study of the Ring Opening of Cycloalkanes by CBS-QB3 Calculations. J. Phys. Chem. A 2006, 110, 12693–12704. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.-M.; Li, Z.-R.; Li, X.-Y. Theoretical Kinetic Study of Thermal Decomposition of Cyclohexane. Energy Fuels 2012, 26, 2811–2820. [Google Scholar] [CrossRef]
- Aribike, D.S.; Susu, A.A.; Ogunye, A.F. Mechanistic and mathematical modeling of the thermal decompostion of cyclohexane. Thermochim. Acta 1981, 51, 113–127. [Google Scholar] [CrossRef]
- Zsély, I.G.G.; Varga, T.; Nagy, T.; Cserháti, M.; Turányi, T.; Peukert, S.; Braun-Unkhoff, M.; Naumann, C.; Riedel, U. Determination of rate parameters of cyclohexane and 1-hexene decomposition reactions. Energy 2012, 43, 85–93. [Google Scholar] [CrossRef]
- Knepp, A.M.; Meloni, G.; Jusinski, L.E.; Taatjes, C.A.; Cavalotti, C.; Klippenstein, S.J. Theory, measurements, and modeling of OH and HO2 formation in the reaction of cyclohexyl radicals with O2. Phys. Chem. Chem. Phys. 2007, 9, 4315–4331. [Google Scholar] [CrossRef]
- Wang, Z.; Ye, L.; Yuan, W.; Zhang, L.; Wang, Y.; Cheng, Z.; Zhang, F.; Qi, F. Experimental and kinetic modeling study on methylcyclohexane pyrolysis and combustion. Combust. Flame 2014, 161, 84–100. [Google Scholar] [CrossRef]
- Sikes, T.; Banyon, C.; Schwind, R.A.; Lynch, P.T.; Comandini, A.; Sivaramakrishnan, R.; Tranter, R.S. Initiation reactions in the high temperature decomposition of styrene. Phys. Chem. Chem. Phys. 2021, 23, 18432–18448. [Google Scholar] [CrossRef]
- Tranter, R.S.; Lynch, P.T. A miniature high repetition rate shock tube. Rev. Sci. Instrum. 2013, 84, 094102. [Google Scholar] [CrossRef]
- Lynch, P.T.; Troy, T.P.; Ahmed, M.; Tranter, R.S. Probing combustion chemistry in a miniature shock tube with synchrotron VUV photo ionization mass spectrometry. Anal. Chem. 2015, 87, 2345–2352. [Google Scholar] [CrossRef]
- Randazzo, J.B.; Sivaramakrishnan, R.; Jasper, A.W.; Sikes, T.; Lynch, P.T.; Tranter, R.S. An experimental and theoretical study of the high temperature reactions of the four butyl radical isomers. Phys. Chem. Chem. Phys. 2020, 22, 18304–18319. [Google Scholar] [CrossRef] [PubMed]
- Banyon, C.; Sikes, T.; Tranter, R.S. Reactions of propyl radicals: A shock tube–VUV photoionization mass spectrometry study. Combust. Flame 2021, 224, 14–23. [Google Scholar] [CrossRef]
- Gaydon, A.G.; Hurle, I.R. The Shock Tube in High-Temperature Chemical Physics; Chapman and Hall: London, UK, 1963. [Google Scholar]
- Anderson, J.D. Modern Compressible Flow: With Historical Perspective, 3rd ed.; McGraw-Hill Higher Education: Chicago, IL, USA, 2003. [Google Scholar]
- Bandyopadhyay, B.; Kostko, O.; Fang, Y.; Ahmed, M. Probing Methanol Cluster Growth by Vacuum Ultraviolet Ionization. J. Phys. Chem. A 2015, 119, 4083–4092. [Google Scholar] [CrossRef] [PubMed]
- Cool, T.A.; McIlroy, A.; Qi, F.; Westmoreland, P.R.; Poisson, L.; Peterka, D.S.; Ahmed, M. Photoionization mass spectrometer for studies of flame chemistry with a synchrotron light source. Rev. Sci. Instrum. 2005, 76, 094102. [Google Scholar] [CrossRef]
- Yang, B.; Wang, J.; Cool, T.A.; Hansen, N.; Skeen, S.; Osborn, D.L. Absolute photoionization cross-sections of some combustion intermediates. Int. J. Mass Spectrom. 2012, 309, 118–128. [Google Scholar] [CrossRef]
- Person, J.C.; Nicole, P.P. Isotope Effects in the Photoionization Yields and in the Absorption Cross Sections for Methanol, Ethanol, Methyl Bromide, and Ethyl Bromide. J. Chem. Phys. 2003, 55, 3390–3397. [Google Scholar] [CrossRef]
- Robinson, J.C.; Sveum, N.E.; Neumark, D.M. Determination of absolute photoionization cross sections for isomers of C3H5: Allyl and 2-propenyl radicals. Chem. Phys. Lett. 2004, 383, 601–605. [Google Scholar] [CrossRef]
- Holland, D.M.P.; Shaw, D.A. A study of the valence-shell photoabsorption, photodissociation and photoionisation cross-sections of allene. Chem. Phys. 1999, 243, 333–339. [Google Scholar] [CrossRef]
- Zhou, Z.; Xie, M.; Wang, Z.; Qi, F. Determination of absolute photoionization cross-sections of aromatics and aromatic derivatives. Rapid Commun. Mass Spectrom. 2009, 23, 3994–4002. [Google Scholar] [CrossRef]
- Lias, S.G.; Bartmess, J.E.; Liebman, J.F.; Holmes, J.L.; Levin, R.D.; Mallard, W.G. Ion Energetics Data. In NIST Chemistry WebBook, NIST Standard Reference Database Number 69; Linstrom, J., Mallard, W.G., Eds.; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2023. [Google Scholar] [CrossRef]
- Demeo, D.A.; El-Sayed, M.A. Ionization Potential and Structure of Olefins. J. Chem. Phys. 2003, 52, 2622–2629. [Google Scholar] [CrossRef]
- Li, Y. Available online: http://flame.nsrl.ustc.edu.cn/database/data.php?wid=346 (accessed on 1 June 2023).
- Wang, J.; Yang, B.; Cool, T.A.; Hansen, N.; Kasper, T. Near-threshold absolute photoionization cross-sections of some reaction intermediates in combustion. Int. J. Mass Spectrom. 2008, 269, 210–220. [Google Scholar] [CrossRef]
- Cool, T.A.; Nakajima, K.; Mostefaoui, T.A.; Qi, F.; McIlroy, A.; Westmoreland, P.R.; Law, M.E.; Poisson, L.; Peterka, D.S.; Ahmed, M. Selective detection of isomers with photoionization mass spectrometry for studies of hydrocarbon flame chemistry. J. Chem. Phys. 2003, 119, 8356–8365. [Google Scholar] [CrossRef]
- Schoen, R.I. Absorption, Ionization, and Ion-Fragmentation Cross Sections of Hydrocarbon Vapors under Vacuum-Ultraviolet Radiation. J. Chem. Phys. 1968, 37, 2032–2041. [Google Scholar] [CrossRef]
- Holland, D.M.P.; Shaw, D.A.; Hayes, M.A.; Shpinkova, L.G.; Rennie, E.E.; Karlsson, L.; Baltzer, P.; Wannberg, B. A photoabsorption, photodissociation and photoelectron spectroscopy study of C2H4 and C2D4. Chem. Phys. 1997, 219, 91–116. [Google Scholar] [CrossRef]
- Dalmiya, A.; Mehta, J.M.; Tranter, R.S.; Lynch, P.T. High pressure, high flow rate batch mixing apparatus for high throughput experiments. Rev. Sci. Instrum. 2021, 92, 114104. [Google Scholar] [CrossRef] [PubMed]
- Sikes, T.; Tranter, R.S. Frhodo: A program for simulating chemical kinetic measurements and optimizing kinetic mechanisms. Combust. Flame 2022, 257, 112509. [Google Scholar] [CrossRef]
- Wang, Q.-D. Theoretical studies of unimolecular thermal decomposition reactions of n-hexane and n-hexene isomers. Comput. Theor. Chem. 2017, 1115, 45–55. [Google Scholar] [CrossRef]
- Lockhart, J.P.A.; Goldsmith, C.F.; Randazzo, J.B.; Ruscic, B.; Tranter, R.S. An Experimental and Theoretical Study of the Thermal Decomposition of C4H6 Isomers. J. Phys. Chem. A 2017, 121, 3827–3850. [Google Scholar] [CrossRef]
- Miller, J.A.; Klippenstein, S.J. Dissociation of propyl radicals and other reactions on a C3H7 potential. J. Phys. Chem. A 2013, 117, 2718–2727. [Google Scholar] [CrossRef]
- Goodwin, D.G.; Speth, R.L.; Moffat, H.K.; Weber, B.W. Cantera: An Object-Oriented Software Toolkit for Chemical Kinetics, Thermodynamics, and Transport Processes, Version 2.6.0. 2022. Available online: https://zenodo.org/records/6387882 (accessed on 1 June 2023).
- PyDot. Available online: https://pypi.org/project/pydot/ (accessed on 1 June 2023).
- GraphViz. Available online: https://www.graphviz.org/ (accessed on 1 June 2023).
- Kiefer, J.H.; Mudipalli, P.S.; Sidhu, S.S.; Kern, R.D.; Jursic, B.S.; Xie, K.; Chen, H. Unimolecular Dissociation in Allene and Propyne: The Effect of Isomerization on the Low-Pressure Rate. J. Phys. Chem. A 1997, 101, 4057–4071. [Google Scholar] [CrossRef]
- Houle, F.A.; Beauchamp, J.L. Detection and investigation of allyl and benzyl radicals by photoelectron spectroscopy. J. Am. Chem. Soc. 1978, 100, 3290–3294. [Google Scholar] [CrossRef]
- Tsang, W. Chemical kinetic database for combustion chemistry part V. Propene. J. Phys. Chem. Ref. Data 1991, 20, 222–274. [Google Scholar] [CrossRef]
- Hansen, N.; Merchant, S.S.; Harper, M.R.; Green, W.H. The predictive capability of an automatically generated combustion chemistry mechanism: Chemical structures of premixed iso-butanol flames. Combust. Flame 2013, 160, 2343–2351. [Google Scholar] [CrossRef]
- Fernandes, R.X.; Girl, B.R.; Hippler, H.; Kachiani, C.; Striebel, F. Shock wave study on the thermal unimolecular decomposition of allyl radicals. J. Phys. Chem. A 2005, 109, 1063–1070. [Google Scholar] [CrossRef]
- Lynch, P.T.; Annesley, C.J.; Aul, C.J.; Yang, X.; Tranter, R.S. Recombination of allyl radicals in the high temperature fall-off regime. J. Phys. Chem. A 2013, 117, 4750–4761. [Google Scholar] [CrossRef] [PubMed]
- Georgievskii, Y.; Miller, J.A.; Klippenstein, S.J. Association rate constants for reactions between resonance-stabilized radicals: C3H3 + C3H3, C3H3 + C3H5, and C3H5 + C3H5. Phys. Chem. Chem. Phys. 2007, 9, 4259. [Google Scholar] [CrossRef]
- Harding, L.B.; Klippenstein, S.J.; Georgievskii, Y. On the combination reactions of hydrogen atoms with resonance-stabilized hydrocarbon radicals. J. Phys. Chem. A 2007, 111, 3789–3801. [Google Scholar] [CrossRef]
- Knyazev, V.D.; Slagle, I.R. Kinetics of the Reactions of Allyl and Propargyl Radicals with CH3. J. Phys. Chem. A 2001, 105, 3196–3204. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T5/K (Nominal) | T5/K (Mean) | σ T5/K a | P5/bar | σ P5/bar a | MgF2 Filter |
|---|---|---|---|---|---|
| 1270 | 1264 | 16.37 | 6.87 | 0.15 | N |
| 1277 | 15.60 | 6.96 | 0.15 | Y | |
| 1340 | 1333 | 14.30 | 7.46 | 0.14 | N |
| 1340 | 13.04 | 7.54 | 0.13 | Y | |
| 1400 | 1394 | 16.06 | 8.01 | 0.15 | N |
| 1408 | 16.73 | 8.12 | 0.16 | Y | |
| 1480 | 1487 | 17.60 | 8.84 | 0.18 | N |
| 1472 | 18.64 | 8.71 | 0.18 | Y | |
| 1550 | 1541 | 20.43 | 9.39 | 0.20 | N |
| 1563 | 20.96 | 9.56 | 0.20 | Y |
| T5/K (Nominal) | T5/K (Mean) | σ T5/K a | P5/bar | σ P5/bar a |
|---|---|---|---|---|
| 1160 | 1157 | 19.7 | 4.41 | 0.14 |
| 1360 | 1364 | 16.76 | 5.80 | 0.13 |
| 1470 | 1445 | 17.97 | 6.39 | 0.14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tranter, R.S.; Banyon, C.; Hawtof, R.E.; Kim, K. Pyrolysis of Cyclohexane and 1-Hexene at High Temperatures and Pressures—A Photoionization Mass Spectrometry Study. Energies 2023, 16, 7929. https://doi.org/10.3390/en16247929
Tranter RS, Banyon C, Hawtof RE, Kim K. Pyrolysis of Cyclohexane and 1-Hexene at High Temperatures and Pressures—A Photoionization Mass Spectrometry Study. Energies. 2023; 16(24):7929. https://doi.org/10.3390/en16247929
Chicago/Turabian StyleTranter, Robert S., Colin Banyon, Ryan E. Hawtof, and Keunsoo Kim. 2023. "Pyrolysis of Cyclohexane and 1-Hexene at High Temperatures and Pressures—A Photoionization Mass Spectrometry Study" Energies 16, no. 24: 7929. https://doi.org/10.3390/en16247929
APA StyleTranter, R. S., Banyon, C., Hawtof, R. E., & Kim, K. (2023). Pyrolysis of Cyclohexane and 1-Hexene at High Temperatures and Pressures—A Photoionization Mass Spectrometry Study. Energies, 16(24), 7929. https://doi.org/10.3390/en16247929